Optimisation of Surface-Initiated Photoiniferter-Mediated Polymerisation under Confinement, and the Formation of Block Copolymers in Mesoporous Films


Abstract
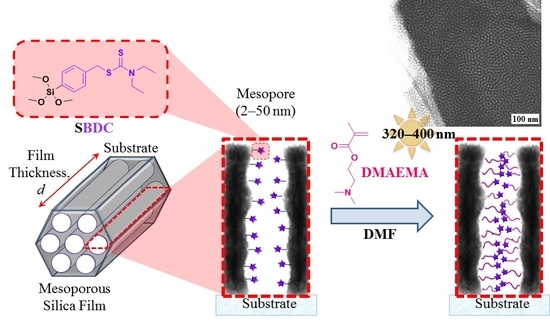
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterisation
2.2.1. Ellipsometry
2.2.2. Cyclic Voltammetry (CV)
2.2.3. Static Contact Angle (CA)
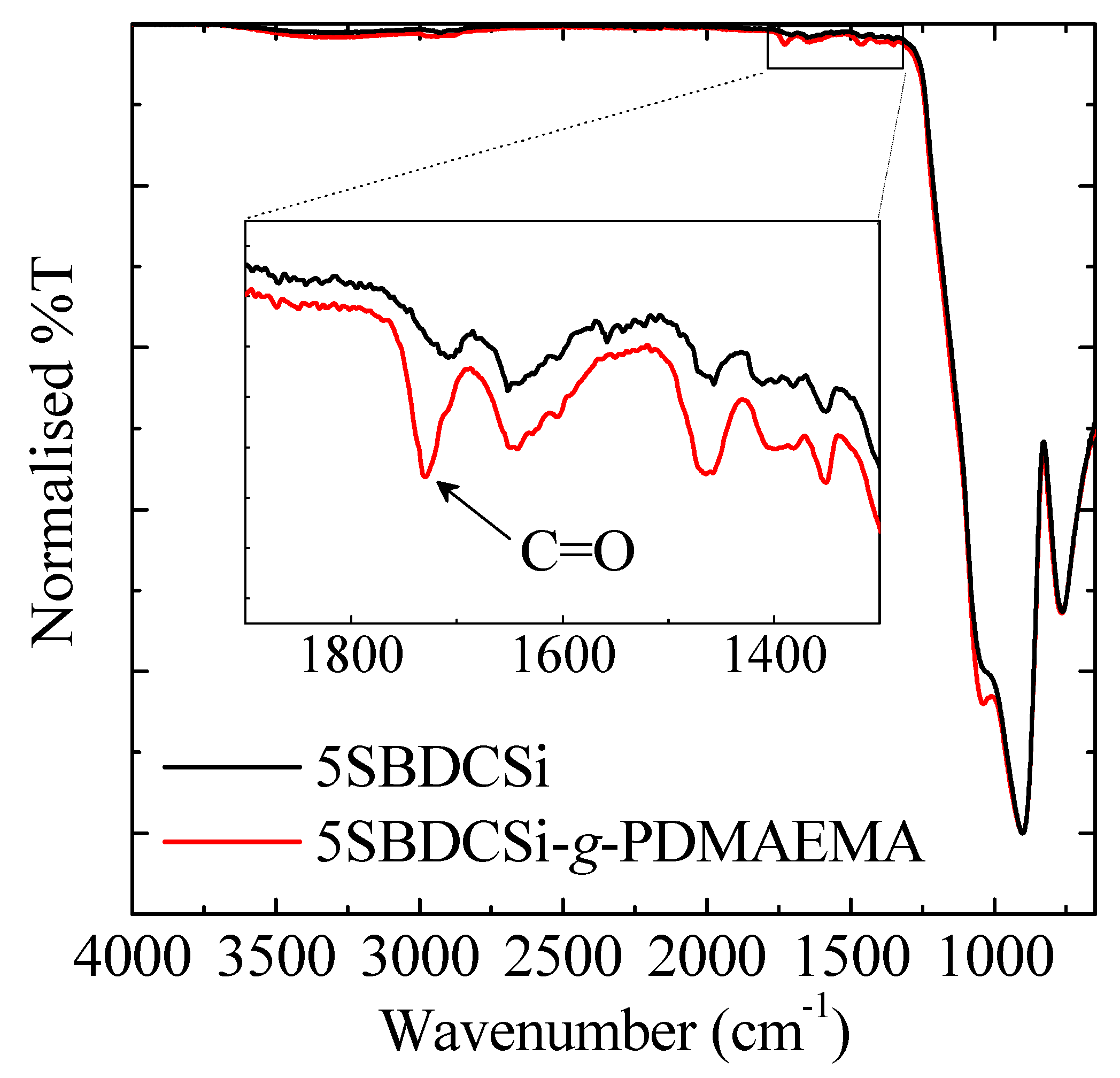
2.2.4. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.2.5. Transmission Electron Microscopy (TEM)
2.2.6. Scanning Electron Microscopy (SEM)
2.2.7. 1H Nuclear Magnetic Resonance (1H NMR) Spectroscopy
2.2.8. Size Exclusion Chromatography (SEC)
2.2.9. Krypton BET Adsorption
2.3. Experimental
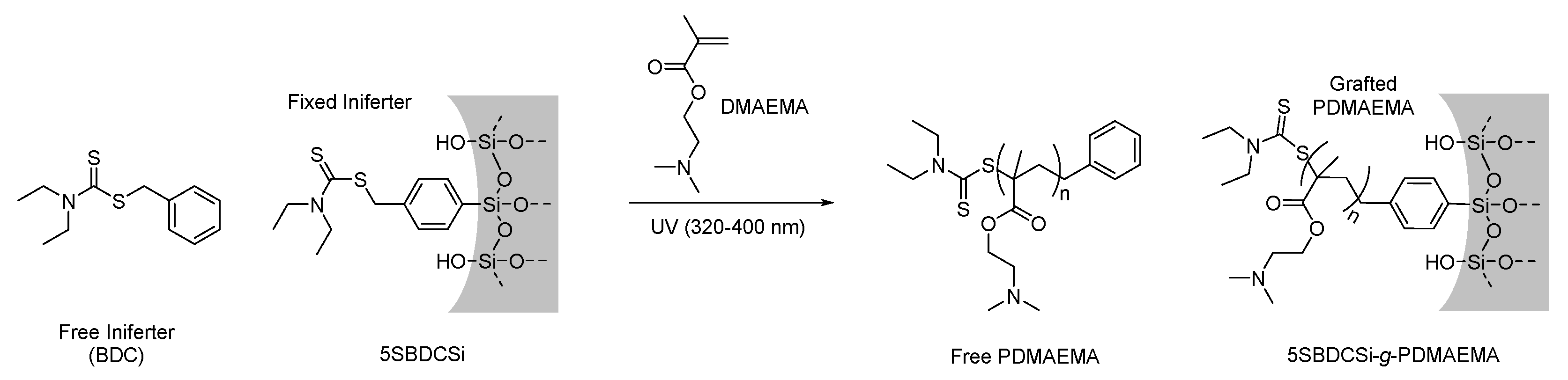
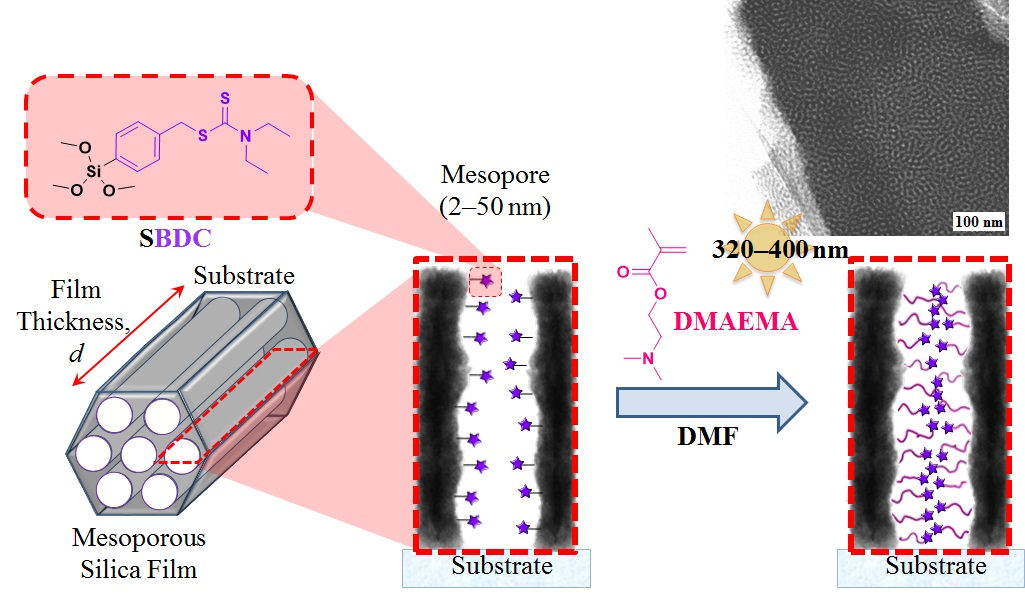
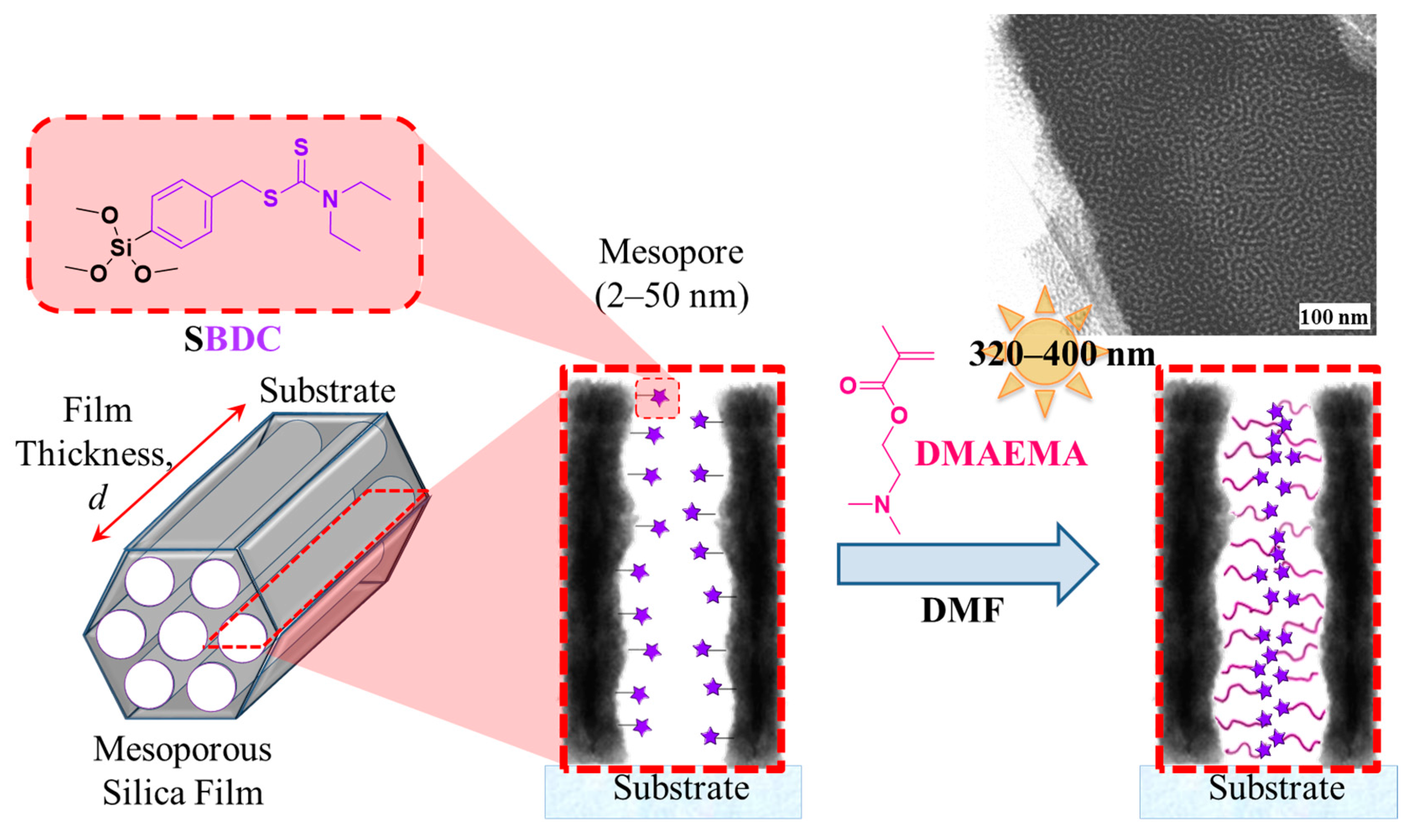
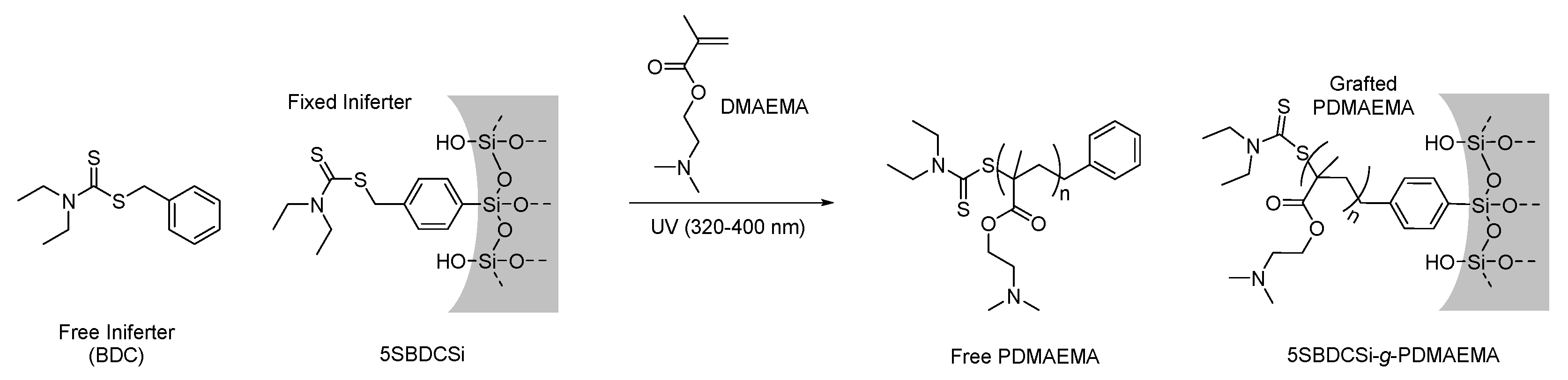
2.3.1. Synthesis of N,N-(Diethylamino)dithiocarbamoyl-benzyl(trimethoxy)silane) (SBDC), a Silane Terminated Iniferter
2.3.2. Synthesis of Benzyl Diethyldithiocarbamate (BDC), a Sacrificial Iniferter and SBDC Mimic
2.3.3. Preparation of Mesoporous Silica Thin Films
2.3.4. Surface-Initiated Photoiniferter-Mediated Polymerisation (SI-PIMP) from SBDC-Functionalised Mesoporous Silica
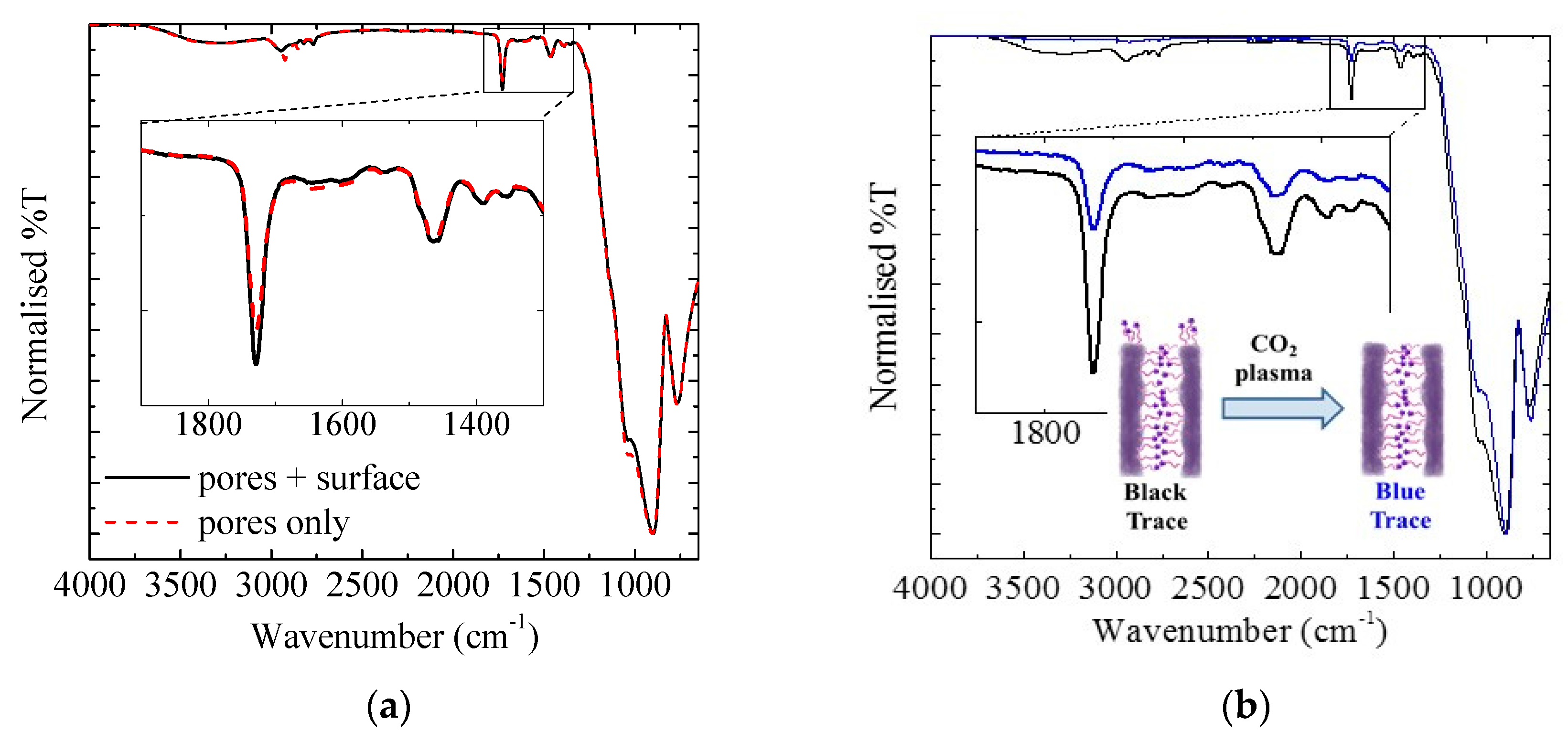
2.3.5. CO2 Plasma Treatment
3. Results and Discussion
3.1. Controlling a Confined Polymerisation
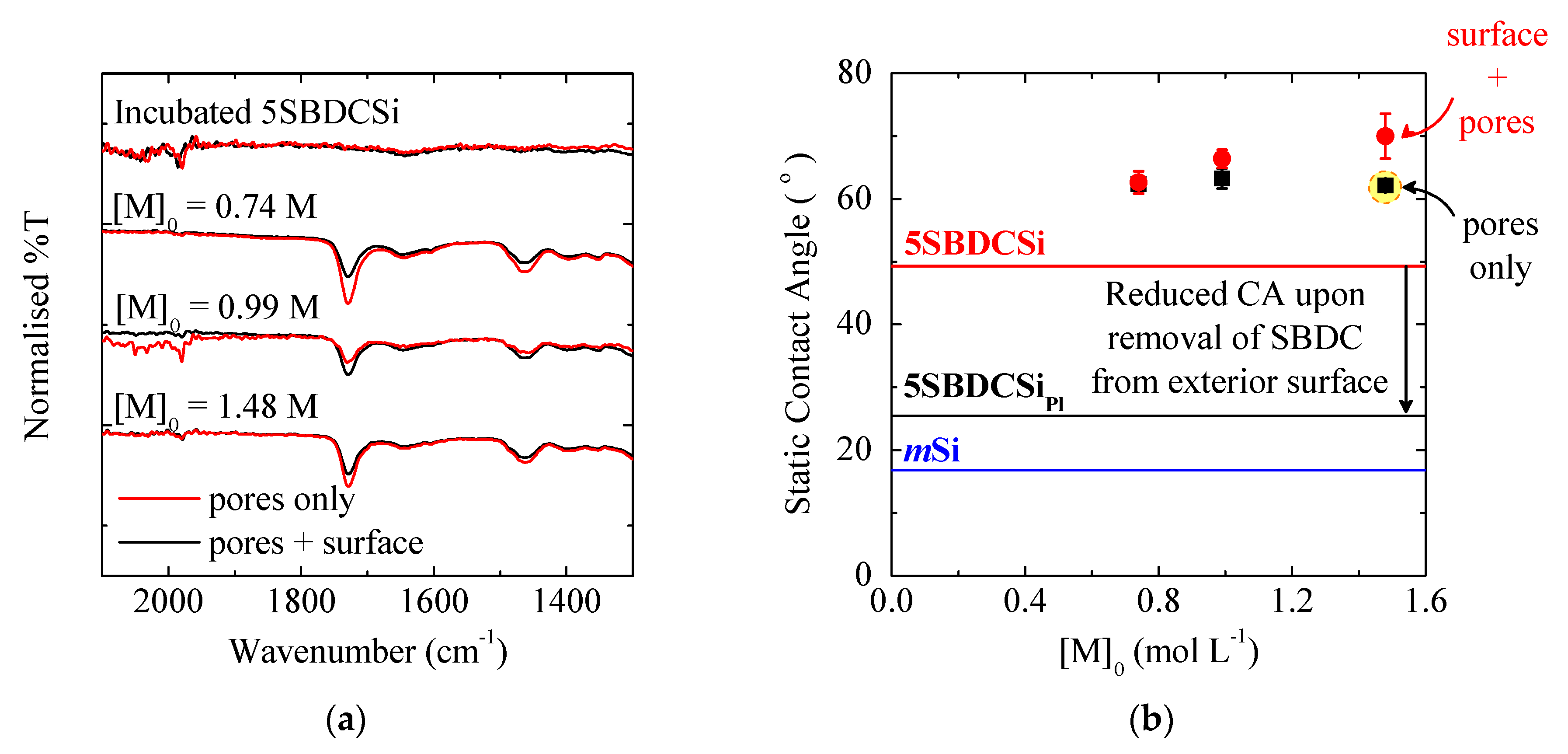
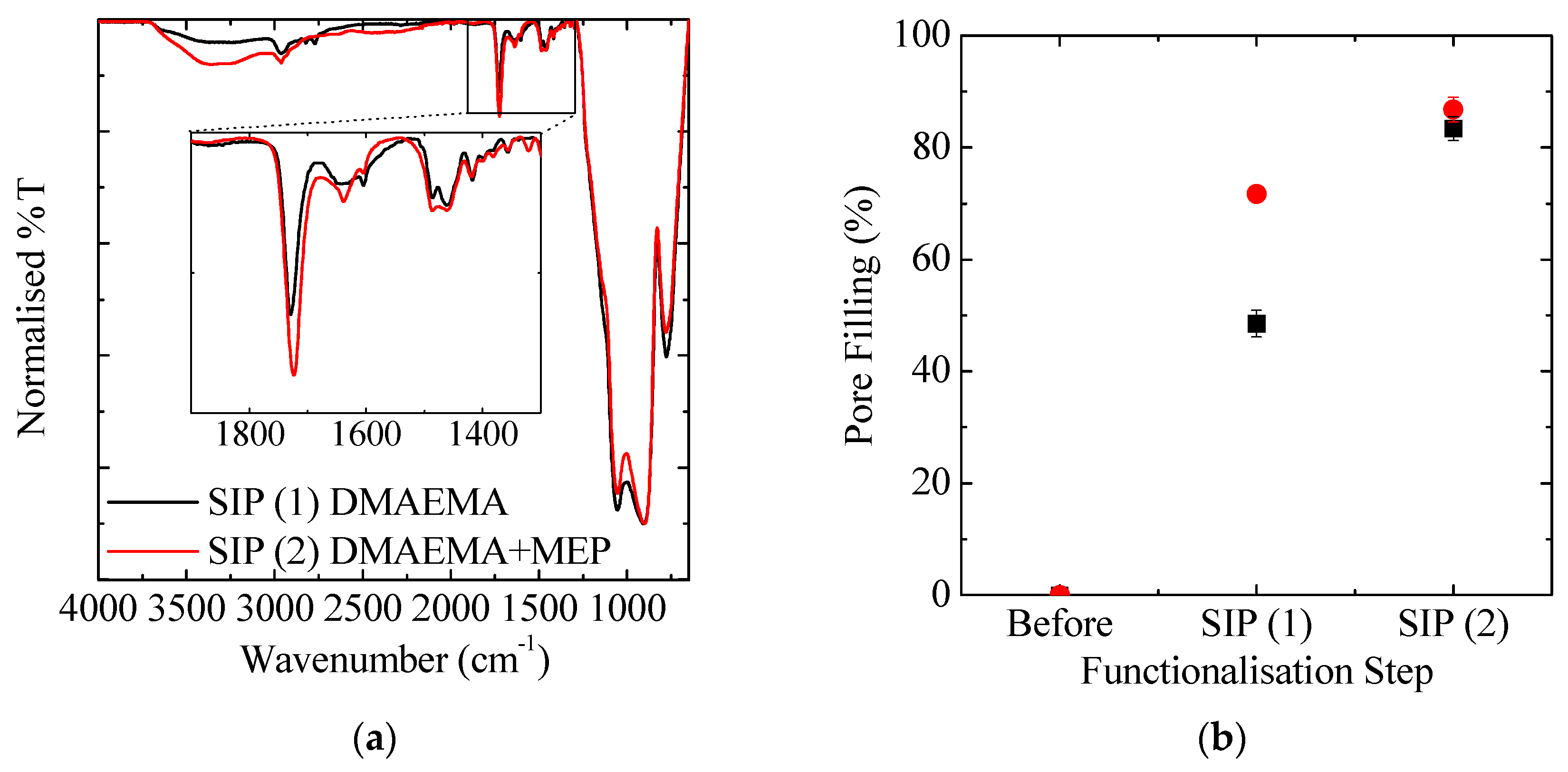
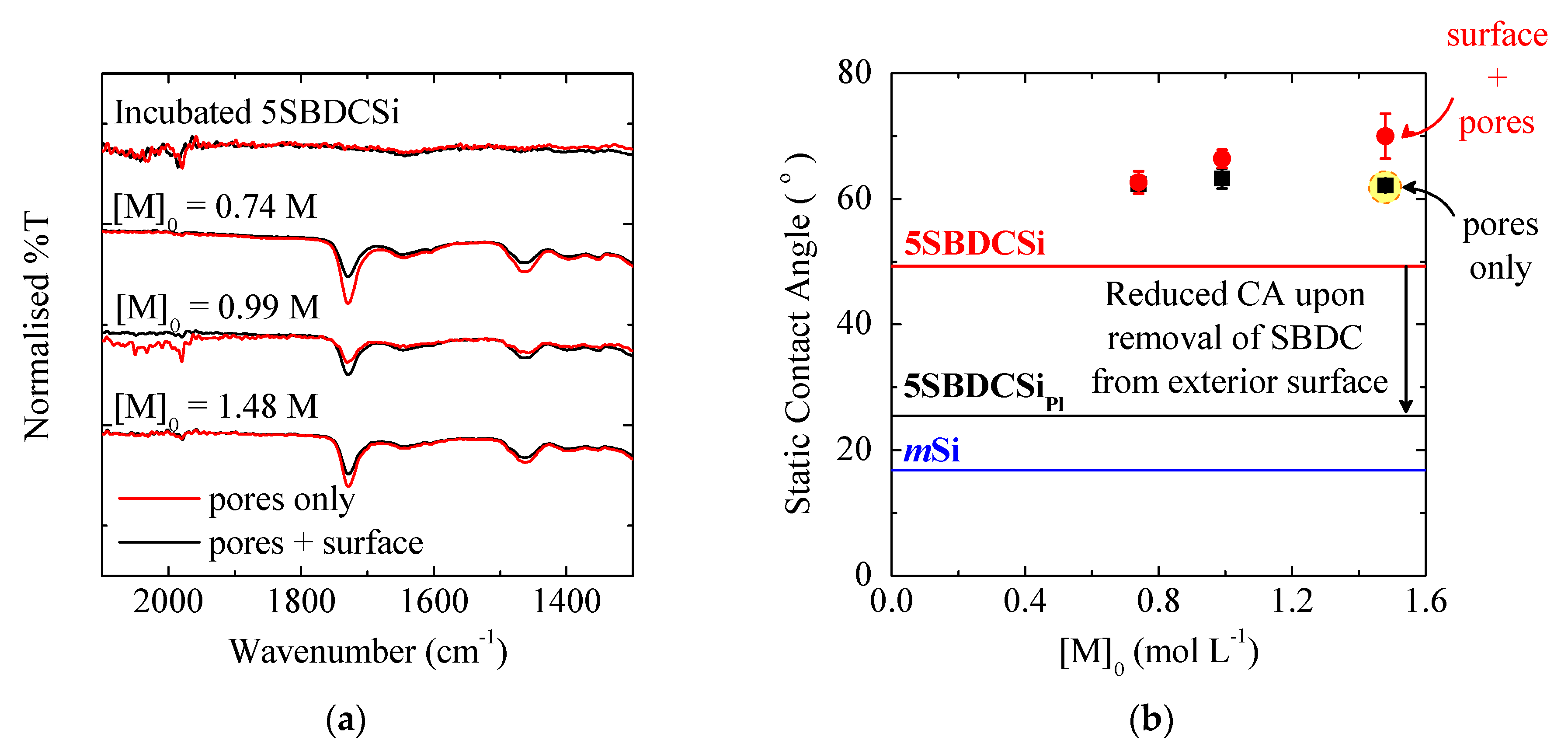
3.1.1. Pore Accessibility
3.1.2. Monomer Concentration
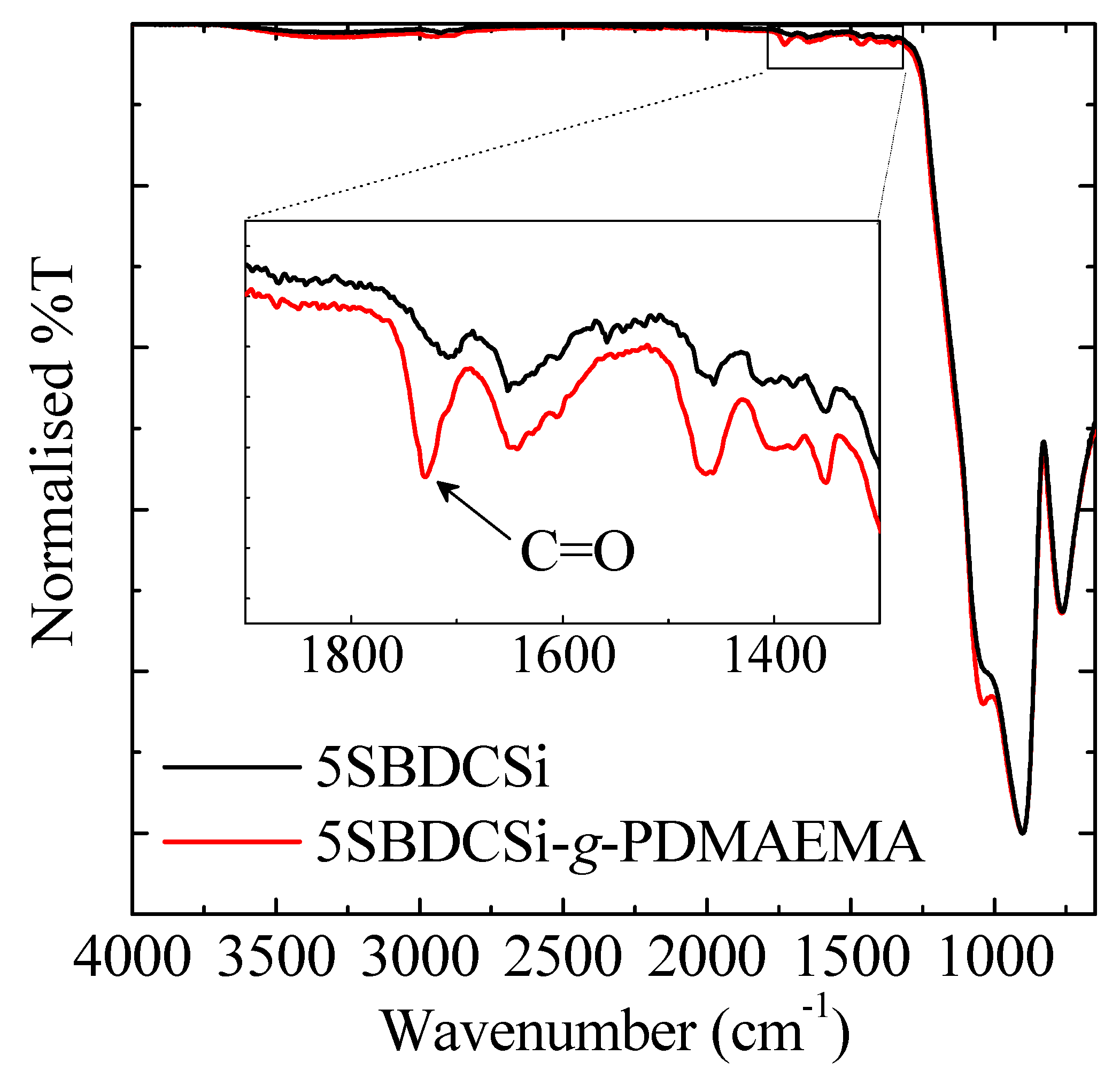
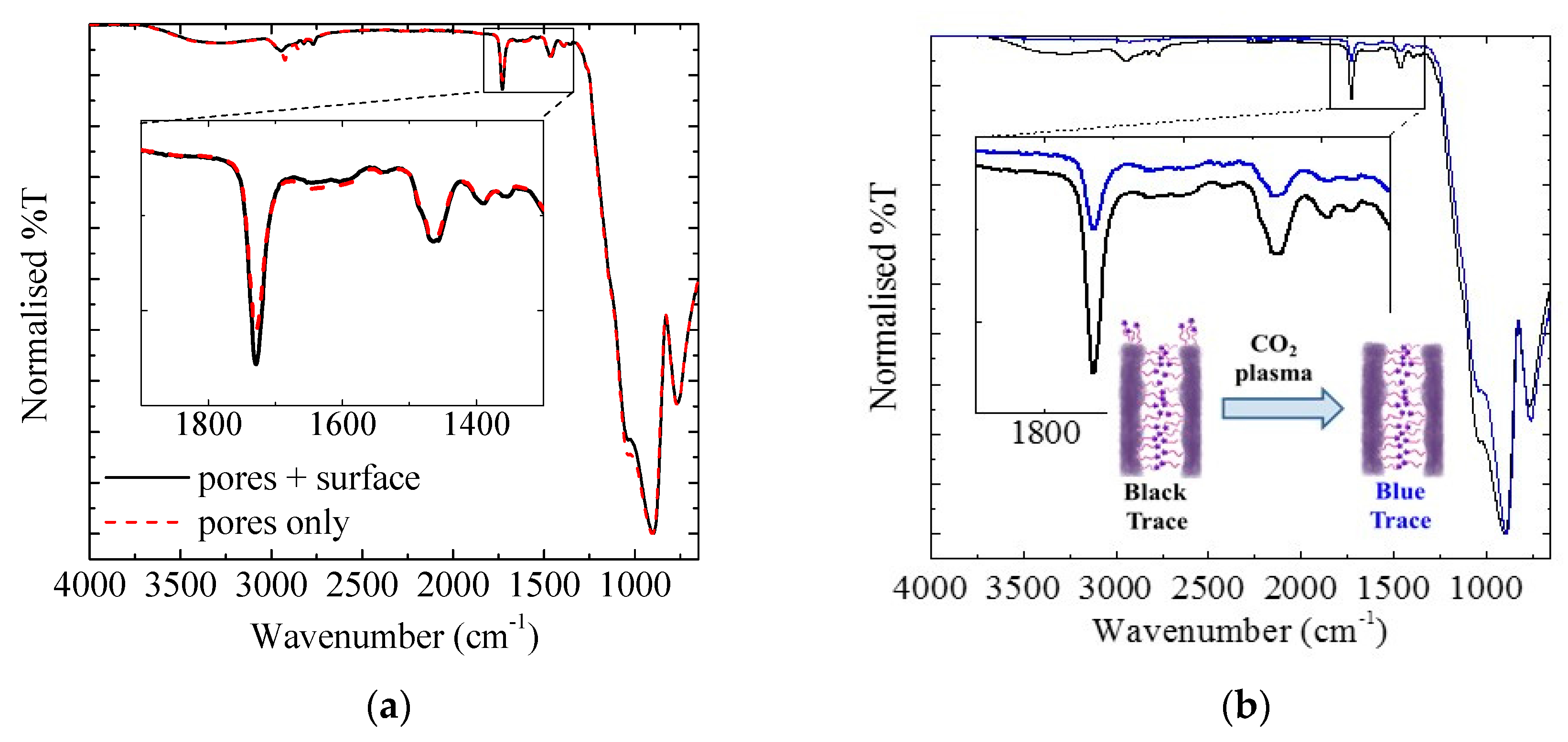
3.1.3. Exterior Surface
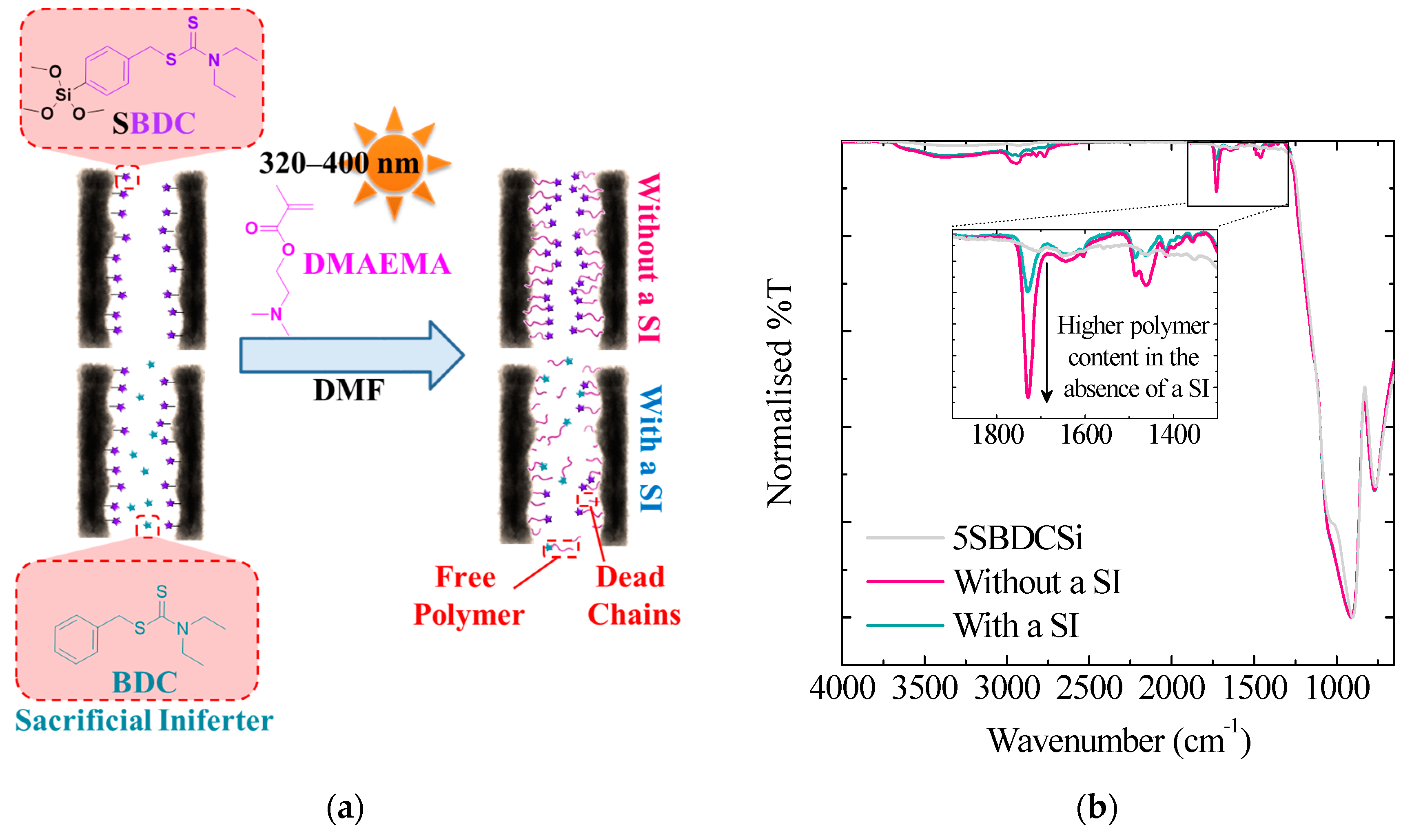
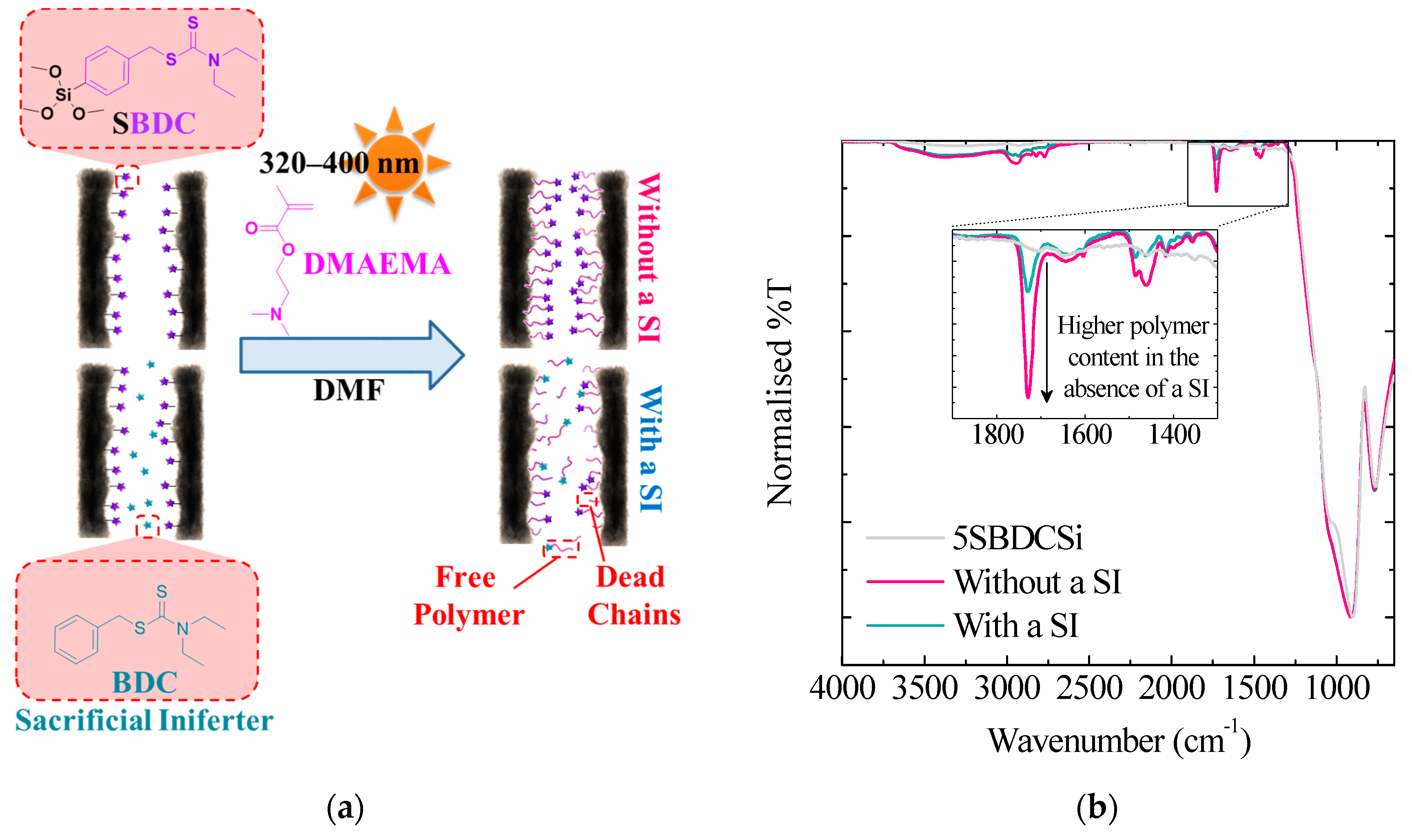
3.1.4. Sacrificial Iniferters
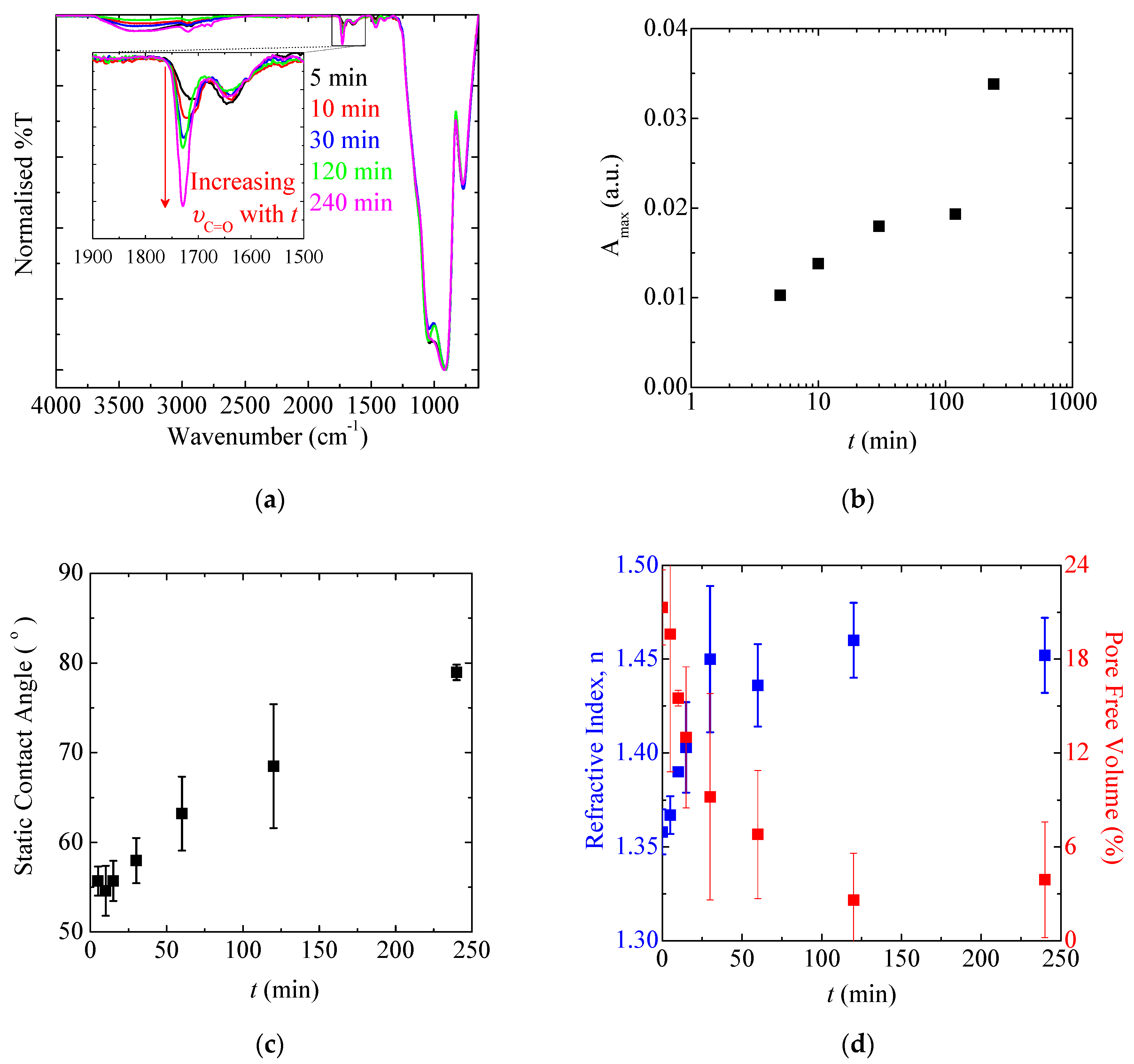
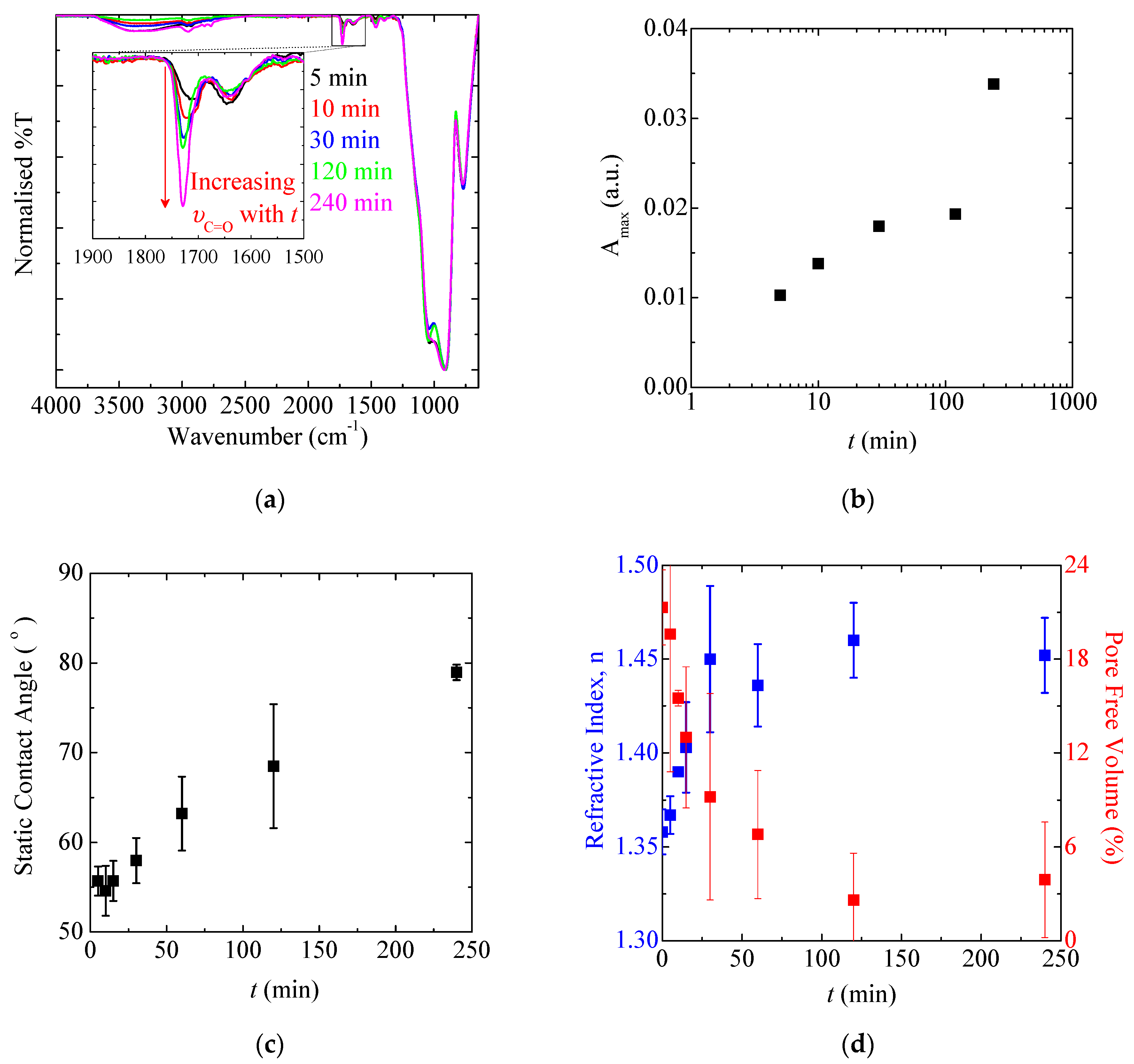
3.1.5. Polymerisation Time and SIP Kinetics
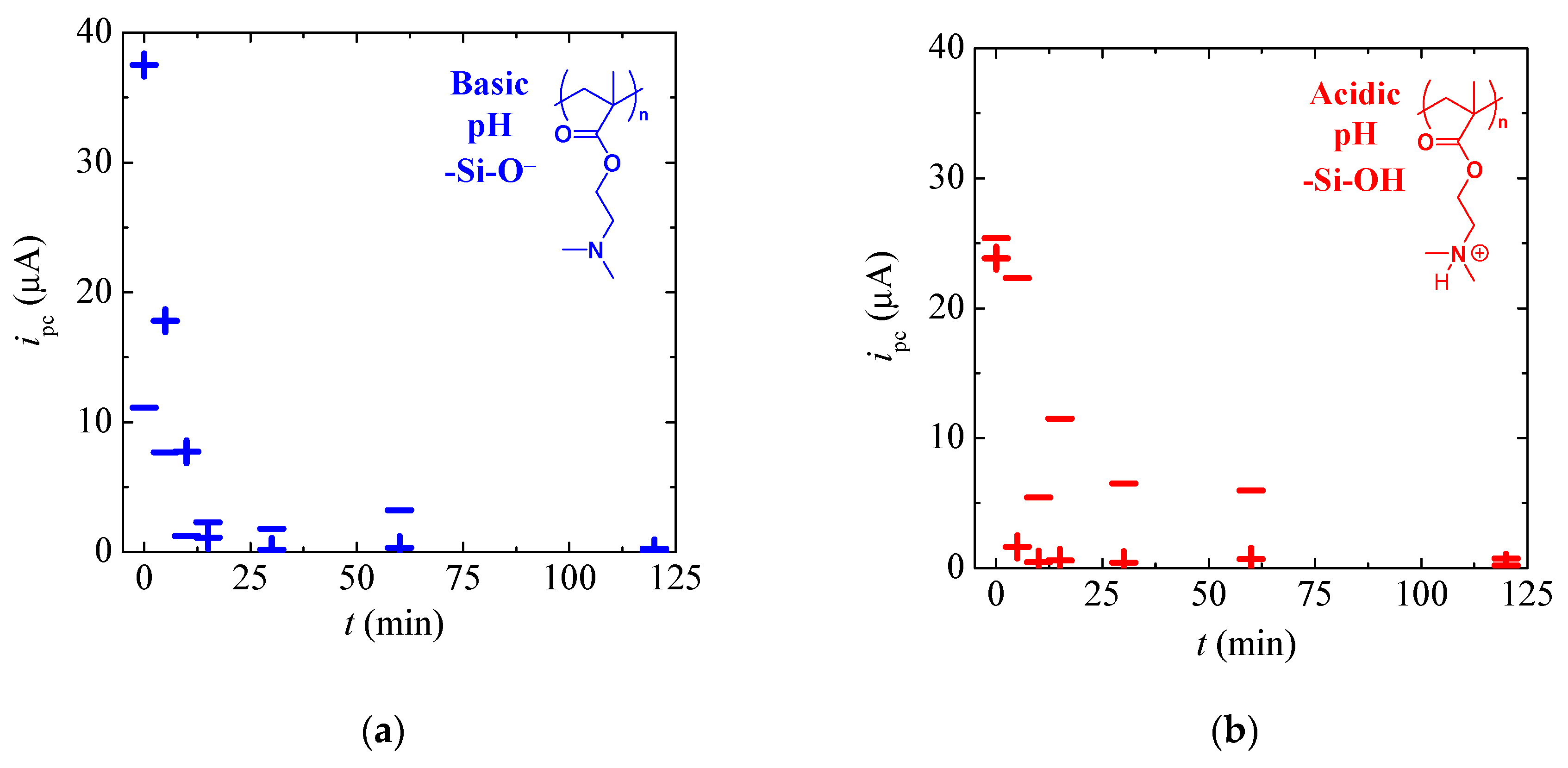
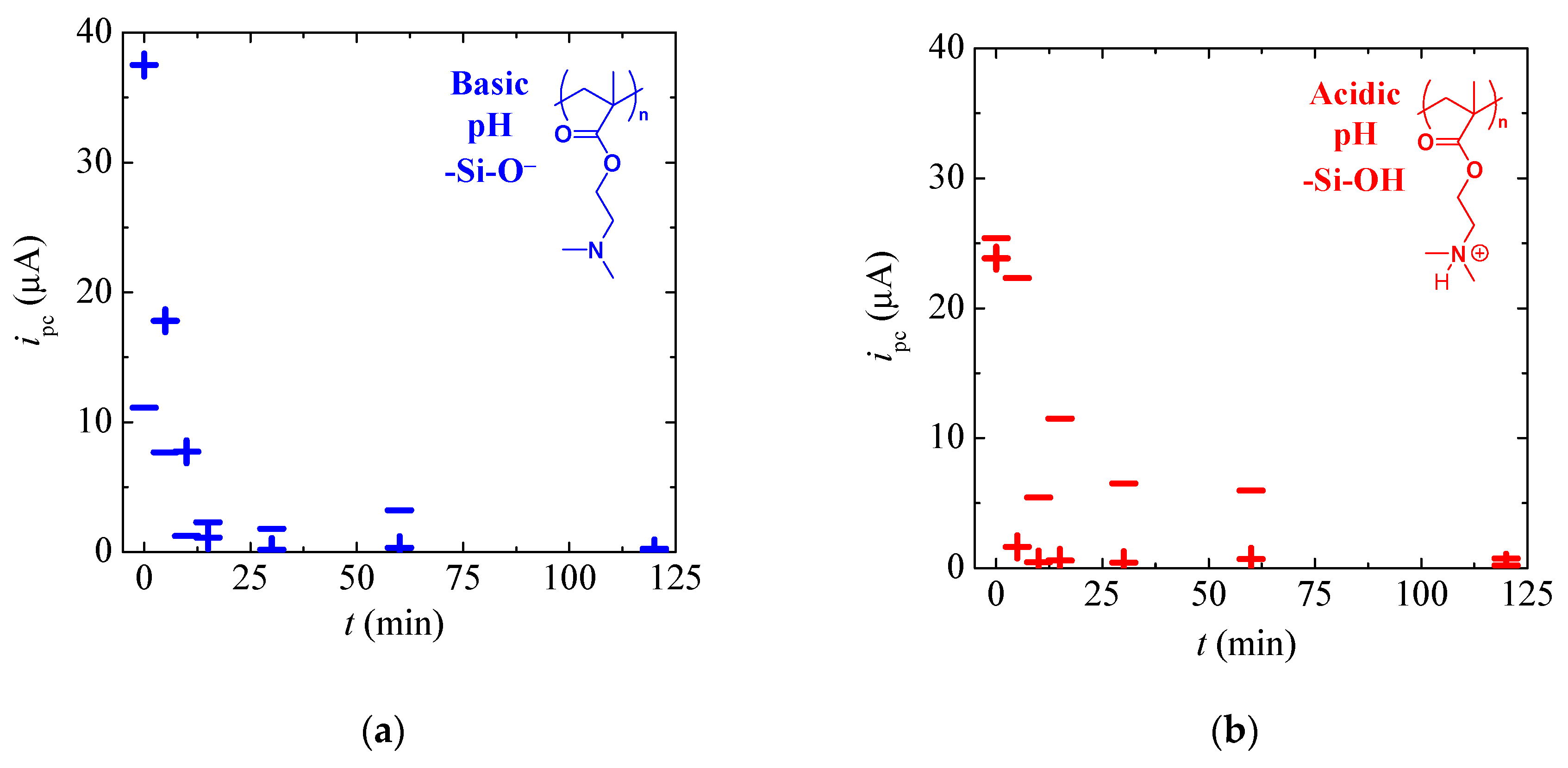
3.2. Tuneable Ionic Permselectivity
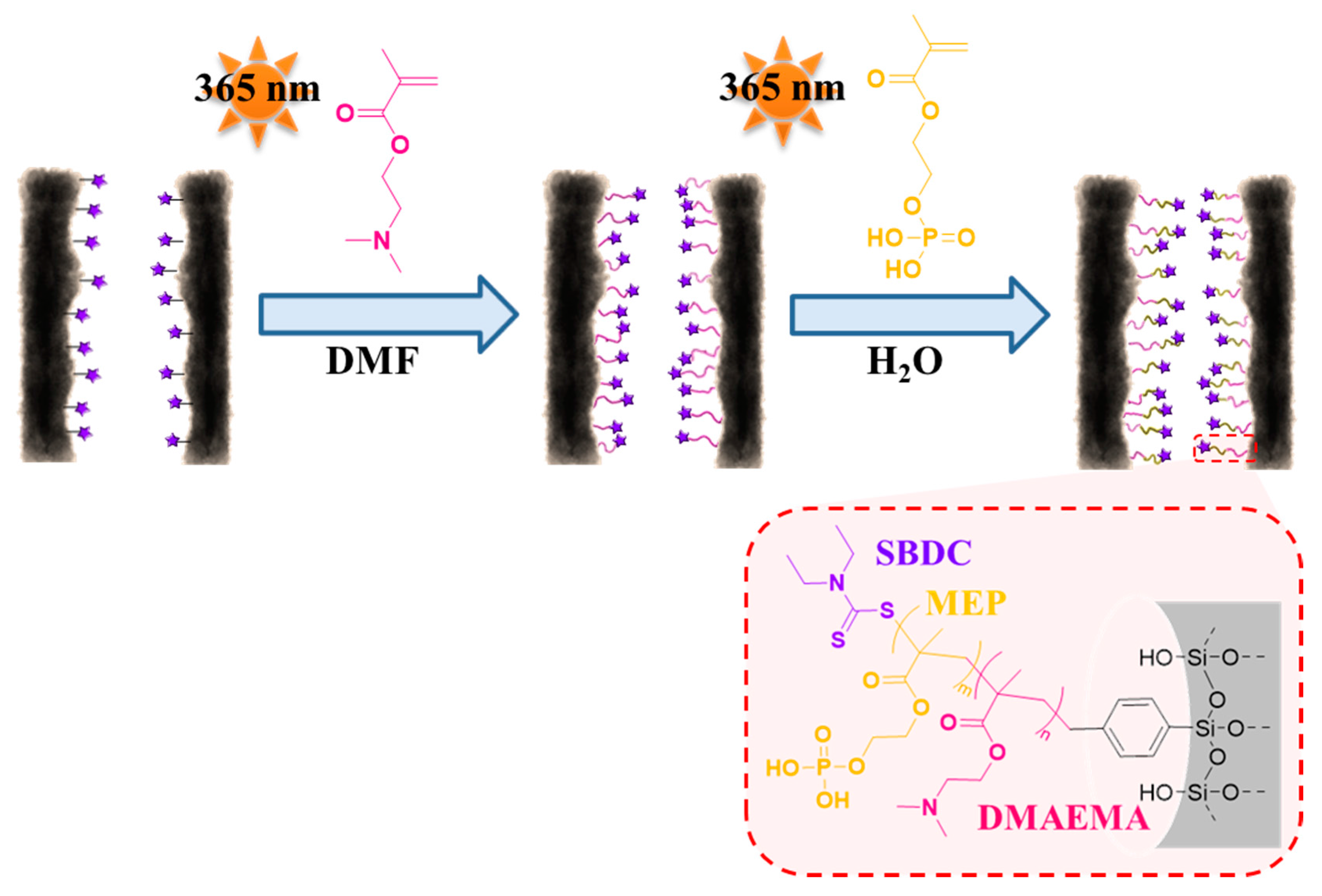
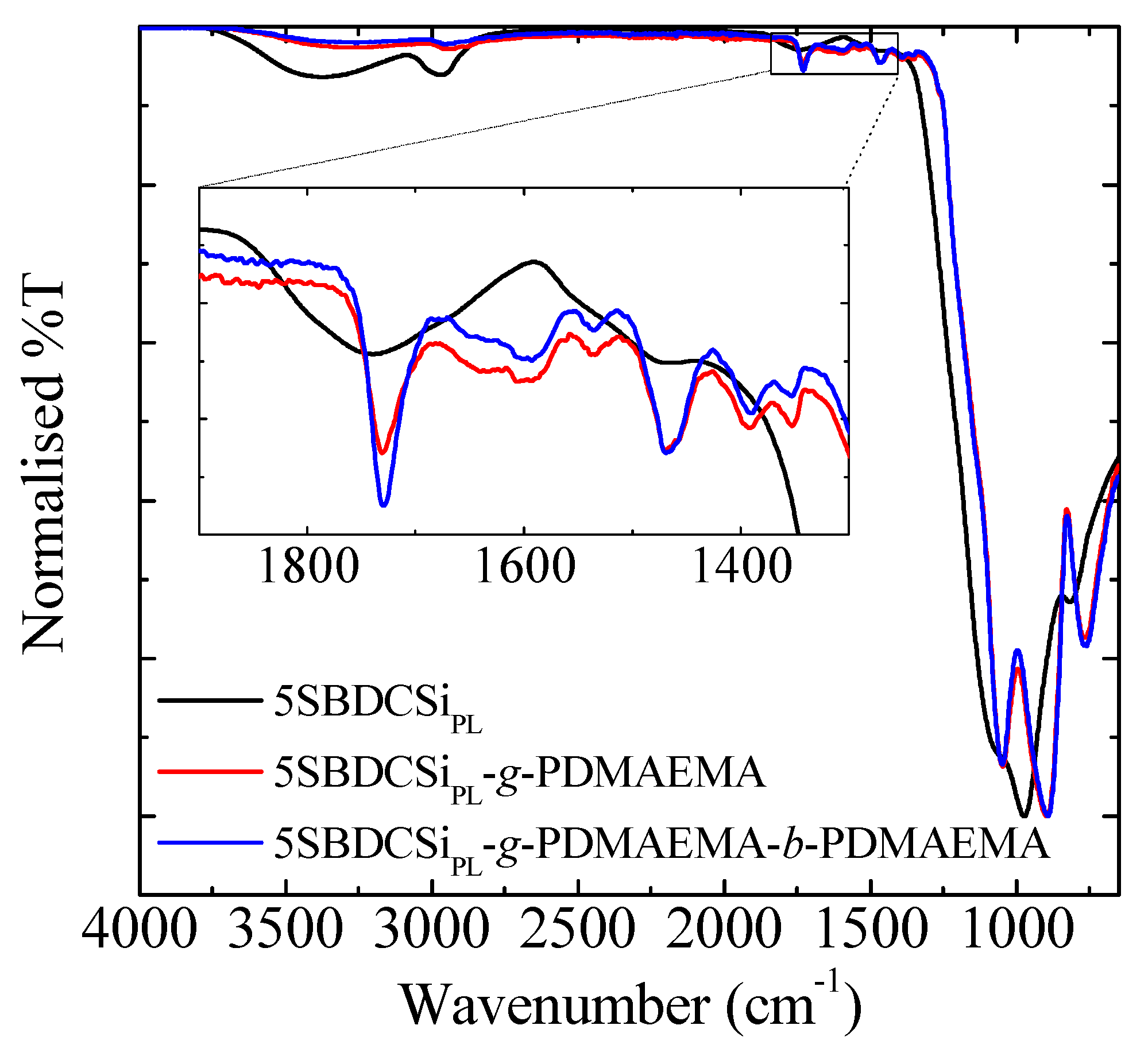
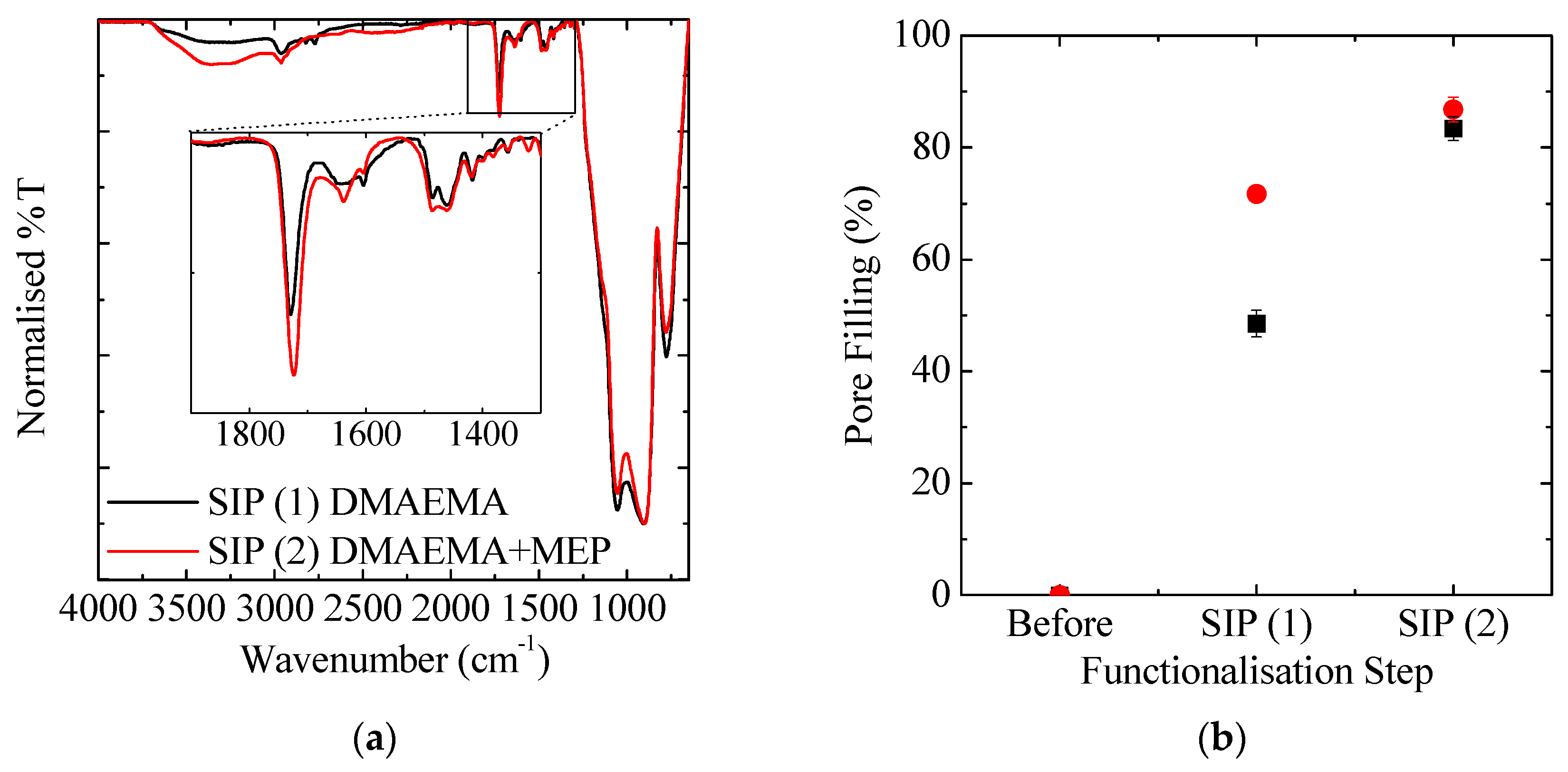
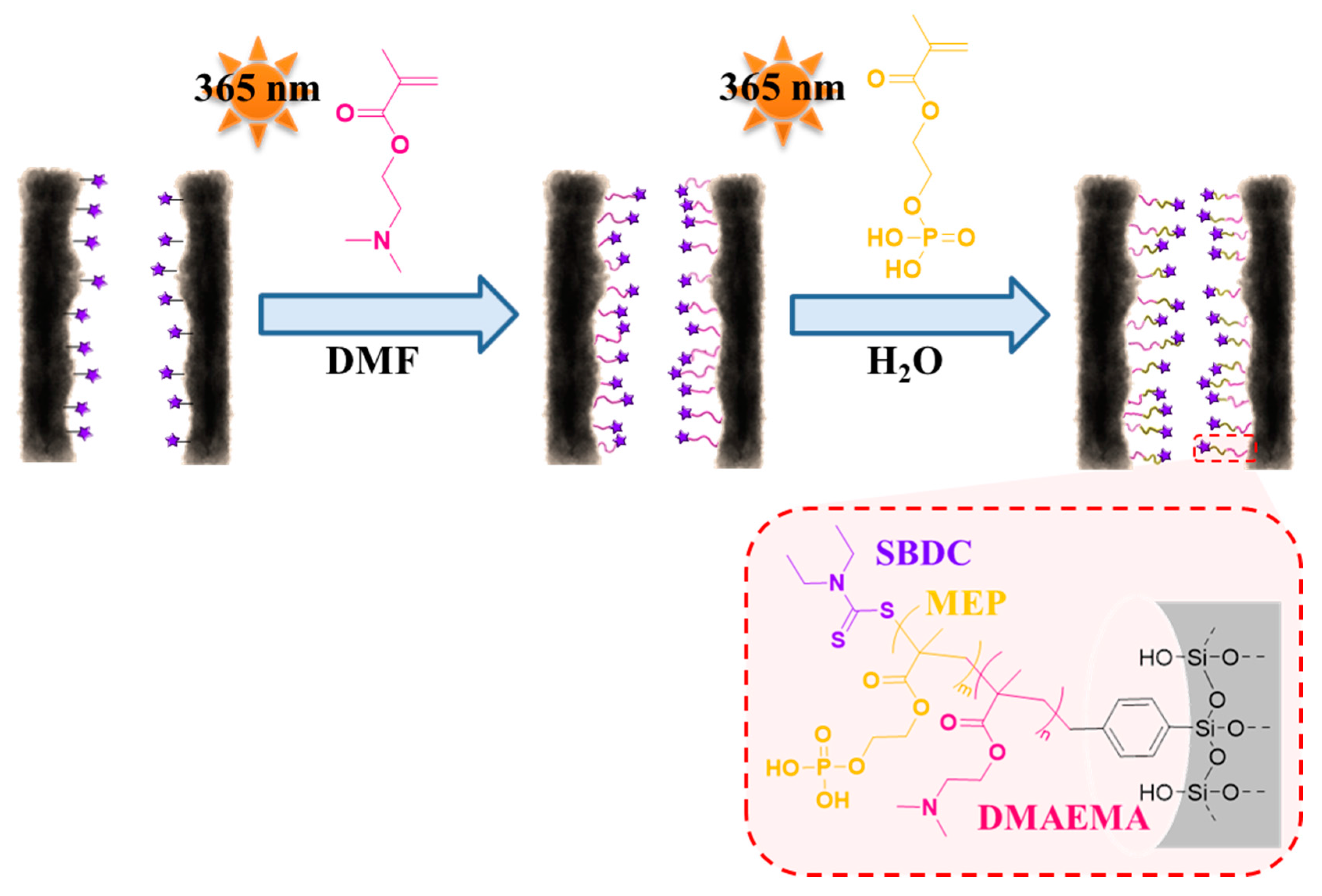
3.3. Re-Initiation and the Potential to form Block Copolymers under Confinement
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tagliazucchi, M.; Szleifer, I. Transport mechanisms in nanopores and nanochannels: Can we mimic nature? Mater. Today 2015, 18, 131–142. [Google Scholar] [CrossRef]
- Perez-Mitta, G.; Albesa, A.G.; Trautmann, C.; Toimil-Molares, M.E.; Azzaroni, O. Bioinspired integrated nanosystems based on solid-state nanopores: “Iontronic” transduction of biological, chemical and physical stimuli. Chem. Sci. 2017, 8, 890–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Ye, G.; Song, Y.; Liu, F.; Huo, X.; Chen, J. Well-defined functional mesoporous silica/polymer hybrids prepared by an ICAR ATRP technique integrated with bio-inspired polydopamine chemistry for lithium isotope separation. Dalton Trans. 2017, 46, 6117–6127. [Google Scholar] [CrossRef] [PubMed]
- Walcarius, A. Mesoporous materials and electrochemistry. Chem. Soc. Rev. 2013, 42, 4098–4140. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-H.; Huang, S.-Z.; Chen, L.-H.; Li, Y.; Yang, X.-Y.; Yuan, Z.-Y.; Su, B.-L. Applications of hierarchically structured porous materials from energy storage and conversion, catalysis, photocatalysis, adsorption, separation, and sensing to biomedicine. Chem. Soc. Rev. 2016, 45, 3479–3563. [Google Scholar] [CrossRef] [PubMed]
- Pal, N.; Bhaumik, A. Soft templating strategies for the synthesis of mesoporous materials: Inorganic, organic–inorganic hybrid and purely organic solids. Adv. Colloid Interface Sci. 2013, 189–190, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Boissière, C.; Grosso, D.; Laberty, C.; Nicole, L. Design, synthesis, and properties of inorganic and hybrid thin films having periodically organized nanoporosity. Chem. Mater. 2008, 20, 682–737. [Google Scholar] [CrossRef]
- Soler-Illia, G.J.A.A.; Azzaroni, O. Multifunctional hybrids by combining ordered mesoporous materials and macromolecular building blocks. Chem. Soc. Rev. 2011, 40, 1107–1150. [Google Scholar] [CrossRef] [PubMed]
- Olivier, A.; Meyer, F.; Raquez, J.-M.; Damman, P.; Dubois, P. Surface-initiated controlled polymerization as a convenient method for designing functional polymer brushes: From self-assembled monolayers to patterned surfaces. Prog. Polym. Sci. 2012, 37, 157–181. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, H.; Zhou, L.; Zhang, F. Confined polymerization: ARGET ATRP of MMA in the nanopores of modified SBA-15. Polymer 2017, 114, 180–188. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Z.-Y.; Su, B.-L. Hierarchically structured porous materials for energy conversion and storage. Adv. Funct. Mater. 2012, 22, 4634–4667. [Google Scholar] [CrossRef]
- Fu, K.; Bohn, P.W. Nanochannel arrays for molecular sieving and electrochemical analysis by nanosphere lithography templated graphoepitaxy of block copolymers. ACS Appl. Mater. Interfaces 2017, 9, 24908–24916. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Lin, X.; Su, B. Vertically ordered silica mesochannel films: Electrochemistry and analytical applications. Analyst 2016, 141, 3482–3495. [Google Scholar] [CrossRef] [PubMed]
- Vilà, N.; André, E.; Ciganda, R.; Ruiz, J.; Astruc, D.; Walcarius, A. Molecular sieving with vertically aligned mesoporous silica films and electronic wiring through isolating nanochannels. Chem. Mater. 2016, 28, 2511–2514. [Google Scholar] [CrossRef]
- Yang, M.-Y.; Tan, L.; Wu, H.-X.; Liu, C.-J.; Zhuo, R.-X. Dual-stimuli-responsive polymer-coated mesoporous silica nanoparticles used for controlled drug delivery. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Xu, P.; Chen, C.; Li, X. Mesoporous-silica nanofluidic channels for quick enrichment/extraction of trace pesticide molecules. Sci. Rep. 2015, 5, 17171. [Google Scholar] [CrossRef] [PubMed]
- Kruk, M. Surface-initiated controlled radical polymerization in ordered mesoporous silicas. Isr. J. Chem. 2012, 52, 246–255. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Ataman, N.C.; Mocny, P.; Wang, J.; Moraes, J.; Klok, H.-A. Surface-initiated controlled radical polymerization: State-of-the-art, opportunities, and challenges in surface and interface engineering with polymer brushes. Chem. Rev. 2017, 117, 1105–1318. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-W.; Higaki, Y.; Cheng, C.-H.; Cheng, M.-H.; Chang, C.-W.; Chen, J.-T.; Takahara, A. Zwitterionic polymer brush grafting on anodic aluminum oxide membranes by surface-initiated atom transfer radical polymerization. Polym. Chem. 2017, 8, 2309–2316. [Google Scholar] [CrossRef]
- Benetti, E.M.; Kang, C.; Mandal, J.; Divandari, M.; Spencer, N.D. Modulation of surface-initiated ATRP by confinement: Mechanism and applications. Macromolecules 2017, 50, 5711–5718. [Google Scholar] [CrossRef]
- Kruk, M.; Dufour, B.; Celer, E.B.; Kowalewski, T.; Jaroniec, M.; Matyjaszewski, K. Grafting monodisperse polymer chains from concave surfaces of ordered mesoporous silicas. Macromolecules 2008, 41, 8584–8591. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, X.; Kuang, T.; Chang, L.; Fu, D.; Yang, J.; Fan, P.; Zhong, M. Polyelectrolyte/mesoporous silica hybrid materials for the high performance multiple-detection of pH value and temperature. Polym. Chem. 2015, 6, 3529–3536. [Google Scholar] [CrossRef]
- Pan, X.; Tasdelen, M.A.; Laun, J.; Junkers, T.; Yagci, Y.; Matyjaszewski, K. Photomediated controlled radical polymerization. Prog. Polym. Sci. 2016, 62, 73–125. [Google Scholar] [CrossRef]
- Silies, L.; Didzoleit, H.; Hess, C.; Stühn, B.; Andrieu-Brunsen, A. Mesoporous thin films, zwitterionic monomers, and iniferter-initiated polymerization: Polymerization in a confined space. Chem. Mater. 2015, 27, 1971–1981. [Google Scholar] [CrossRef]
- Huang, L.; Dolai, S.; Raja, K.; Kruk, M. “Click” grafting of high loading of polymers and monosaccharides on surface of ordered mesoporous silica. Langmuir 2010, 26, 2688–2693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, W.; Xie, R.; Ju, X.-J.; Chu, L.-Y. Stimuli-responsive smart gating membranes. Chem. Soc. Rev. 2016, 45, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Gorman, C.B.; Petrie, R.J.; Genzer, J. Effect of substrate geometry on polymer molecular weight and polydispersity during surface-initiated polymerization. Macromolecules 2008, 41, 4856–4865. [Google Scholar] [CrossRef]
- Chen, M.; Zhong, M.; Johnson, J.A. Light-controlled radical polymerization: Mechanisms, methods, and applications. Chem. Rev. 2016, 116, 10167–10211. [Google Scholar] [CrossRef] [PubMed]
- De Boer, B.; Simon, H.K.; Werts, M.P.L.; van der Vegte, E.W.; Hadziioannou, G. “Living” free radical photopolymerization initiated from surface-grafted iniferter monolayers. Macromolecules 2000, 33, 349–356. [Google Scholar] [CrossRef]
- Krause, J.E.; Brault, N.D.; Li, Y.; Xue, H.; Zhou, Y.; Jiang, S. Photoiniferter-mediated polymerization of zwitterionic carboxybetaine monomers for low-fouling and functionalizable surface coatings. Macromolecules 2011, 44, 9213–9220. [Google Scholar] [CrossRef]
- Karesoja, M.; McKee, J.; Karjalainen, E.; Hietala, S.; Bergman, L.; Linden, M.; Tenhu, H. Mesoporous silica particles grafted with poly(ethyleneoxide-block-n-vinylcaprolactam). J. Polym. Sci. Part A Polym. Chem. 2013, 51, 5012–5020. [Google Scholar] [CrossRef]
- Boissiere, C.; Grosso, D.; Lepoutre, S.; Nicole, L.; Bruneau, A.B.; Sanchez, C. Porosity and mechanical properties of mesoporous thin films assessed by environmental ellipsometric porosimetry. Langmuir 2005, 21, 12362–12371. [Google Scholar] [CrossRef] [PubMed]
- Brunsen, A.; Calvo, A.; Williams, F.J.; Soler-Illia, G.J.A.A.; Azzaroni, O. Manipulation of molecular transport into mesoporous silica thin films by the infiltration of polyelectrolytes. Langmuir 2011, 27, 4328–4333. [Google Scholar] [CrossRef] [PubMed]
- Babu, D.J.; Yadav, S.; Heinlein, T.; Cherkashinin, G.; Schneider, J.J. Carbon dioxide plasma as a versatile medium for purification and functionalization of vertically aligned carbon nanotubes. J. Phys. Chem. C 2014, 118, 12028–12034. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Computer simulation of controlled radical polymerization: Effect of chain confinement due to initiator grafting density and solvent quality in “grafting from” method. Macromolecules 2010, 43, 9567–9577. [Google Scholar] [CrossRef]
- Suteewong, T.; Sai, H.; Bradbury, M.; Estroff, L.A.; Gruner, S.M.; Wiesner, U. Synthesis and formation mechanism of aminated mesoporous silica nanoparticles. Chem. Mater. 2012, 24, 3895–3905. [Google Scholar] [CrossRef]
- Matheron, M.; Bourgeois, A.; Brunet-Bruneau, A.; Albouy, P.-A.; Biteau, J.; Gacoin, T.; Boilot, J.-P. Highly ordered ctab-templated organosilicate films. J. Mater. Chem. 2005, 15, 4741–4745. [Google Scholar] [CrossRef]
- Matheron, M.; Gacoin, T.; Boilot, J.-P. Stabilization of well-organized transient micellar phases in CTAB-templated silica and organosilica thin films. Soft Matter 2007, 3, 223–229. [Google Scholar] [CrossRef]
- Cagnol, F.; Grosso, D.; Sanchez, C. A general one-pot process leading to highly functionalised ordered mesoporous silica films. Chem. Commun. 2004, 1742–1743. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, Y.; Ejaz, M.; Sato, K.; Goto, A.; Fukuda, T. Mechanism and kinetics of raft-mediated graft polymerization of styrene on a solid surface. 1. Experimental evidence of surface radical migration. Macromolecules 2001, 34, 8872–8878. [Google Scholar] [CrossRef]
- Benedikt, S.; Moszner, N.; Liska, R. Benzoyl phenyltelluride as highly reactive visible-light TERP-reagent for controlled radical polymerization. Macromolecules 2014, 47, 5526–5531. [Google Scholar] [CrossRef]
- Zhang, T.; Du, Y.; Muller, F.; Amin, I.; Jordan, R. Surface-initiated Cu(0) mediated controlled radical polymerization (SI-CuCRP) using a copper plate. Polym. Chem. 2015, 6, 2726–2733. [Google Scholar] [CrossRef]
- Barbey, R.; Lavanant, L.; Paripovic, D.; Schuwer, N.; Sugnaux, C.; Tugulu, S.; Klok, H.A. Polymer brushes via surface-initiated controlled radical polymerization: Synthesis, characterization, properties, and applications. Chem. Rev. 2009, 109, 5437–5527. [Google Scholar] [CrossRef] [PubMed]
- Von Werne, T.; Patten, T.E. Atom transfer radical polymerization from nanoparticles: A tool for the preparation of well-defined hybrid nanostructures and for understanding the chemistry of controlled/“living” radical polymerizations from surfaces. J. Am. Chem. Soc. 2001, 123, 7497–7505. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Akashi, T.; Huang, Y.; Tsujii, Y. Surface-initiated living radical polymerization from narrowly size-distributed silica nanoparticles of diameters less than 100 nm. Macromolecules 2010, 43, 8805–8812. [Google Scholar] [CrossRef]
- Ohno, K.; Morinaga, T.; Koh, K.; Tsujii, Y.; Fukuda, T. Synthesis of monodisperse silica particles coated with well-defined, high-density polymer brushes by surface-initiated atom transfer radical polymerization. Macromolecules 2005, 38, 2137–2142. [Google Scholar] [CrossRef]
- Jeyaprakash, J.D.; Samuel, S.; Dhamodharan, R.; Rühe, J. Polymer brushes via ATRP: Role of activator and deactivator in the surface-initiated ATRP of styrene on planar substrates. Macromol. Rapid Commun. 2002, 23, 277–281. [Google Scholar] [CrossRef]
- Dunphy, D.R.; Sheth, P.H.; Garcia, F.L.; Brinker, C.J. Enlarged pore size in mesoporous silica films templated by pluronic F127: Use of poloxamer mixtures and increased template/SiO2 ratios in materials synthesized by evaporation-induced self-assembly. Chem. Mater. 2015, 27, 75–84. [Google Scholar] [CrossRef]
- Gupta, S.; Agrawal, M.; Conrad, M.; Hutter, N.A.; Olk, P.; Simon, F.; Eng, L.M.; Stamm, M.; Jordan, R. Poly(2-(dimethylamino)ethyl methacrylate) brushes with incorporated nanoparticles as a SERS active sensing layer. Adv. Funct. Mater. 2010, 20, 1756–1761. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Location | Static Contact Angle (°) | ||
|---|---|---|---|
| With BDC 3 | Without BDC | Δ | |
| Pores only 2 | 53 ± 1 | 86 ± 1 | 32 |
| Pores + Exterior Surface | 65 ± 2 | 79 ± 4 | 14 |
| Total Polymerisation Time (min) | Static Contact Angle (°) | |
|---|---|---|
| With a SI | Without a SI | |
| 0 | 27 ± 0 | 27 ± 0 |
| 30 | 43 ± 1 | 63 ± 3 |
| 60 | 48 ± 3 | 62 ± 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tom, J.C.; Brilmayer, R.; Schmidt, J.; Andrieu-Brunsen, A. Optimisation of Surface-Initiated Photoiniferter-Mediated Polymerisation under Confinement, and the Formation of Block Copolymers in Mesoporous Films. Polymers 2017, 9, 539. https://doi.org/10.3390/polym9100539
Tom JC, Brilmayer R, Schmidt J, Andrieu-Brunsen A. Optimisation of Surface-Initiated Photoiniferter-Mediated Polymerisation under Confinement, and the Formation of Block Copolymers in Mesoporous Films. Polymers. 2017; 9(10):539. https://doi.org/10.3390/polym9100539
Chicago/Turabian StyleTom, Jessica C., Robert Brilmayer, Johannes Schmidt, and Annette Andrieu-Brunsen. 2017. "Optimisation of Surface-Initiated Photoiniferter-Mediated Polymerisation under Confinement, and the Formation of Block Copolymers in Mesoporous Films" Polymers 9, no. 10: 539. https://doi.org/10.3390/polym9100539