
Carbon Microparticles from Organosolv Lignin as Filler for Conducting Poly(Lactic Acid)




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Lignin Carbonisation
2.3. Characterisation of Lignin and Carbon Particles
2.4. Preparation and Characterisation of Poly(Lactic Acid) Composites Containing Different Amounts of Lignin-Derived Carbon Microparticles
3. Results and Discussion
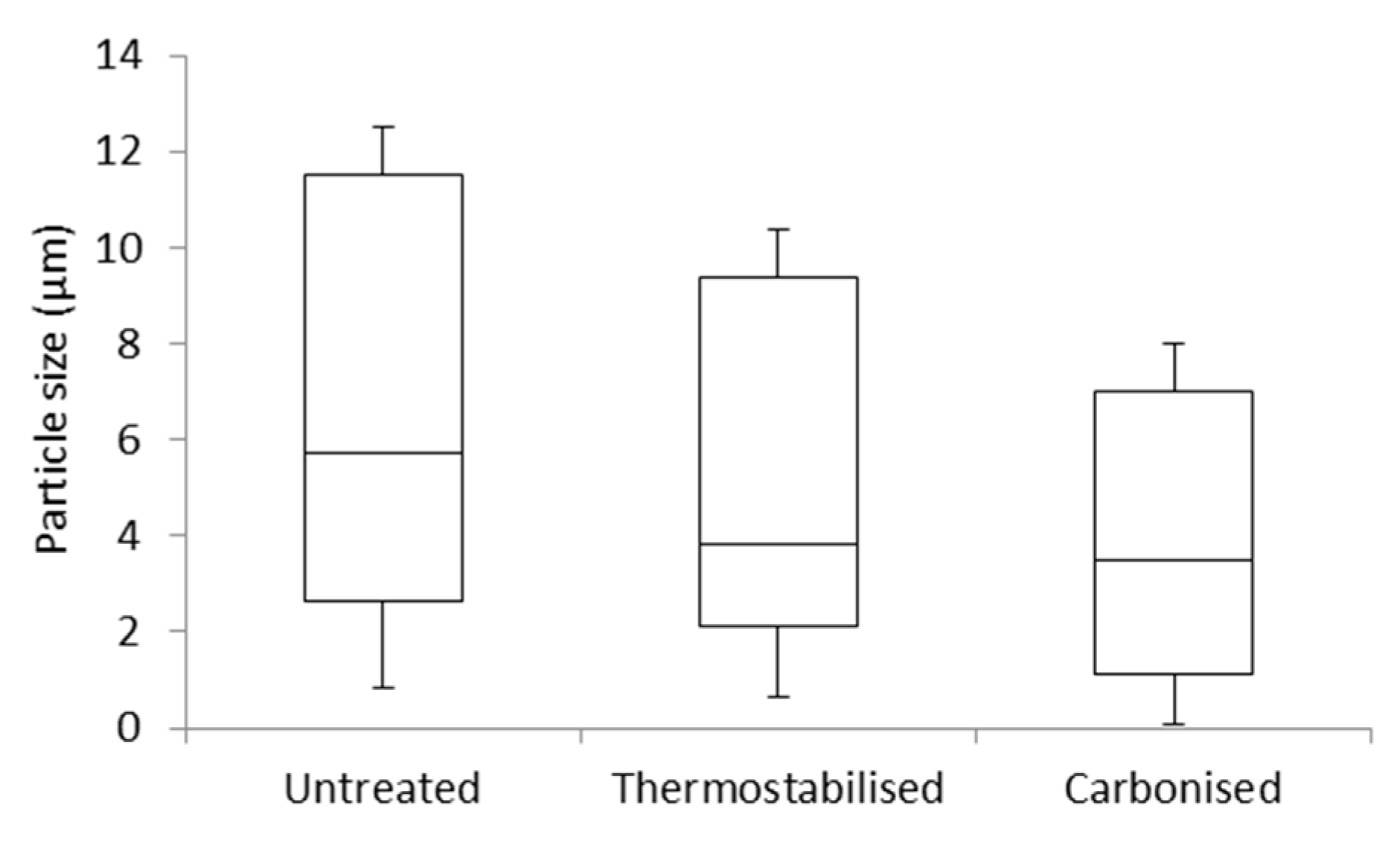
3.1. Scanning Electron Microscopy (SEM)
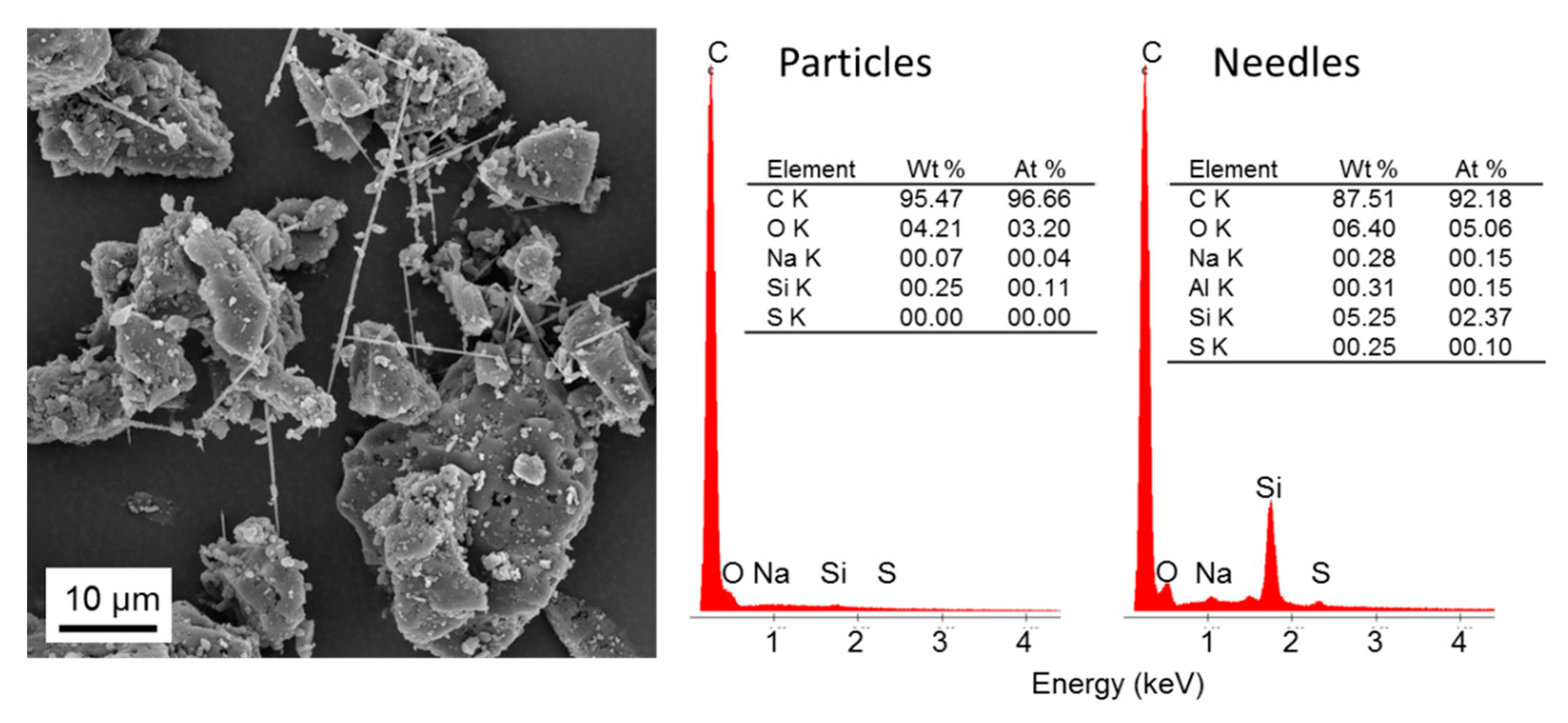
3.2. Energy-Dispersive X-ray Spectroscopy (EDX)
3.3. Attenuated Diffuse Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR)
3.4. Differential Scanning Calorimetry (DSC) and Thermogravinetric Analysis (TGA)
3.5. X-ray Powder Diffraction
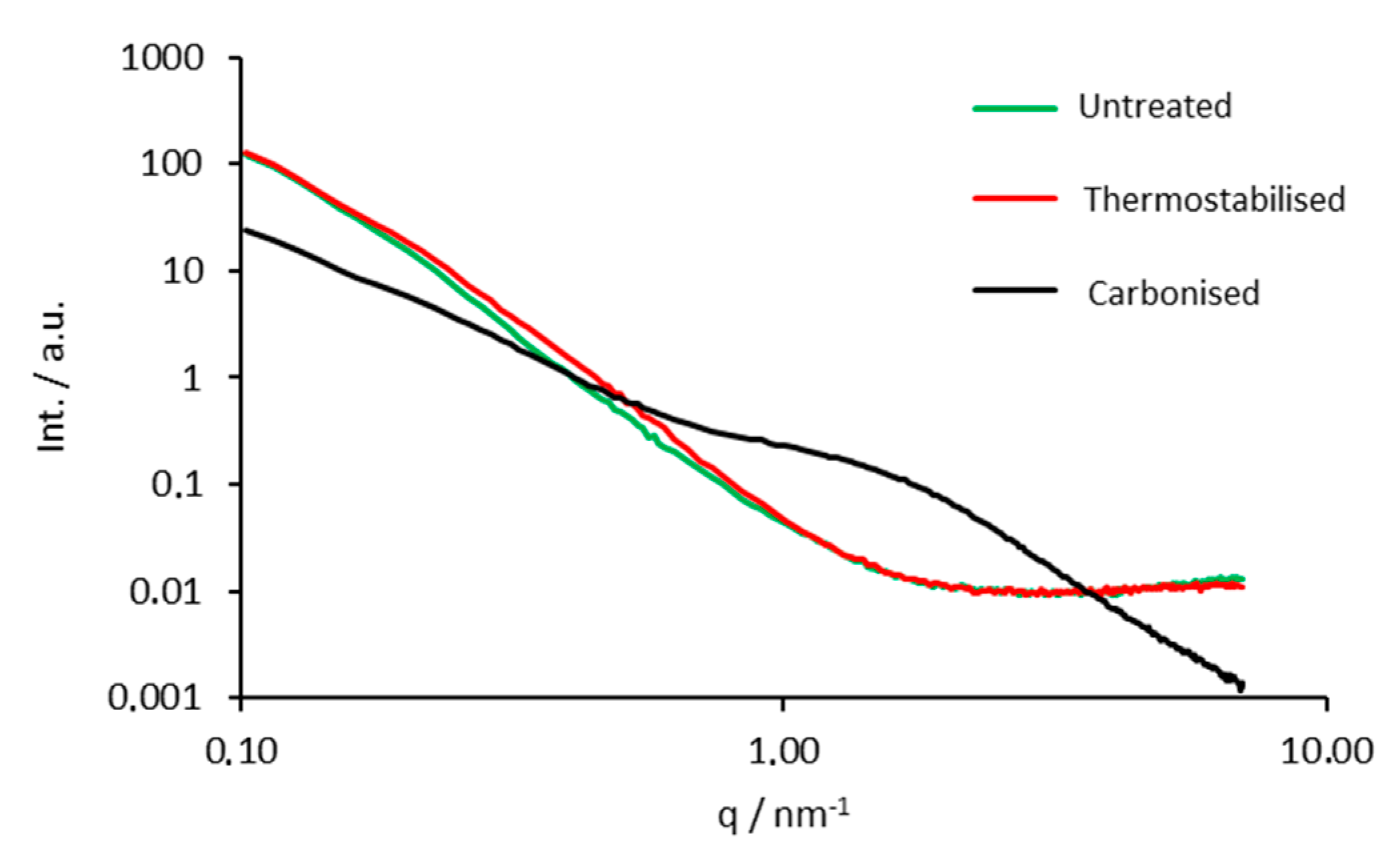
3.6. Small Angle X-ray Scattering
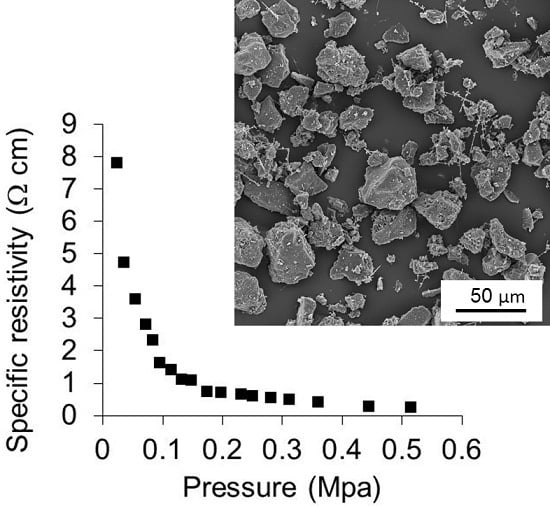
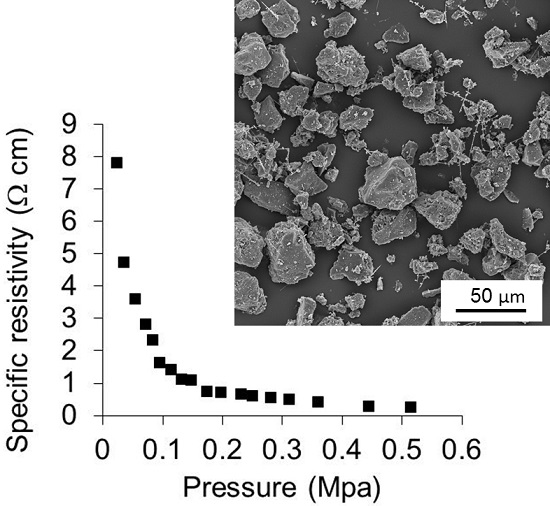
3.7. Specific Electrical Resistivity
3.8. Conducting Poly(Lactic Acid) (PLA) Carbon Composites
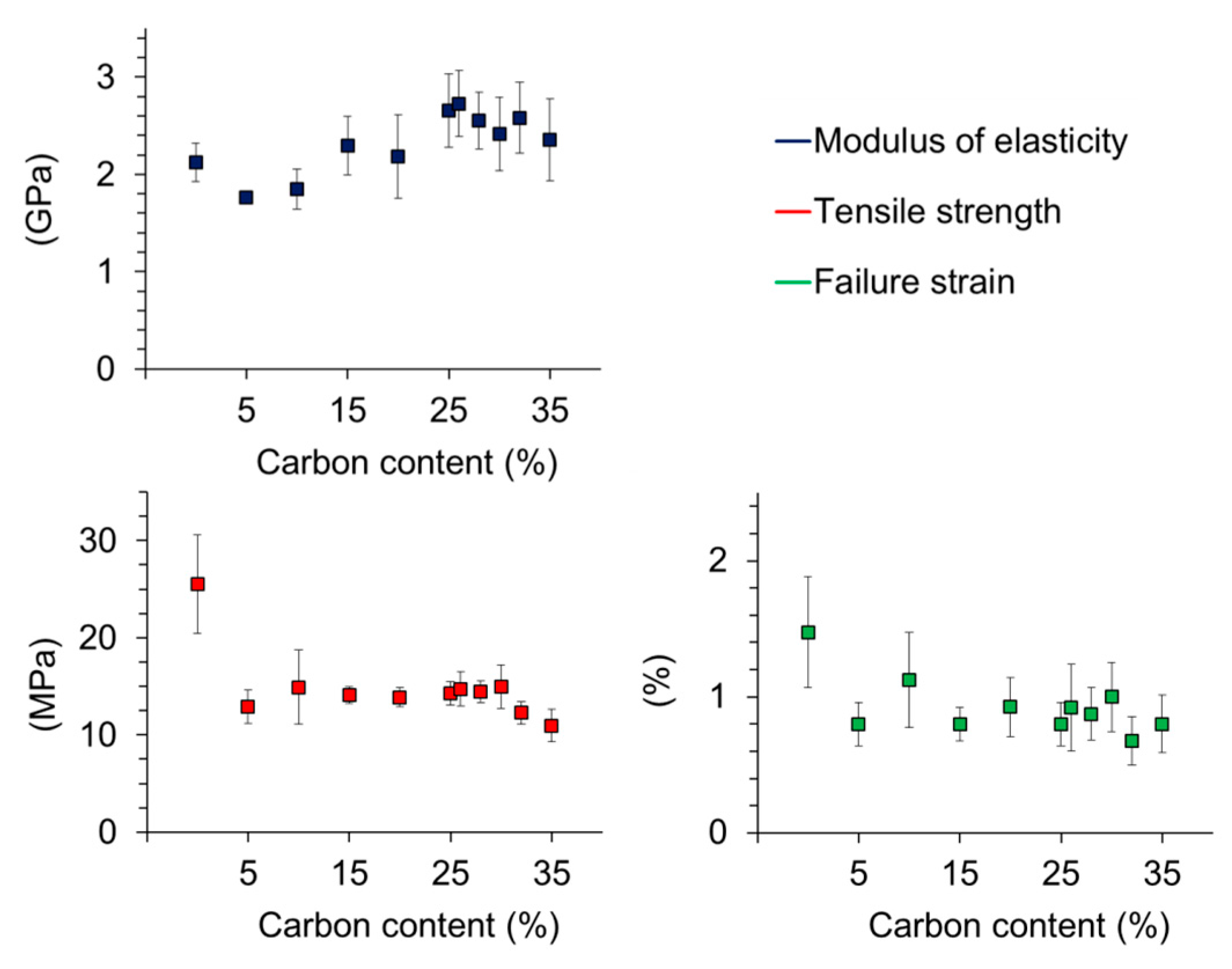
3.9. Mechanical Properties of Poly(Lactic Acid) (PLA) Carbon Composites
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El Mansouri, N.-E.; Salvadó, J. Analytical methods for determining functional groups in various technical lignins. Ind. Crop. Prod. 2007, 26, 116–124. [Google Scholar] [CrossRef]
- Belgacem, M.N.; Gandini, A. Monomers, Polymers and Composites from Renewable Resources; Elsevier Ltd.: London, UK, 2008. [Google Scholar]
- Lin, S.Y.; Dence, C.W. Methods in Lignin Chemistry; Springer: Berlin, Germany, 1992. [Google Scholar]
- Lora, J.H.; Glasser, W.G. Recent industrial applications of lignin: A sustainable alternative to nonrenewable materials. J. Polym. Environ. 2002, 10, 39–48. [Google Scholar] [CrossRef]
- Baker, D.A.; Rials, T.G. Recent advances in low-cost carbon fiber manufacture from lignin. J. Appl. Polym. Sci. 2013, 130, 713–728. [Google Scholar] [CrossRef]
- Chand, S. Carbon fibers for composites. J. Mater. Sci. 2000, 35, 1303–1313. [Google Scholar] [CrossRef]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D.; Compere, A.L.; Griffith, W. Lignin-based carbon fibers for composite fiber applications. Carbon 2002, 40, 2913–2920. [Google Scholar] [CrossRef]
- Ruiz-Rosas, R.; Bedia, J.; Lallave, M.; Loscertales, I.G.; Barrero, A.; Rodríguez-Mirasol, J.; Cordero, T. The production of submicron diameter carbon fibers by the electrospinning of lignin. Carbon 2010, 48, 696–705. [Google Scholar] [CrossRef]
- Balberg, I. A comprehensive picture of the electrical phenomena in carbon black-polymer composites. Carbon 2002, 40, 139–143. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, C.; Zhang, Y. Preparation and properties of styrene-butadiene rubber nanocomposites blended with carbon black-graphene hybrid filler. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Ma, L.-F.; Bao, R.-Y.; Huang, S.-L.; Liu, Z.-Y.; Yang, W.; Xie, B.-H.; Yang, M.-B. Electrical properties and morphology of carbon black filled PP/EPDM blends: Effect of selective distribution of fillers induced by dynamic vulcanization. J. Mater. Sci. 2013, 48, 4942–4951. [Google Scholar] [CrossRef]
- Feller, J.-F.; Petitjean, É. Conductive polymer composites(cpc): Influence of processing conditions, shear rate and temperature on electrical properties of poly(butylene terephthalate)/poly(amide12-b-tetramethyleneglycol)– carbon black blends. Macromol. Symp. 2003, 203, 309–316. [Google Scholar] [CrossRef]
- Schulze, P.; Seidel-Morgenstern, A.; Lorenz, H.; Leschinsky, M.; Unkelbach, G. Advanced process for precipitation of lignin from ethanol organosolv spent liquors. Bioresour. Technol. 2016, 199, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, N.-E.E.; Salvadó, J. Structural characterization of technical lignins for the production of adhesives: Application to lignosulfonate, kraft, soda-anthraquinone, organosolv and ethanol process lignins. Ind. Crops Prod. 2006, 24, 8–16. [Google Scholar] [CrossRef]
- Zhang, K.; Pei, Z.; Wang, D. Organic solvent pretreatment of lignocellulosic biomass for biofuels and biochemicals: A review. Bioresour. Technol. 2016, 199, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- El Hage, R.; Brosse, N.; Chrusciel, L.; Sanchez, C.; Sannigrahi, P.; Ragauskas, A. Characterization of milled wood lignin and ethanol organosolv lignin from miscanthus. Polym. Degrad. Stab. 2009, 94, 1632–1638. [Google Scholar] [CrossRef]
- Sánchez-González, J.; Macías-García, A.; Alexandre-Franco, M.F.; Gómez-Serrano, V. Electrical conductivity of carbon blacks under compression. Carbon 2005, 43, 741–747. [Google Scholar] [CrossRef]
- Gindl-Altmutter, W.; Fürst, C.; Mahendran, A.R.; Obersriebnig, M.; Emsenhuber, G.; Kluge, M.; Veigel, S.; Keckes, J.; Liebner, F. Electrically conductive kraft lignin-based carbon filler for polymers. Carbon 2015, 89, 161–168. [Google Scholar] [CrossRef]
- Dwiyanti, M.S.; Stewart, J.R.; Yamada, T. Forages for feedstocks of biorefineries in temperate environments: Review of lignin research in bioenergy crops and some insight into miscanthus studies. Crop Pasture Sci. 2014, 65, 1199–1206. [Google Scholar] [CrossRef]
- Kuo, D.T.F.; Vander Sande, J.B.; Gschwend, P.M. Characterization of black carbon in geosorbents at the nanometer scale by stem–edx elemental mapping. Org. Geochem. 2013, 56, 81–93. [Google Scholar] [CrossRef]
- Norberg, I.; Nordström, Y.; Drougge, R.; Gellerstedt, G.; Sjöholm, E. A new method for stabilizing softwood kraft lignin fibers for carbon fiber production. J. Appl. Polym. Sci. 2013, 128, 3824–3830. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wooten, J.B.; Baliga, V.L.; Lin, X.; Geoffrey Chan, W.; Hajaligol, M.R. Characterization of chars from pyrolysis of lignin. Fuel 2004, 83, 1469–1482. [Google Scholar] [CrossRef]
- Braun, J.L.; Holtman, K.M.; Kadla, J.F. Lignin-based carbon fibers: Oxidative thermostabilization of kraft lignin. Carbon 2005, 43, 385–394. [Google Scholar] [CrossRef]
- Kubo, S.; Kadla, J.F. Hydrogen bonding in lignin: A fourier transform infrared model compound study. Biomacromolecules 2005, 6, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Britt, P.F.; Buchanan, A.C., III; Malcolm, E.A. Thermolysis of phenethyl phenyl ether: A model for ether linkages in lignin and low rank coal. J. Org. Chem. 1995, 60, 6523–6536. [Google Scholar] [CrossRef]
- Nimz, H. Über eine neuartige umlagerung in der ligninchemie. Angew. Chem. 1966, 78, 821–822. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rye, C.S.; Gurnagul, N. Lignin inhibits autoxidative degradation of cellulose. Polym. Degrad. Stab. 1995, 49, 291–297. [Google Scholar] [CrossRef]
- Drbohlav, J.; Stevenson, W.T.K. The oxidative stabilization and carbonization of a synthetic mesophase pitch, part I: The oxidative stabilization process. Carbon 1995, 33, 693–711. [Google Scholar] [CrossRef]
- Drbohlav, J.; Stevenson, W.T.K. The oxidative stabilization and carbonization of a synthetic mesophase pitch, part II: The carbonization process. Carbon 1995, 33, 713–731. [Google Scholar] [CrossRef]
- Matsumoto, T.; Mochida, I. A structural study on oxidative stabilization of mesophase pitch fibers derived from coaltar. Carbon 1992, 30, 1041–1046. [Google Scholar] [CrossRef]
- Sahoo, S.; Seydibeyoğlu, M.Ö.; Mohanty, A.K.; Misra, M. Characterization of industrial lignins for their utilization in future value added applications. Biomass Bioenergy 2011, 35, 4230–4237. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, W. Synthesis of cationic starch with a high degree of substitution in an ionic liquid. Carbohydr. Polym. 2010, 80, 1172–1177. [Google Scholar] [CrossRef]
- Saito, T.; Perkins, J.H.; Vautard, F.; Meyer, H.M.; Messman, J.M.; Tolnai, B.; Naskar, A.K. Methanol fractionation of softwood kraft lignin: Impact on the lignin properties. Chemsuschem 2014, 7, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Q.; Lu, C.J.; Xia, Z.P.; Zhou, Y.; Luo, Z. X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 2007, 45, 1686–1695. [Google Scholar] [CrossRef]
- Foston, M.; Nunnery, G.A.; Meng, X.; Sun, Q.; Baker, F.S.; Ragauskas, A. NMR a critical tool to study the production of carbon fiber from lignin. Carbon 2013, 52, 65–73. [Google Scholar] [CrossRef]
- Pantea, D.; Darmstadt, H.; Kaliaguine, S.; Sümmchen, L.; Roy, C. Electrical conductivity of thermal carbon blacks. Carbon 2001, 39, 1147–1158. [Google Scholar] [CrossRef]
- Celzard, A.; Marêché, J.F.; Payot, F.; Furdin, G. Electrical conductivity of carbonaceous powders. Carbon 2002, 40, 2801–2815. [Google Scholar] [CrossRef]
- Chung, D.D.L. Electrical applications of carbon materials. J. Mater. Sci. 2004, 39, 2645–2661. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Qi, L.; Yang, J.; Xu, M.; Luo, X.; Ma, D. Polyurethane/conducting carbon black composites: Structure, electric conductivity, strain recovery behavior, and their relationships. J. Appl. Polym. Sci. 2000, 75, 68–77. [Google Scholar] [CrossRef]
- Ren, D.; Zheng, S.; Wu, F.; Yang, W.; Liu, Z.; Yang, M. Formation and evolution of the carbon black network in polyethylene/carbon black composites: Rheology and conductivity properties. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Tsuji, H.; Kawashima, Y.; Takikawa, H.; Tanaka, S. Poly(l-lactide)/nano-structured carbon composites: Conductivity, thermal properties, crystallization, and biodegradation. Polymer 2007, 48, 4213–4225. [Google Scholar] [CrossRef]
- Liu, X.; Kruckel, J.; Schubert, D.W. Building and testing a simple setup to investigate the electrical properties of polymer/carbon black composites. Compos. Struct. 2015, 129, 55–59. [Google Scholar] [CrossRef]
- Li, C.Y.; Thostenson, E.T.; Chou, T.W. Dominant role of tunneling resistance in the electrical conductivity of carbon nanotube-based composites. Appl. Phys. Lett. 2007, 91. [Google Scholar] [CrossRef]
- Huang, J.-C. Carbon black filled conducting polymers and polymer blends. Adv. Polym. Technol. 2002, 21, 299–313. [Google Scholar] [CrossRef]
- Nerantzaki, M.; Papageorgiou, G.Z.; Bikiaris, D.N. Effect of nanofiller’s type on the thermal properties and enzymatic degradation of poly(ε-caprolactone). Polym. Degrad. Stab. 2014, 108, 257–268. [Google Scholar] [CrossRef]
- Kim, K.H.; Jo, W.H. A strategy for enhancement of mechanical and electrical properties of polycarbonate/multi-walled carbon nanotube composites. Carbon 2009, 47, 1126–1134. [Google Scholar] [CrossRef]
- Fu, S.-Y.; Feng, X.-Q.; Lauke, B.; Mai, Y.-W. Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate–polymer composites. Compos. B Eng. 2008, 39, 933–961. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | n | P/Q (m2·cm−3) | R/(nm) |
|---|---|---|---|
| untreated | −3.85(5) | 97(20) | - |
| thermostabilised | −3.87(2) | 130(30) | - |
| carbonised | −3.45(2) | 770(50) | 1.06(4) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köhnke, J.; Fürst, C.; Unterweger, C.; Rennhofer, H.; Lichtenegger, H.C.; Keckes, J.; Emsenhuber, G.; Mahendran, A.R.; Liebner, F.; Gindl-Altmutter, W. Carbon Microparticles from Organosolv Lignin as Filler for Conducting Poly(Lactic Acid). Polymers 2016, 8, 205. https://doi.org/10.3390/polym8060205
Köhnke J, Fürst C, Unterweger C, Rennhofer H, Lichtenegger HC, Keckes J, Emsenhuber G, Mahendran AR, Liebner F, Gindl-Altmutter W. Carbon Microparticles from Organosolv Lignin as Filler for Conducting Poly(Lactic Acid). Polymers. 2016; 8(6):205. https://doi.org/10.3390/polym8060205
Chicago/Turabian StyleKöhnke, Janea, Christian Fürst, Christoph Unterweger, Harald Rennhofer, Helga C. Lichtenegger, Jozef Keckes, Gerhard Emsenhuber, Arunjunai Raj Mahendran, Falk Liebner, and Wolfgang Gindl-Altmutter. 2016. "Carbon Microparticles from Organosolv Lignin as Filler for Conducting Poly(Lactic Acid)" Polymers 8, no. 6: 205. https://doi.org/10.3390/polym8060205