
A Phase Field Technique for Modeling and Predicting Flow Induced Crystallization Morphology of Semi-Crystalline Polymers


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Modeling Nucleation
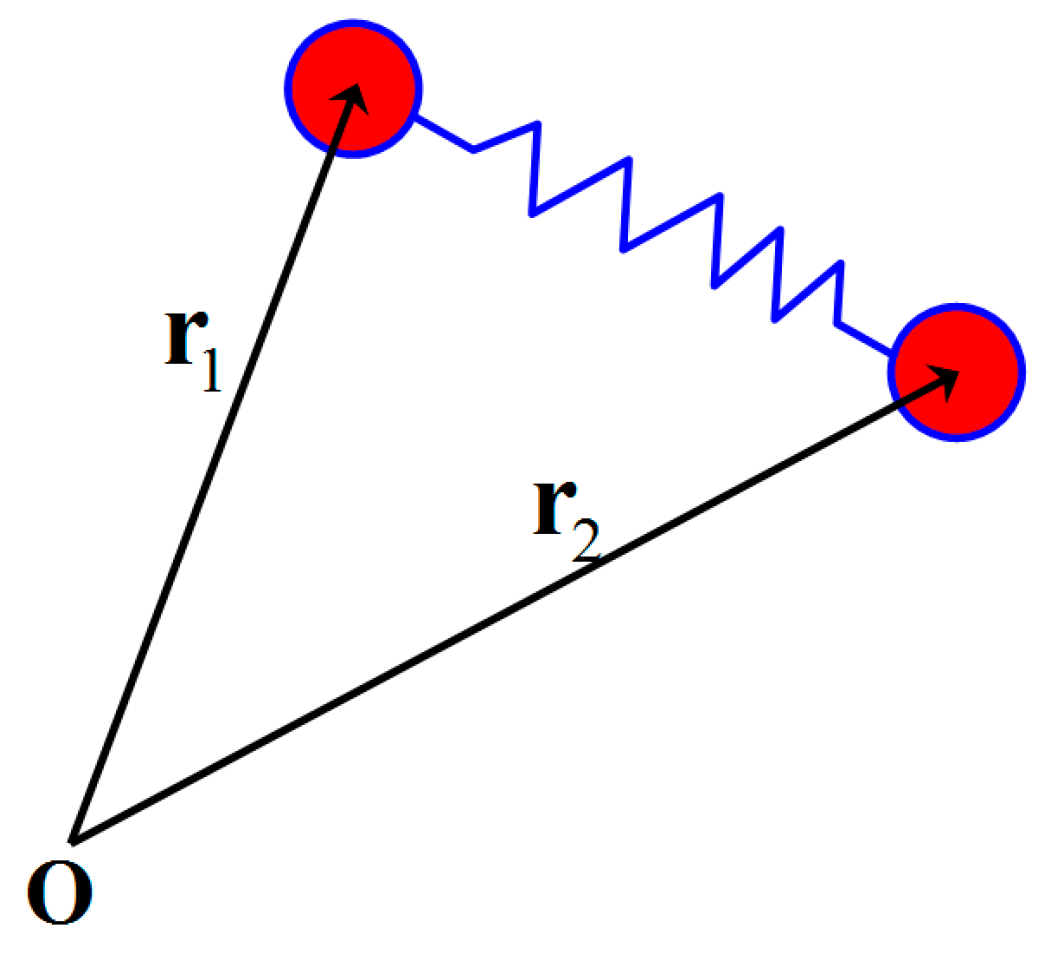
2.1. FENE-P Model for Describing Molecular Orientation and Stretch
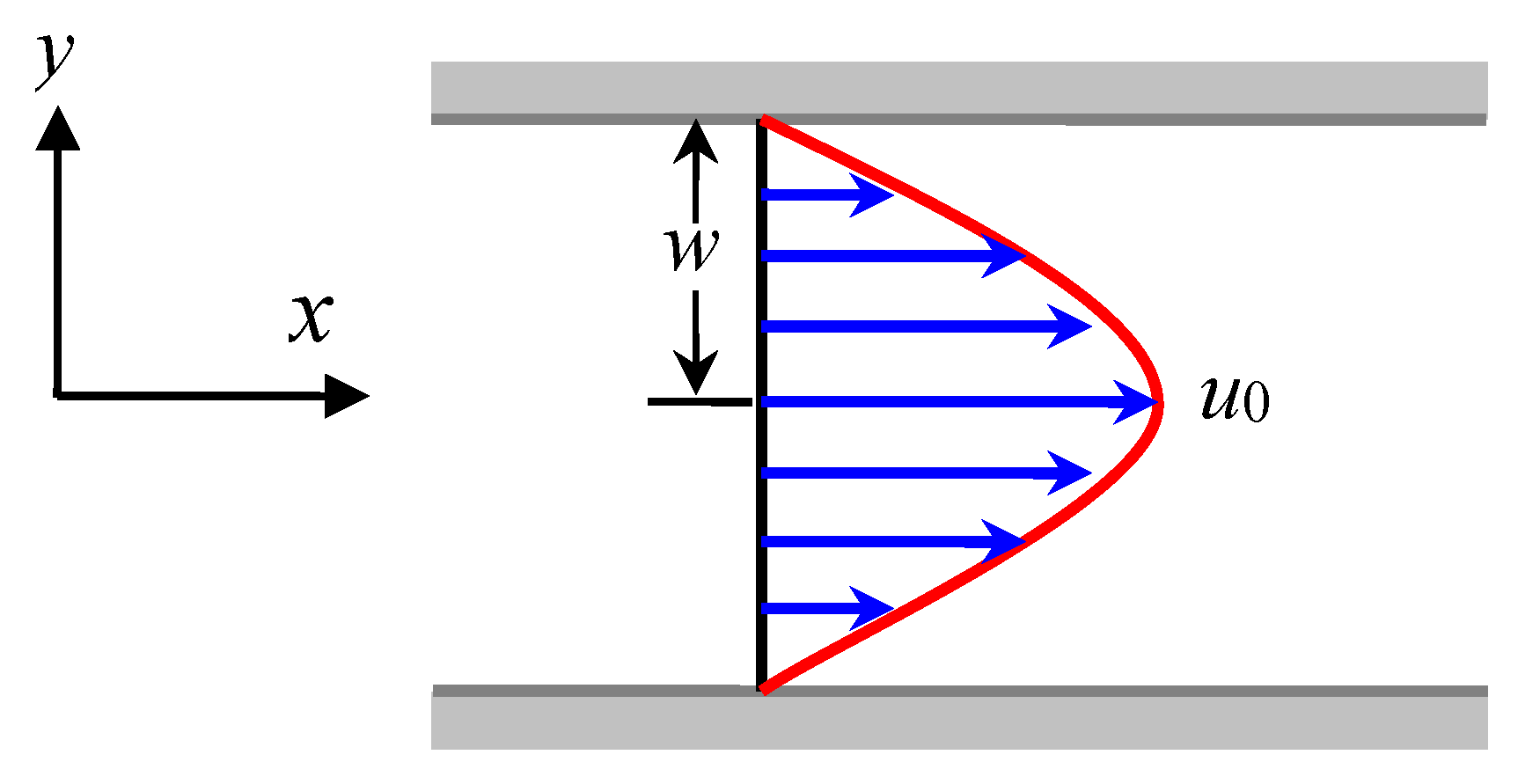
2.2. FENE-P Flow Model for Describing the Macroscopic Flow Field
2.3. Flow Induced Structures
2.4. Oriented Nucleus
3. Modeling Crystal Growth
3.1. Free Energy of the System
3.2. Phase Field Model
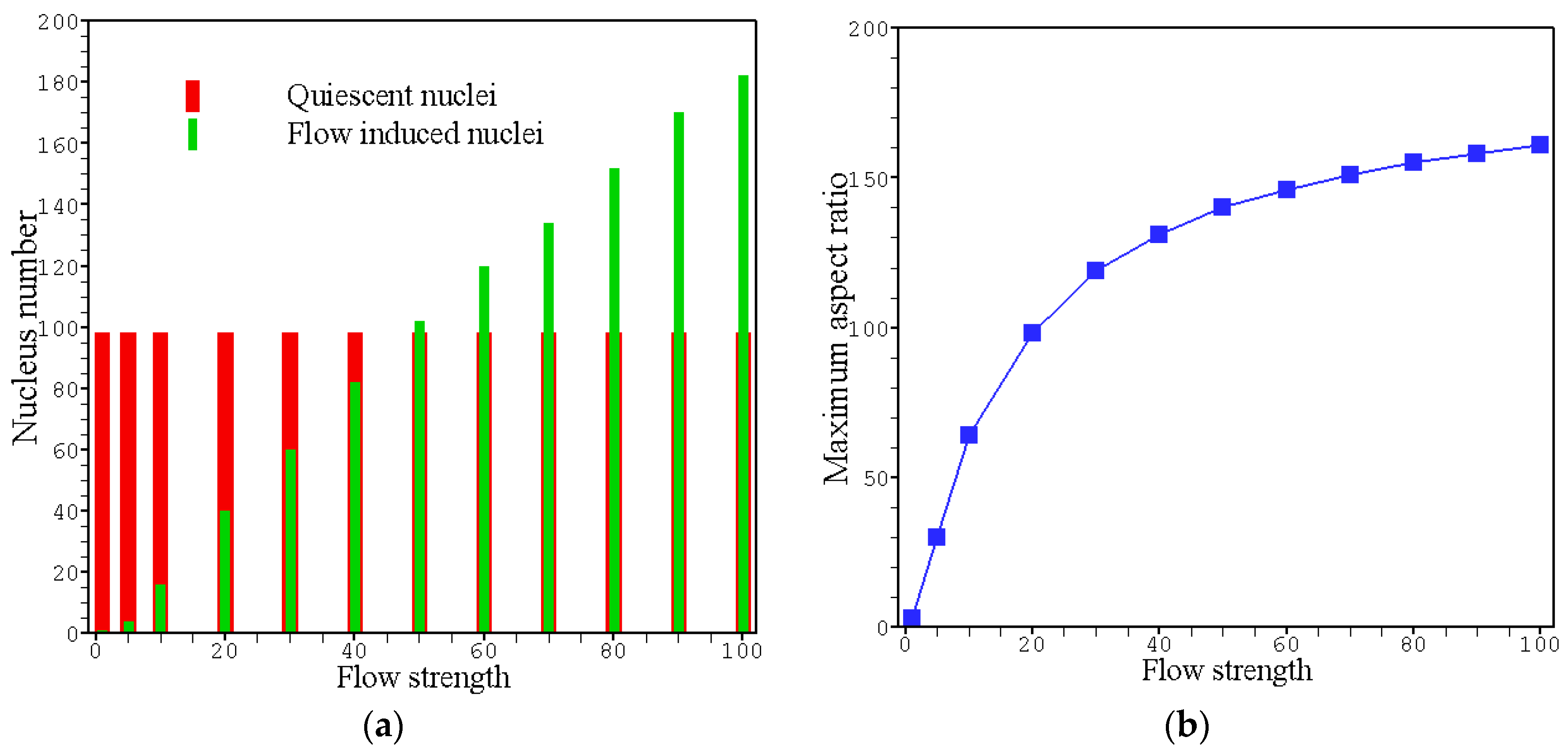
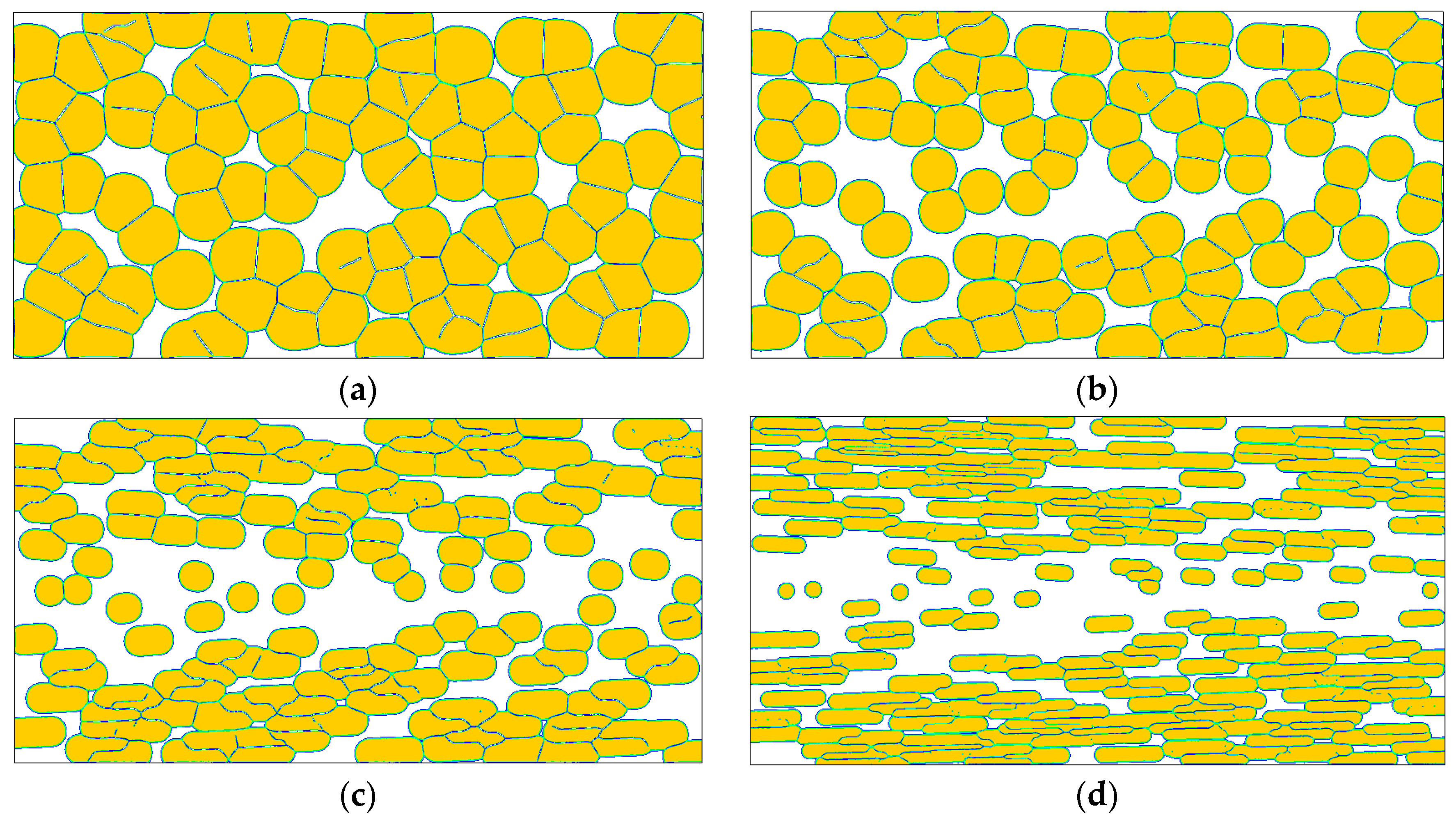
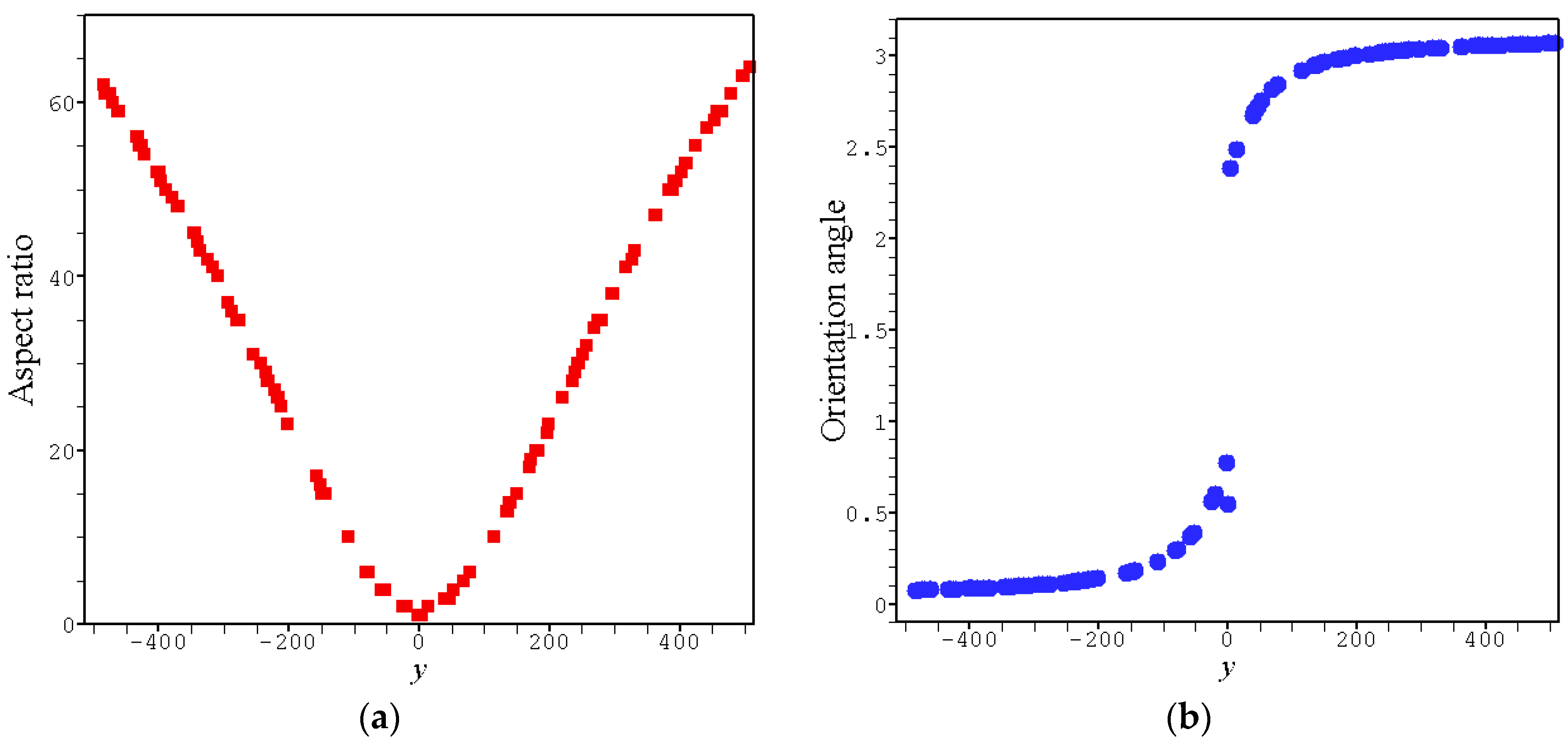
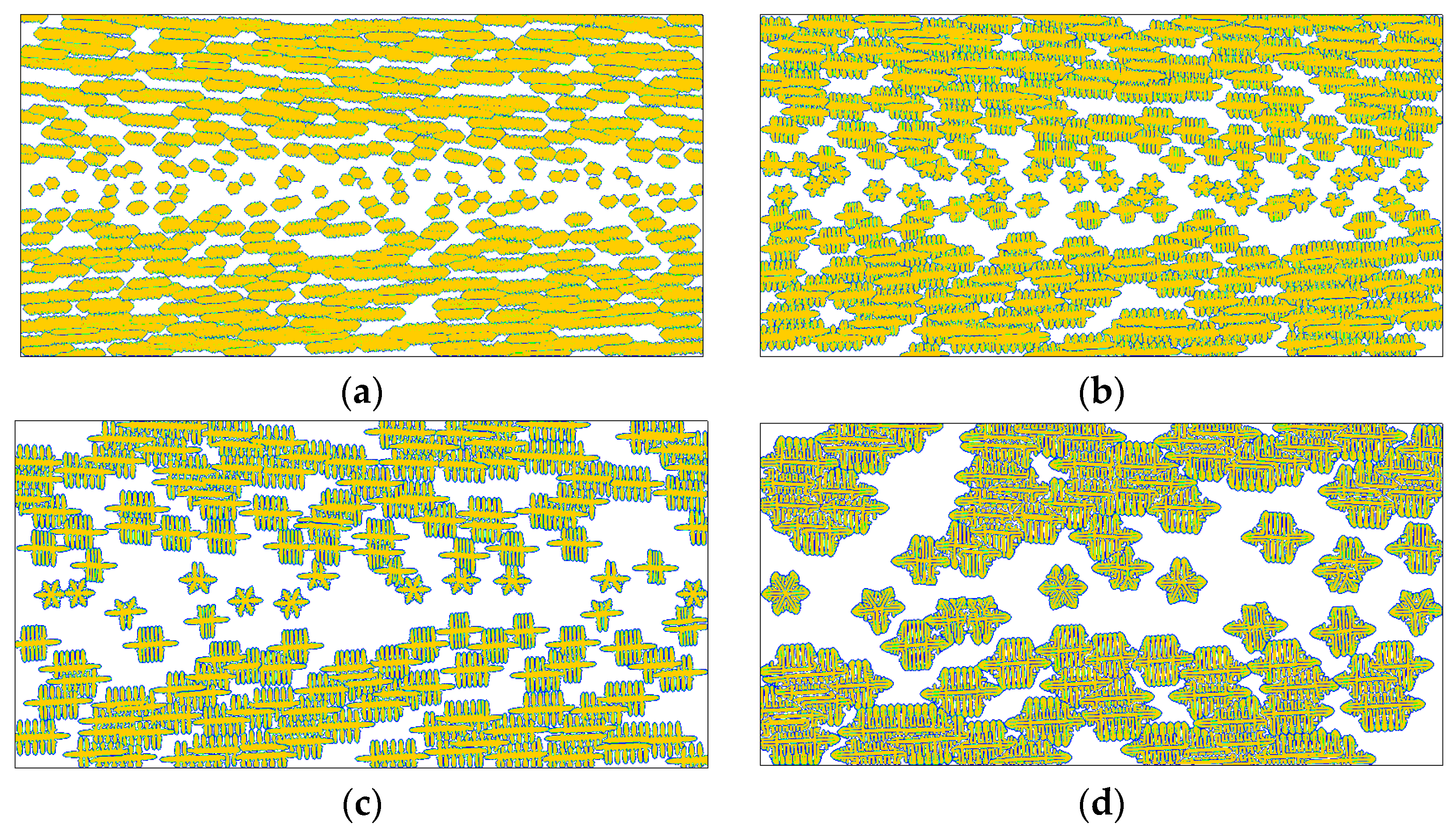
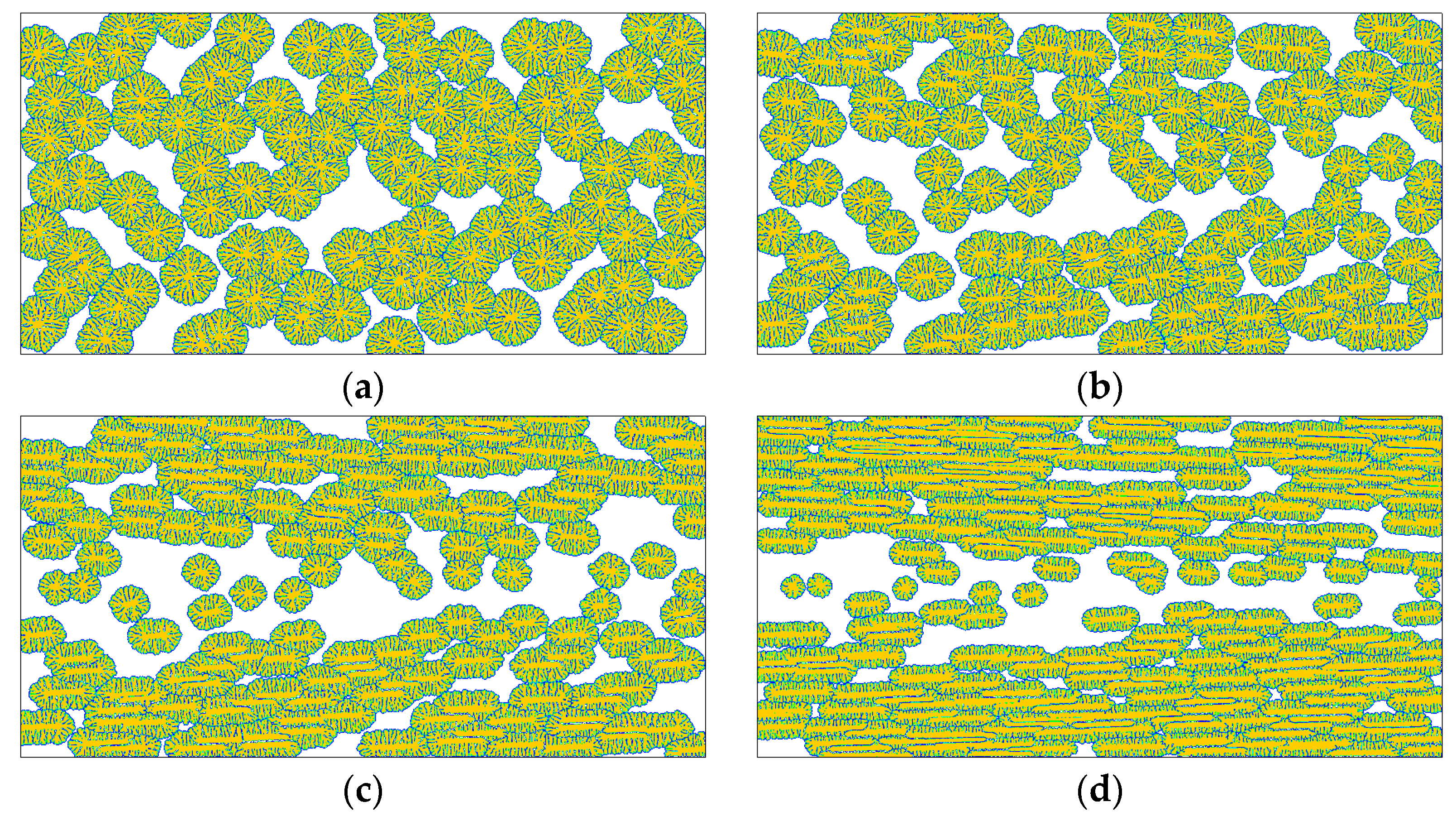
4. Results and Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zuidema, H. Flow Induced Crystallization of Polymers: Application to Injection Moulding. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2000. [Google Scholar]
- Graham, R.S.; Olmsted, P.D. Coarse-grained simulations of flow-induced nucleation in semicrystalline polymers. Phys. Rev. Lett. 2009, 103, 115702. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S. On Polyethylene crystals grown from flowing solutions in xylene. Bull. Text. Res. Inst. 1963, 66, 1–9. [Google Scholar]
- Yan, T.Z.; Zhao, B.J.; Cong, Y.H.; Fang, Y.Y.; Cheng, S.W.; Li, L.B.; Pan, G.Q.; Wang, Z.J.; Li, X.H.; Bian, F.G. Critical strain for shish-kebab formation. Macromolecules 2010, 43, 602–605. [Google Scholar] [CrossRef]
- Hayashi, Y.; Matsuba, G.; Zhao, Y.F.; Nishida, K.; Kanaya, T. Precursor of shish-kebab in isotactic polystyrene under shear flow. Polymer 2009, 50, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Kimata, S.; Sakurai, T.; Nozue, Y.; Kasahara, T.; Yamaguchi, N.; Karino, T.; Shibayama, M.; Kornfield, J.A. Molecular basis of the shish-kebab morphology in polymer crystallization. Science 2007, 316, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, B.S.; Yang, L.; Somani, R.H.; Avila-Orta, C.A.; Zhu, L. Unexpected shish-kebab structure in a sheared polyethylene melt. Phys. Rev. Lett. 2005, 94, 117802. [Google Scholar] [CrossRef] [PubMed]
- Janeschitz-Kriegl, H.; Ratajski, E. Some fundamental aspects of the kinetics of flow-induced crystallization of polymers. Colloid Polym. Sci. 2010, 288, 1525–1537. [Google Scholar] [CrossRef]
- Steenbakkers, R.J.A.; Peters, G.W.M. Suspension-based rheological modeling of crystallizing polymer melts. Rheol. Acta 2008, 47, 643–665. [Google Scholar] [CrossRef]
- Coppola, S.; Grizzuti, N.; Maffettone, P.L. Microrheological modeling of flow-induced crystallization. Macromolecules 2001, 34, 5030–5036. [Google Scholar] [CrossRef]
- Piorkowska, E.; Billon, N.; Haudin, J.M.; Gadzinowska, K. Spherulitic structure development during crystallization in confined space II. Effect of spherulite nucleation at borders. J. Appl. Polym. Sci. 2005, 97, 2319–2329. [Google Scholar] [CrossRef]
- Raabe, D.; Godara, A. Mesoscale simulation of the kinetics and topology of spherulite growth during crystallization of isotactic polypropylene (iPP) by using a cellular automaton. Model. Simul. Mater. Sci. Eng. 2005, 13, 733–751. [Google Scholar] [CrossRef]
- Huang, T.; Kamal, M.R. Morphological modeling of polymer solidification. Polym. Eng. Sci. 2000, 40, 1796–1808. [Google Scholar] [CrossRef]
- Ruan, C.; Ouyang, J.; Liu, S. Multi-scale modeling and simulation of crystallization during cooling in short fiber reinforced composites. Int. J. Heat Mass Tran. 2012, 55, 1911–1921. [Google Scholar] [CrossRef]
- Eder, G.; Janeschitz-Kriegl, H. Crystallization in processing of polymers. Mater. Sci. Technol. 1997, 18, 189–268. [Google Scholar]
- Peters, G.W.M.; Swartjes, F.H.M.; Meijer, H.E.H. A Recoverable strain based model for flow-induced crystallization. Macromol. Symp. 2002, 185, 277–292. [Google Scholar] [CrossRef]
- Hu, W.; Frenkel, D.; Mathot, V.B.F. Simulation of shish-kebab crystallite induced by a single prealigned macromolecule. Macromolecules 2002, 35, 7172–7174. [Google Scholar] [CrossRef]
- Yamamoto, T. Computer modeling of polymer crystallization–Toward computer-assisted materials’ design. Polymer 2009, 50, 1975–1985. [Google Scholar] [CrossRef]
- Baig, C.; Edwards, B.J. Atomistic simulation of crystallization of a polyethylene melt in steady uniaxial extension. J. Non-Newton. Fluid Mech. 2010, 165, 992–1004. [Google Scholar] [CrossRef]
- Nie, Y.J.; Zhang, R.J.; Zheng, K.S.; Zhou, Z.P. Nucleation details of nanohybrid shish-kebabs in polymer solutions studied by molecular simulations. Polymer 2015, 76, 1–7. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Turng, L.S.; Shen, C.Y. Modeling and prediction of morphology and crystallinity for cylindrical-shaped crystals during polymer processing. Polym. Eng. Sci. 2010, 50, 1226–1235. [Google Scholar] [CrossRef]
- Criscione, A.; Kintea, D.; Tuković, Ž.; Jakirlić, S.; Roisman, I.V.; Tropea, C. Crystallization of supercooled water: A level-set-based modeling of the dendrite tip velocity. Int. J. Heat Mass Tran. 2013, 66, 830–837. [Google Scholar] [CrossRef] [Green Version]
- López, J.; Gómez, P.; Hernández, J. A volume of fluid approach for crystal growth simulation. J. Comput. Phys. 2010, 229, 6663–6672. [Google Scholar] [CrossRef]
- Liu, Z.J.; Ouyang, J.; Zhou, W.; Wang, X.D. Numerical simulation of the polymer crystallization during cooling stage by using level set method. Comput. Mater. Sci. 2015, 97, 245–253. [Google Scholar] [CrossRef]
- Xu, H.J.; Matkar, R.; Kyu, T. Phase-field modeling on morphological landscape of isotactic polystyrene single crystals. Phys. Rev. E 2005, 72, 011804. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shi, T.F.; Chen, J.Z. Simulated morphological landscape of polymer single crystals by phase field model. J. Chem. Phys. 2008, 129, 194903. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jin, Z.K.; Xing, Y.; Gao, H.; Wang, X.K. Simulated rhythmic growth of targeted single crystal by polymer phase-field model. Comput. Mater. Sci. 2013, 68, 23–26. [Google Scholar] [CrossRef]
- Wang, X.D.; Ouyang, J.; Su, J.; Zhou, W. A phase-field model for simulating various spherulite morphologies of semi-crystalline polymers. Chin. Phys. B 2013, 22, 106103. [Google Scholar] [CrossRef]
- Tong, X.; Beckermann, C.; Karma, A.; Li, Q. Phase-field simulations of dendritic crystal growth in a forced flow. Phys. Rev. E 2001, 63, 061601. [Google Scholar] [CrossRef] [PubMed]
- Asta, M.; Beckermann, C.; Karma, A.; Kurz, W.; Napolitano, R.; Plapp, M.; Purdy, G.; Rappaz, M.; Trivedi, R. Solidification microstructures and solid-state parallels: Recent developments, future directions. Acta Mater. 2009, 57, 941–971. [Google Scholar] [CrossRef]
- De Gennes, P.G. Coil-stretch transition of dilute flexible polymers under ultrahigh velocity gradients. J. Chem. Phys. 1974, 60, 5030–5042. [Google Scholar] [CrossRef]
- Azzurri, F.; Alfonso, G.C. Insights into formation and relaxation of shearinduced nucleation precursors in isotactic polystyrene. Macromolecules 2008, 41, 1377–1383. [Google Scholar] [CrossRef]
- Cavallo, D.; Azzurri, F.; Balzano, L.; Funari, S.S.; Alfonso, G.C. Flow memory and stability of shear-induced nucleation precursors in isotactic polypropylene. Macromolecules 2010, 43, 9394–9400. [Google Scholar] [CrossRef]
- Pantani, R.; de Santis, F.; Speranza, V.; Titomanlio, G. Modelling morphology evolution during solidification of IPP in processing conditions. AIP Conf. Proc. 2014, 1593, 636–640. [Google Scholar]
- Upadhyay, R.K.; Isayev, A.I.; Shen, S.F. Transient shear flow behavior of polymeric fluids according to the Leonov model. Rheol. Acta 1981, 20, 443–457. [Google Scholar] [CrossRef]
- Herrchen, M.; Öttinger, H.C. A detailed comparison of various FENE dumbbell models. J. Non-Newton. Fluid Mech. 1997, 68, 17–42. [Google Scholar] [CrossRef]
- Su, J.; Ouyang, J.; Wang, X.D.; Yang, B.X. Lattice Boltzmann method coupled with the Oldroyd-B constitutive model for a viscoelastic fluid. Phys. Rev. E 2013, 88, 053304. [Google Scholar] [CrossRef] [PubMed]
- Koscher, E.; Fulchiron, R. Influence of shear on polypropylene crystallization: morphology development and kinetics. Polymer 2002, 43, 6931–6942. [Google Scholar] [CrossRef]
- Charbon, C.; Swaminarayan, S. A multiscale model for polymer crystallization. II. Solidification of a macroscopic part. Polym. Eng. Sci. 1998, 38, 644–656. [Google Scholar] [CrossRef]
- Yu, F.Y.; Zhang, H.B.; Wang, Z.G.; Yu, W.; Zhou, C.X. Overshoots in stress and free energy change during the flow-induced crystallization of polymeric melt in shear flow. Chin. J. Polym. Sci. 2010, 28, 657–666. [Google Scholar] [CrossRef]
- Jarecki, L. Kinetic theory of crystal nucleation under transient molecular orientation. Lect. Notes Phys. 2007, 714, 65–86. [Google Scholar]
- Wang, X.D.; Ouyang, J.; Su, J.; Zhou, W. Investigating the role of oriented nucleus in polymer shish-kebab crystal growth via phase-field method. J. Chem. Phys. 2014, 140, 114102. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Balzano, L.; Portale, G.; Peters, G.W.M. Flow induced crystallization in isotactic polypropylene during and after flow. Polymer 2014, 55, 6140–6151. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Weeks, J.J. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J. Res. Natl. Bur. Stand. A 1962, 66, 13–28. [Google Scholar] [CrossRef]
- Housmans, J.W.; Peters, G.W.M.; Meijer, H.E.H. Flow-induced crystallization of propylene/ethylene random copolymers. J. Therm. Anal. Calorim. 2009, 98, 693–705. [Google Scholar] [CrossRef]
- Pantani, R.; Coccorullo, I.; Speranza, V.; Titomanlio, G. Modeling of morphology evolution in the injection molding process of thermoplastic polymers. Prog. Polym. Sci. 2005, 30, 1185–1222. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Issaian, A.M.; Kornfield, J.A. Shear-enhanced crystallization in isotactic polypropylene. 1. Correspondence between in situ rheo-optics and ex situ structure determination. Macromolecules 1999, 32, 7537–7547. [Google Scholar] [CrossRef]
- Zhang, C.G.; Hu, H.Q.; Wang, D.J.; Yan, S.K.; Han, C.C. In situ optical microscope study of the shear-induced crystallization of isotactic polypropylene. Polymer 2005, 46, 8157–8161. [Google Scholar] [CrossRef]
- Lagasse, R.R.; Maxwell, B. An experimental study of the kinetics of polymer crystallization during shear flow. Polym. Eng. Sci. 1976, 16, 189–199. [Google Scholar] [CrossRef]
- Chai, C.K.; Auzoux, Q.; Randrianatoandro, H.; Navard, P.; Haudin, J. Influence of pre-shearing on the crystallisation of conventional and metallocene polyethylenes. Polymer 2003, 44, 773–782. [Google Scholar]
- Kessler, D.A.; Koplik, J.; Levine, H. Pattern selection in fingered growth phenomena. Adv. Phys. 1988, 37, 255–339. [Google Scholar] [CrossRef]
- Kantz, M.R.; Newman, H.D.; Stigale, F.H. The skin-core morphology and structure–Property relationships in injection-moulded polypropylene. J. Appl. Polym. Sci. 1972, 16, 1249–1260. [Google Scholar] [CrossRef]
- Huang, L.X.; Wang, Z.; Zheng, G.Q.; Guo, J.Z.H.; Dai, K.; Liu, C.T. Enhancing oriented crystals in injection-molded HDPE through introduction of pre-shear. Mater. Des. 2015, 78, 12–18. [Google Scholar] [CrossRef]
- Matsuba, G.; Sakamoto, S.; Ogino, Y.; Nishida, K.; Kanaya, T. Crystallization of polyethylene blends under shear flow. Effects of crystallization temperature and ultrahigh molecular weight component. Macromolecules 2007, 40, 7270–7275. [Google Scholar] [CrossRef]
- Kessler, D.A.; Levine, H. Fluctuation-induced diffusive instabilities. Nature 1998, 394, 556–558. [Google Scholar] [CrossRef]
- Wang, X.D.; Ouyang, J.; Su, J.; Zhou, W. Phase field modeling of the ring-banded spherulites of crystalline polymers: The role of thermal diffusion. Chin. Phys. B 2014, 23, 126103. [Google Scholar] [CrossRef]
- Taguchi, K.; Miyaji, H.; Izumi, K.; Hoshino, A.; Miyamoto, Y.; Kokawa, R. Crystal growth of isotactic polystyrene in ultrathin films: film thickness dependence. J. Macromol. Sci. B 2001, 41, 1033–1042. [Google Scholar] [CrossRef]










© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Ouyang, J.; Zhou, W.; Liu, Z. A Phase Field Technique for Modeling and Predicting Flow Induced Crystallization Morphology of Semi-Crystalline Polymers. Polymers 2016, 8, 230. https://doi.org/10.3390/polym8060230
Wang X, Ouyang J, Zhou W, Liu Z. A Phase Field Technique for Modeling and Predicting Flow Induced Crystallization Morphology of Semi-Crystalline Polymers. Polymers. 2016; 8(6):230. https://doi.org/10.3390/polym8060230
Chicago/Turabian StyleWang, Xiaodong, Jie Ouyang, Wen Zhou, and Zhijun Liu. 2016. "A Phase Field Technique for Modeling and Predicting Flow Induced Crystallization Morphology of Semi-Crystalline Polymers" Polymers 8, no. 6: 230. https://doi.org/10.3390/polym8060230
APA StyleWang, X., Ouyang, J., Zhou, W., & Liu, Z. (2016). A Phase Field Technique for Modeling and Predicting Flow Induced Crystallization Morphology of Semi-Crystalline Polymers. Polymers, 8(6), 230. https://doi.org/10.3390/polym8060230