
Chemical Modification of Butyl Rubber with Maleic Anhydride via Nitroxide Chemistry and Its Application in Polymer Blends

 ,
,
Abstract
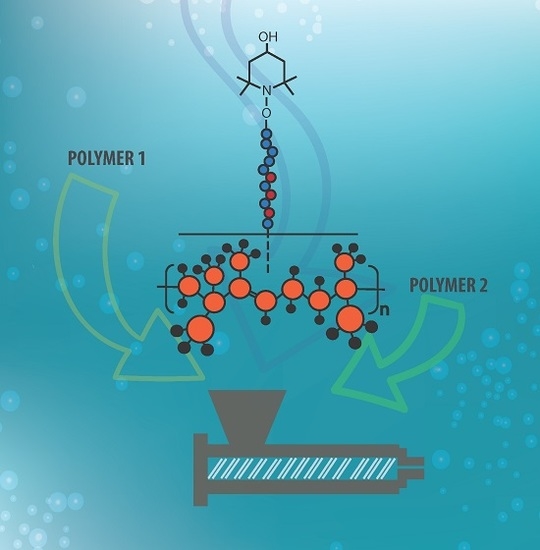
:
1. Introduction
Background
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Functionalization of IIR with Nitroxide Moieties at Lab Scale: IIRF1
2.4. Synthesis of Model Compatibilizer IIR-g-MA at Lab Scale
2.5. Functionalization of IIR with Nitroxide Moieties at a Large Scale: IIRF2
2.6. Synthesis of Compatibilizer IIR-g-SMA at a Large Scale
2.7. Melt Processing
2.8. Preparation of Clay Nanocomposites
2.9. Microtoming Procedure
2.10. Staining Procedure
3. Results and Discussions
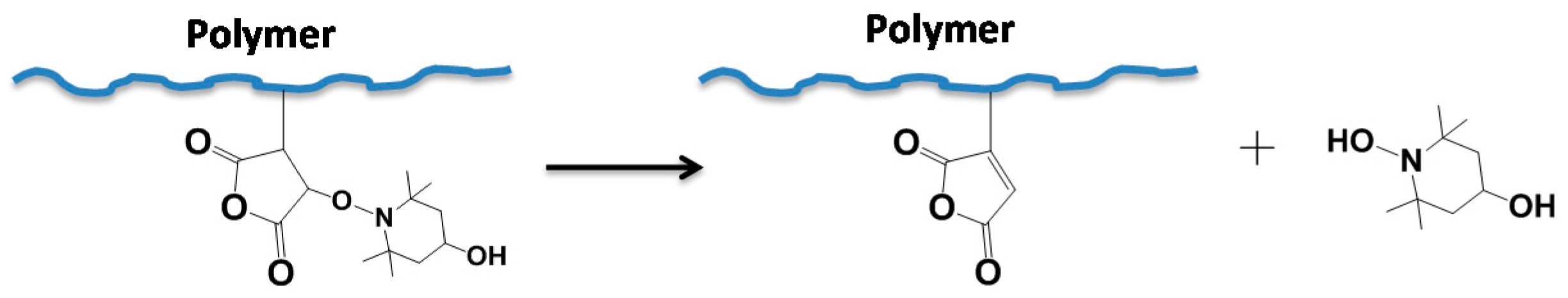
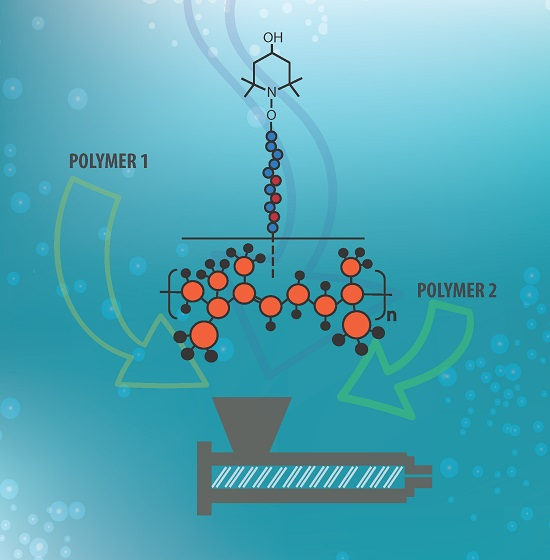
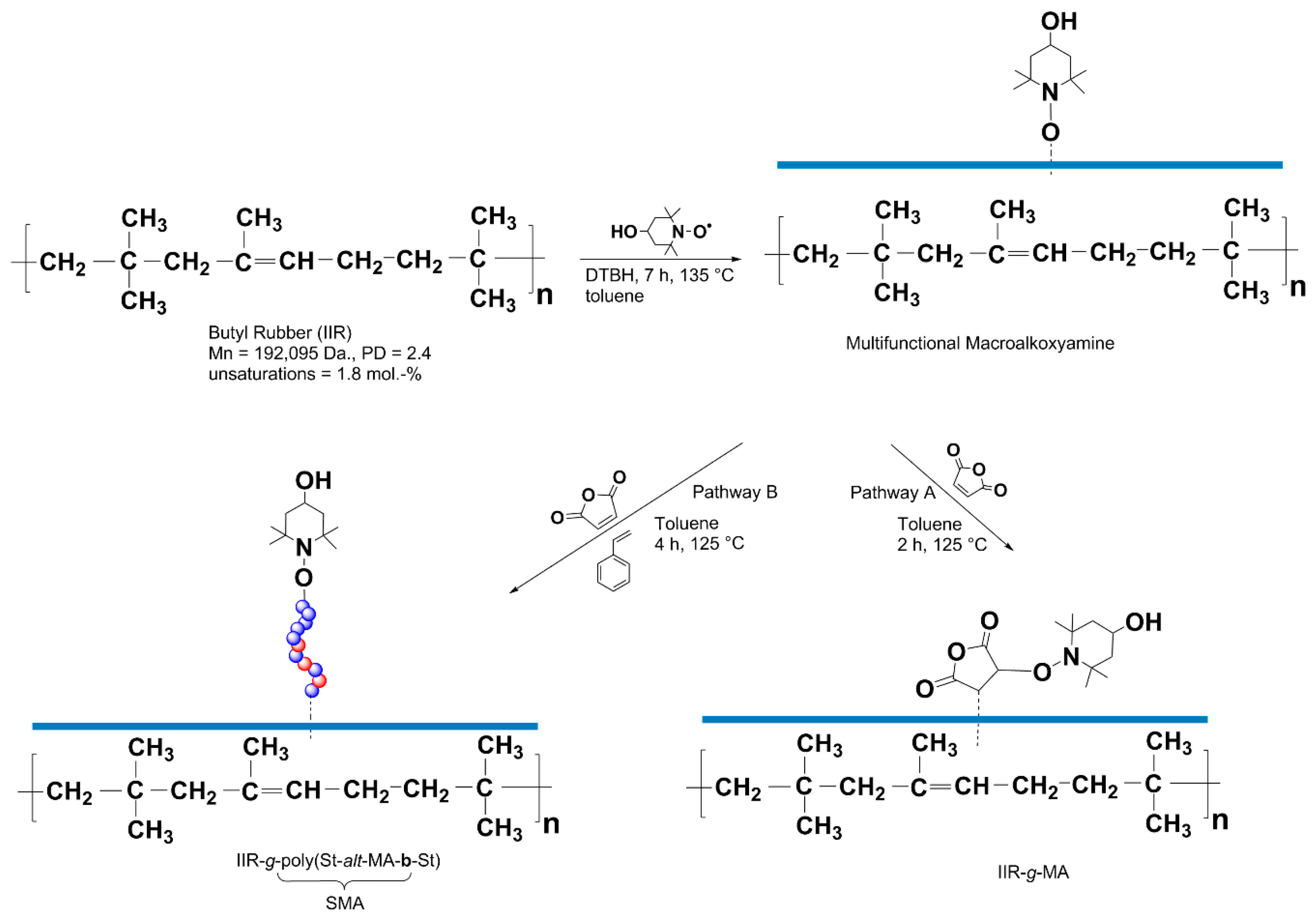
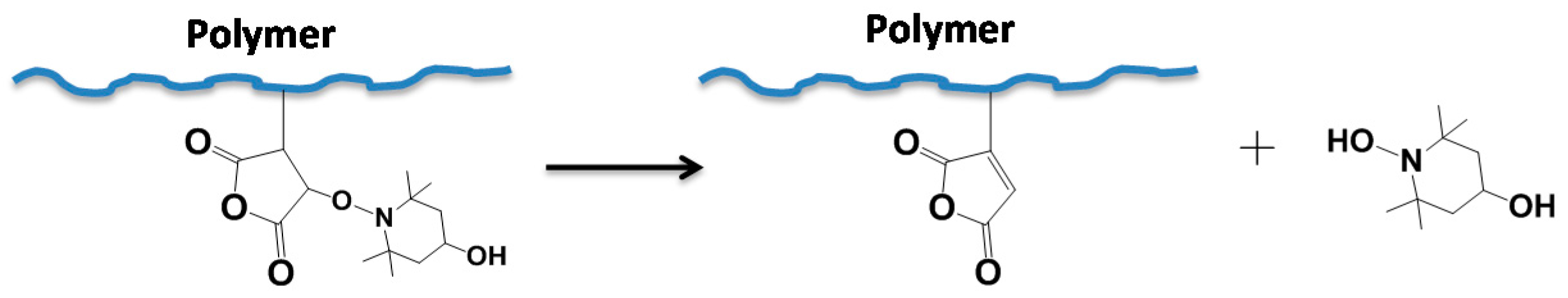
3.1. Synthesis Procedures and Possible Mechanisms
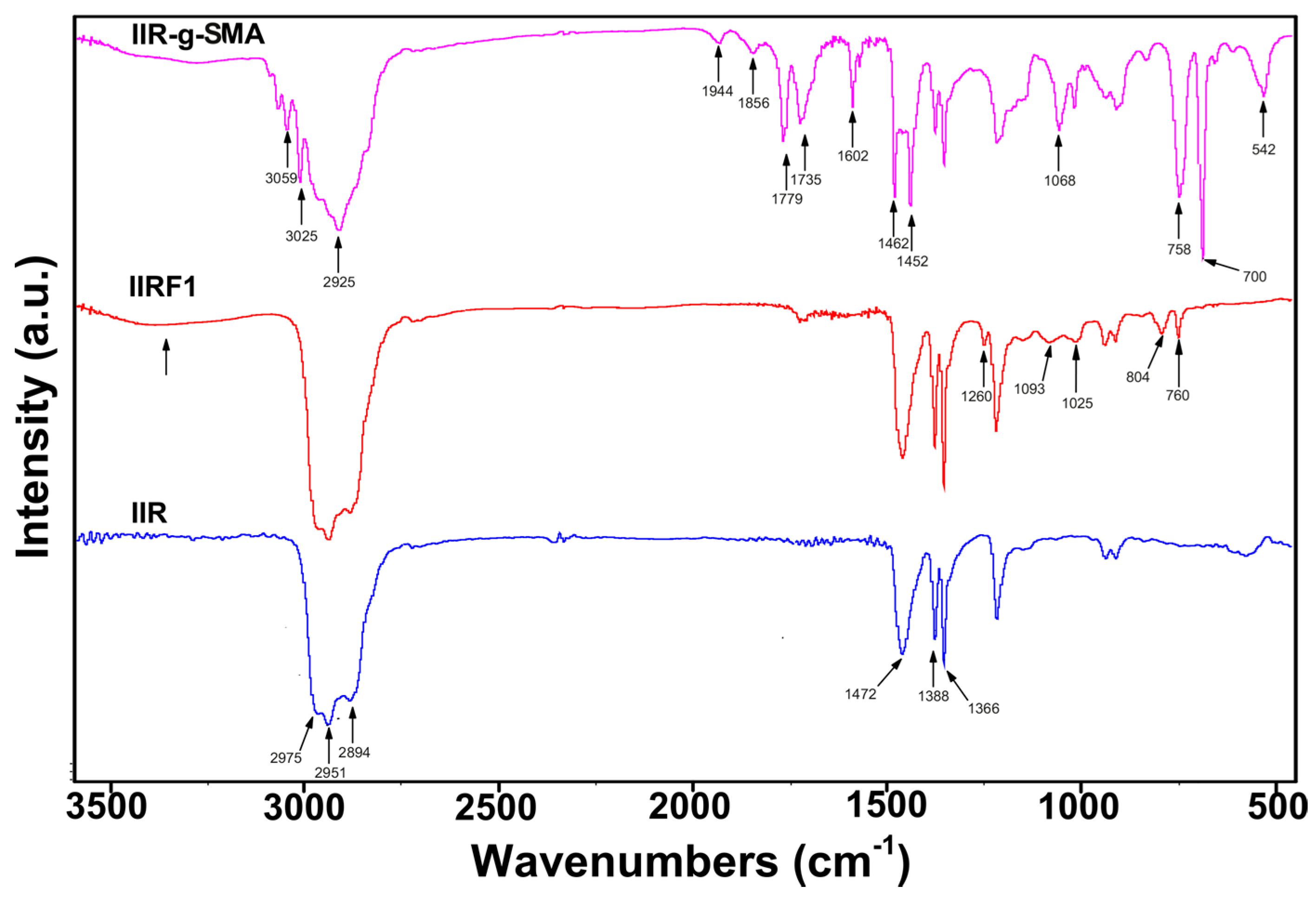
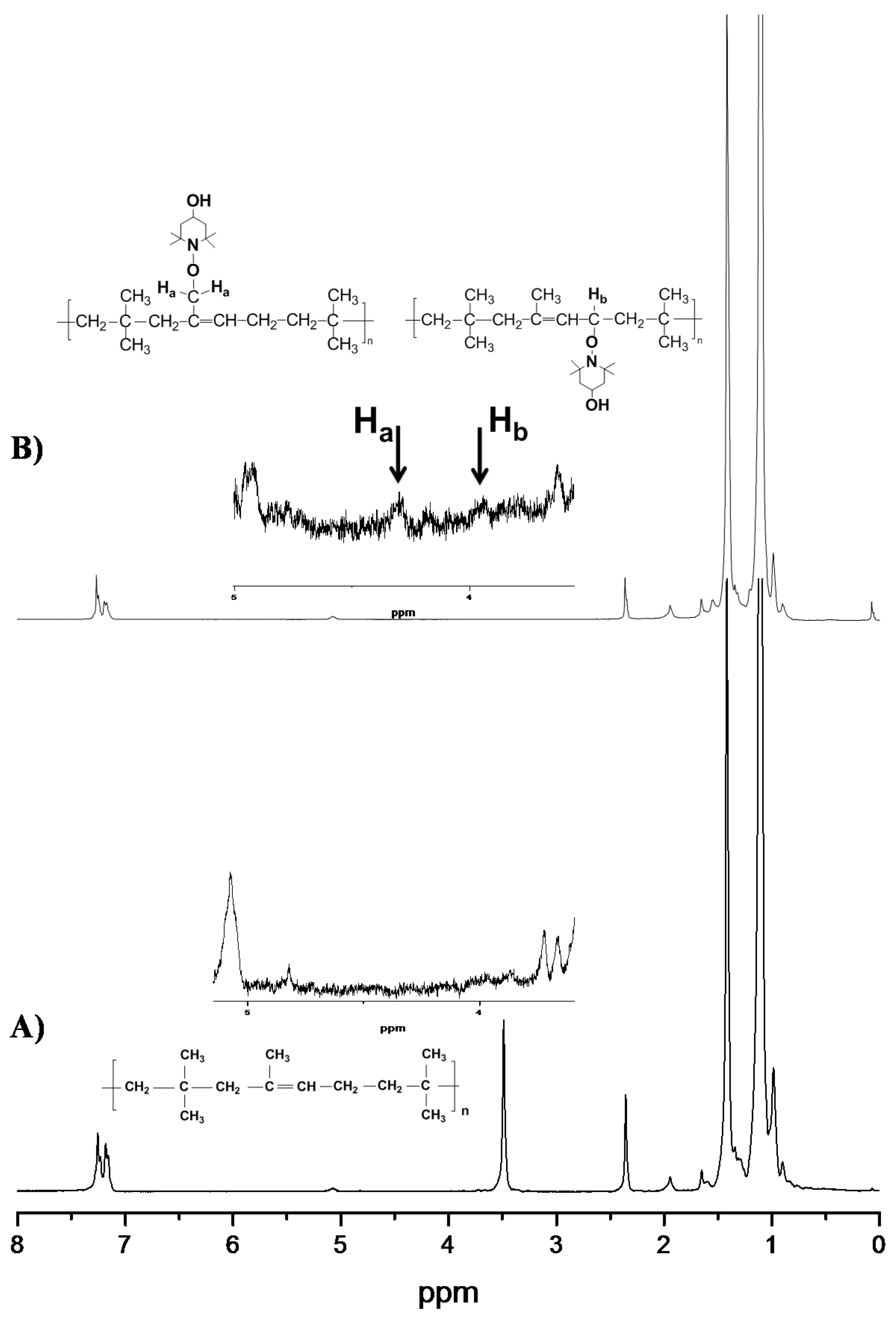
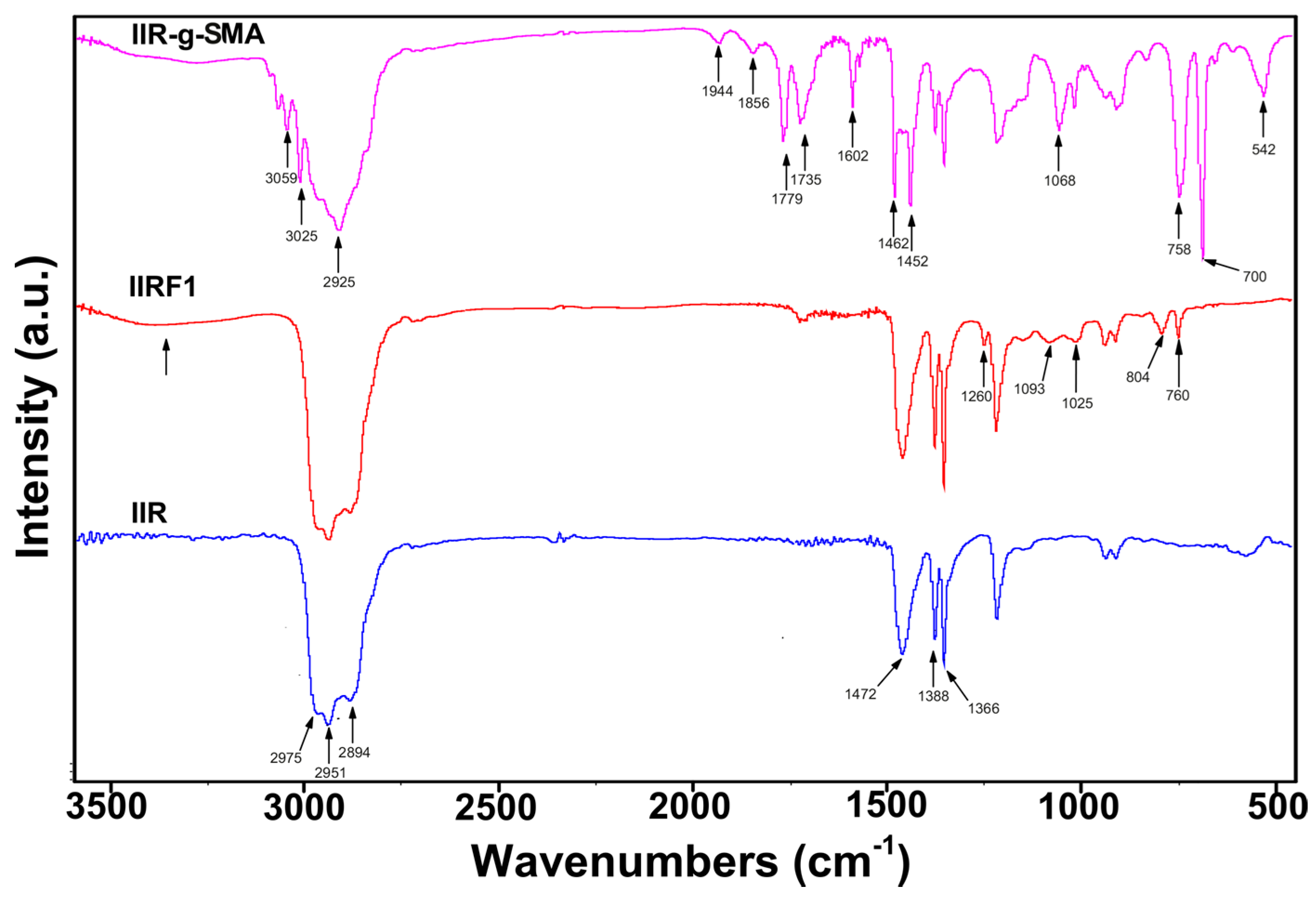
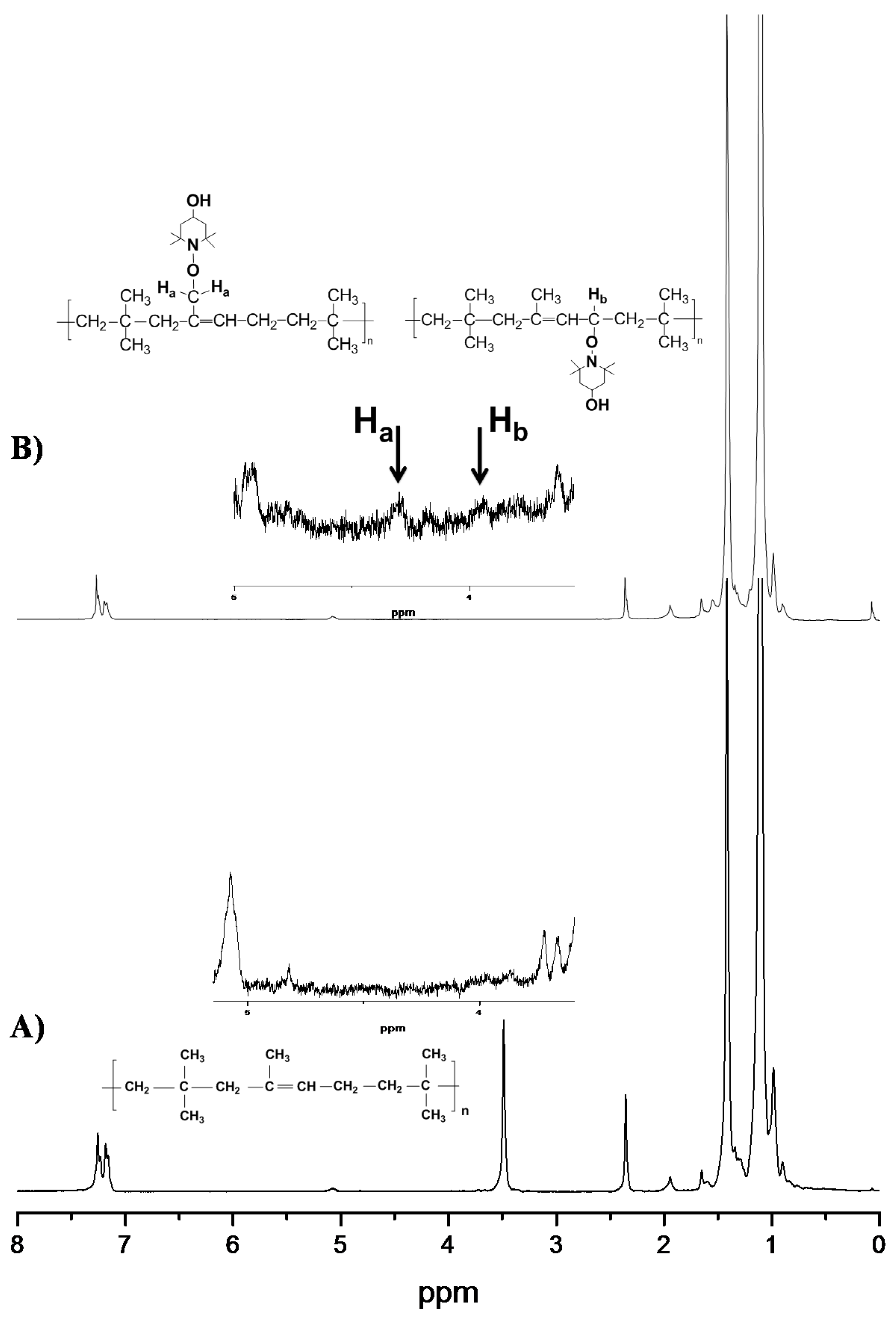
3.2. Spectroscopic Analysis
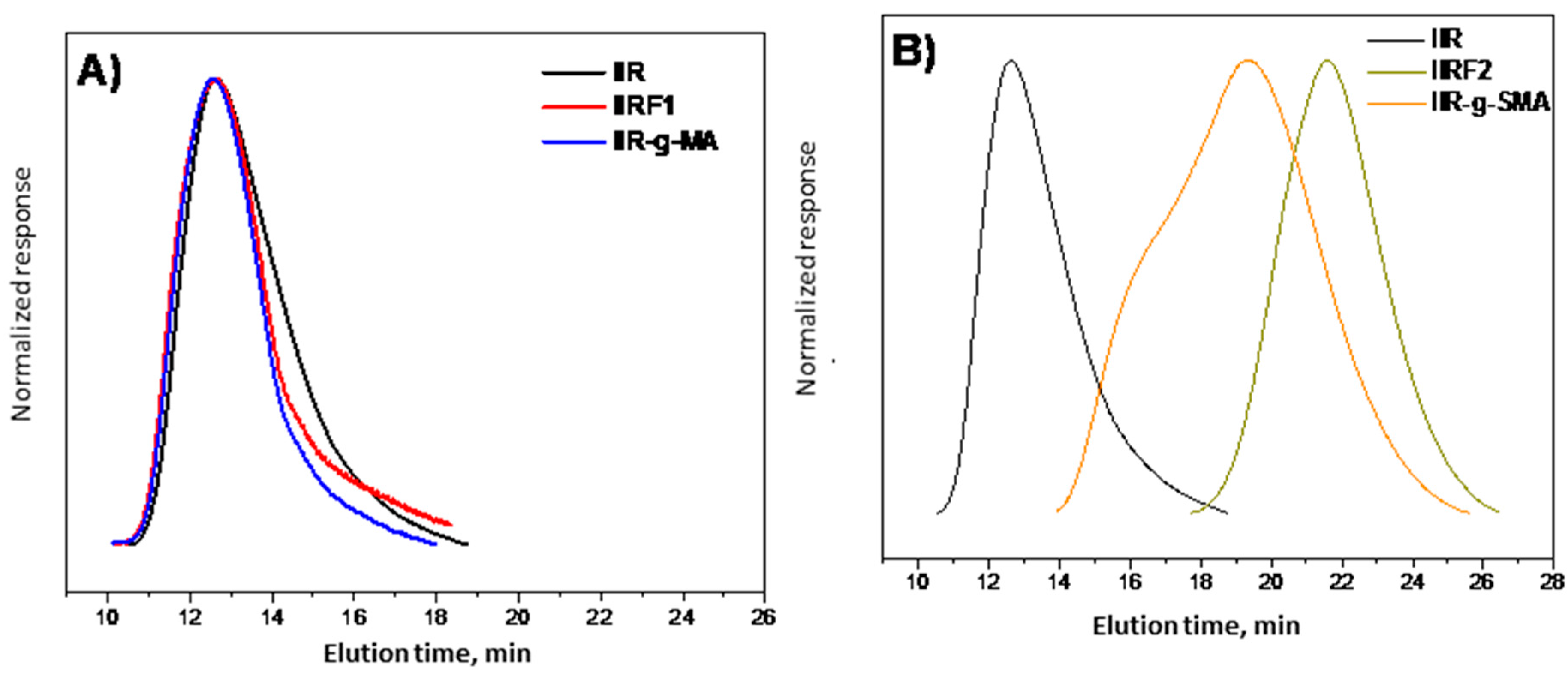
3.3. GPC Analysis
3.4. Blend Compatibilization
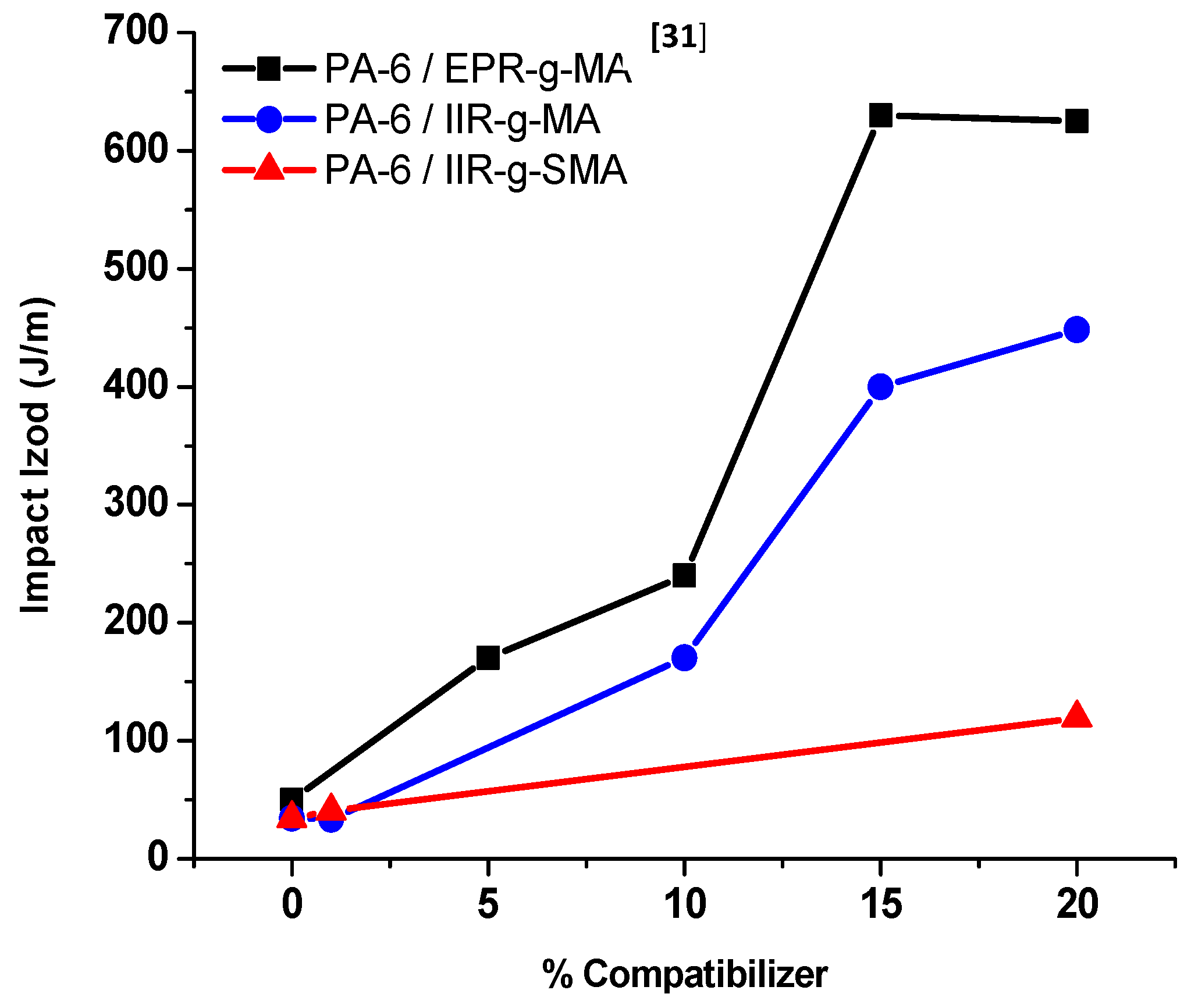
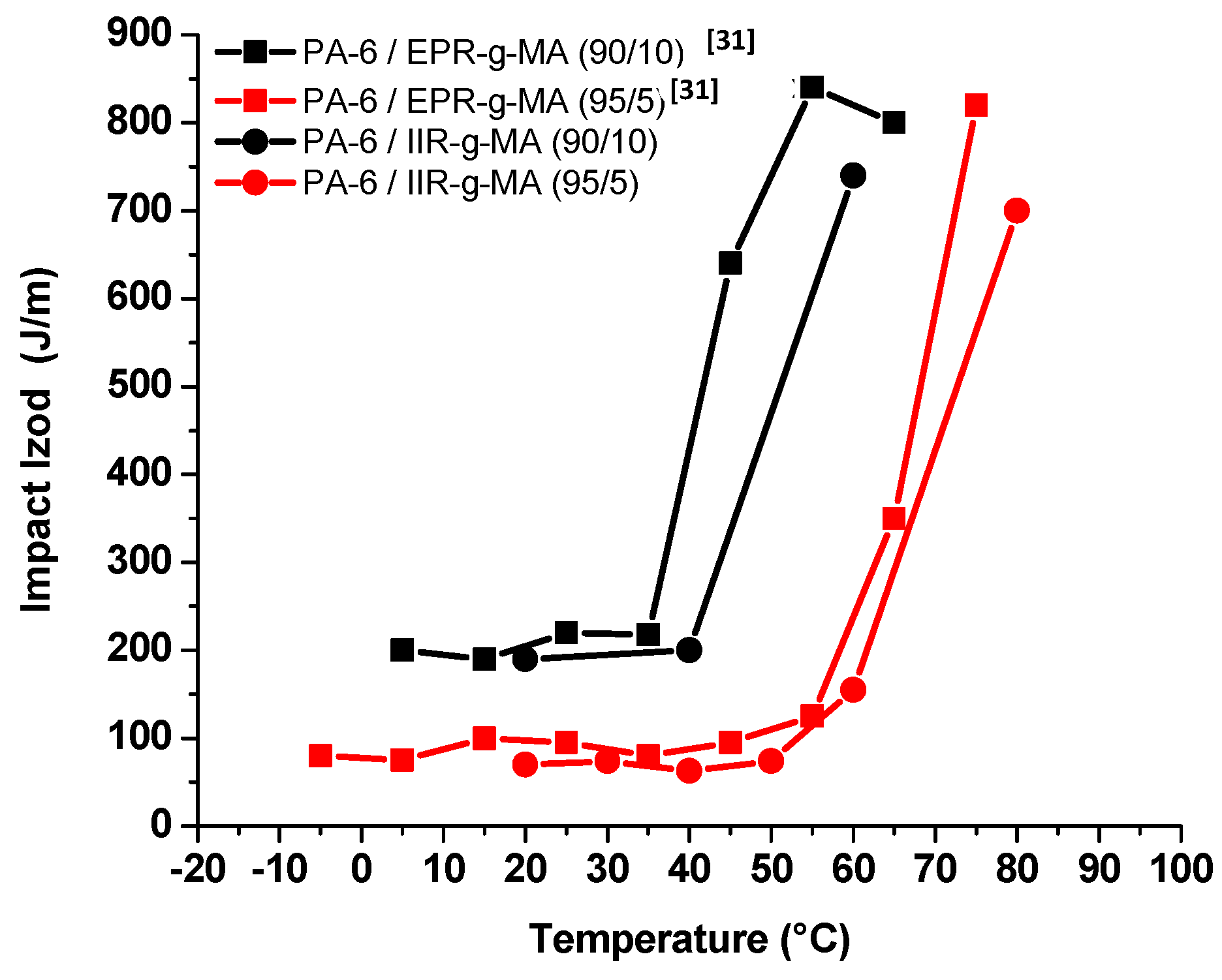
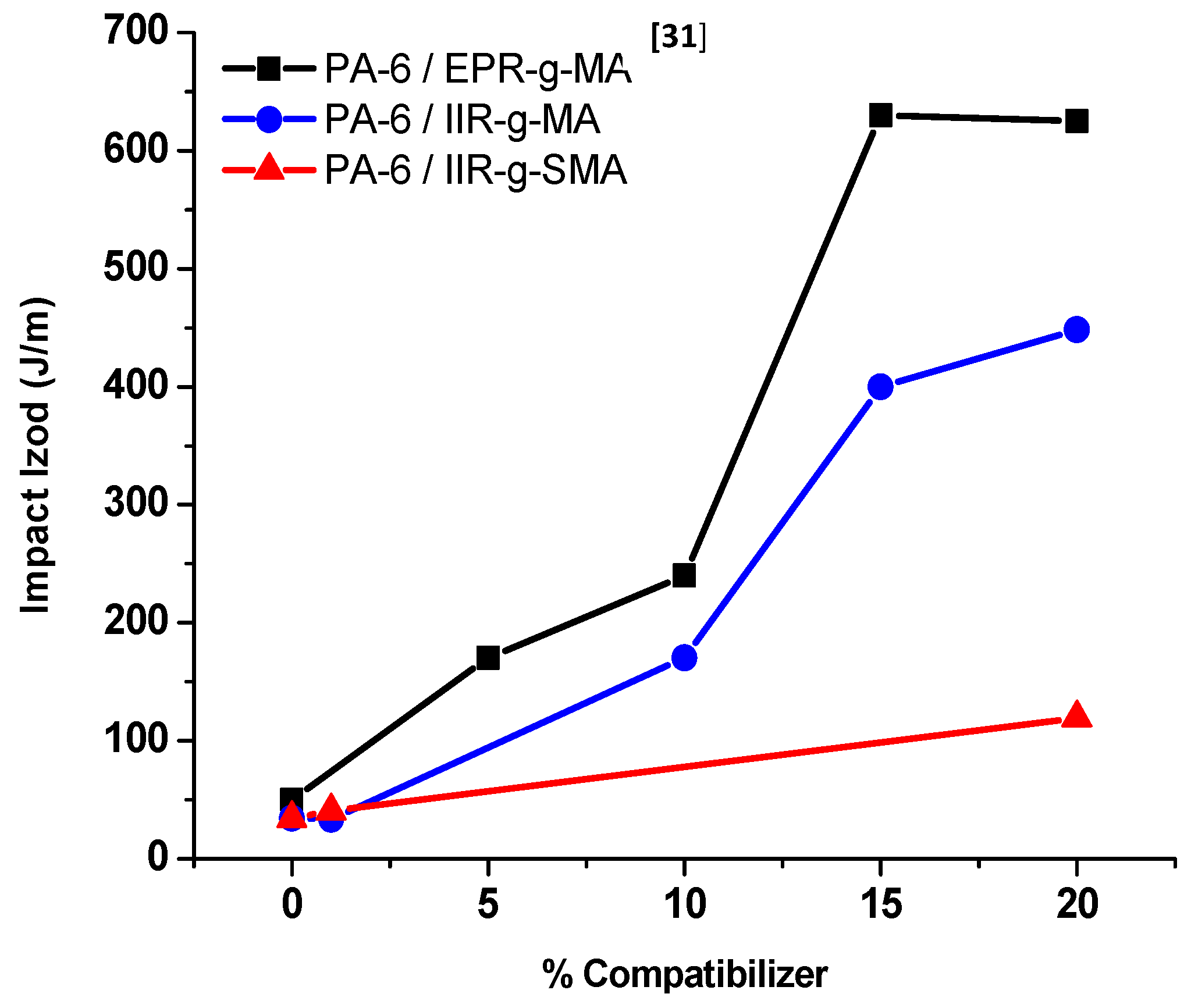
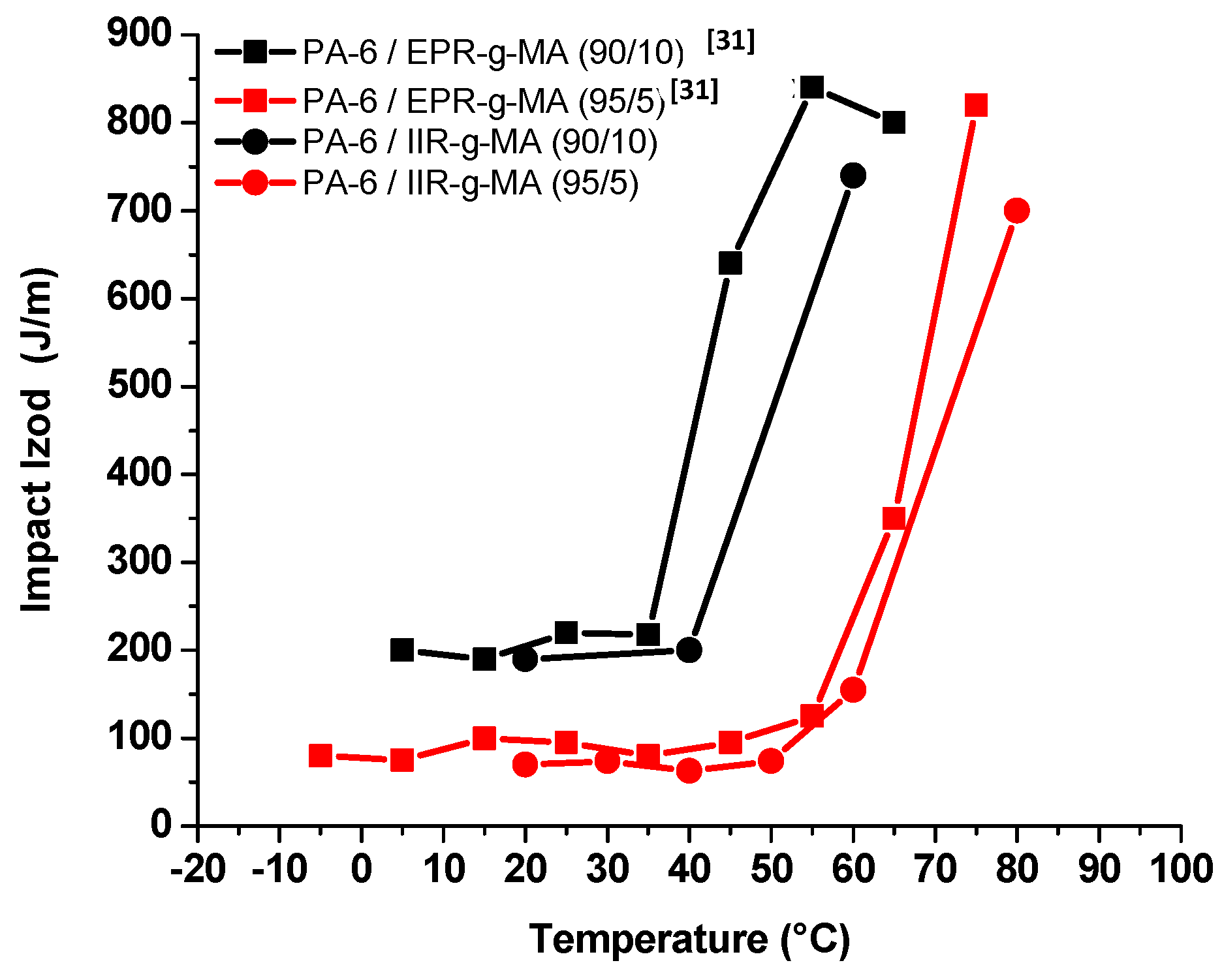
3.4.1. Izod Impact Strength
3.4.2. Blend Morphology
3.5. Nanocomposite Preparation and Testing
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cho, J.W.; Paul, D.R. Glass fiber-reinforced polyamide composites toughened with ABS and EPR-g-MA. J. Appl. Polym. Sci. 2001, 80, 484–497. [Google Scholar]
- Lee, D.-Y.; Subramanian, N.; Fellows, C.M.; Gilbert, R.G. Structure-property relationships in modified natural rubber latexes grafted with methyl methacrylate and vinyl neo-decanoate. J. Polym. Sci. Part A 2002, 40, 809–822. [Google Scholar] [CrossRef]
- González-Montiel, A.; Keskkula, H.; Paul, D.R. Morphology of nylon 6/polypropylene blends compatibilized with maleated polypropylene. J. Polym. Sci. B Polym. Phys. 1995, 33, 1751–1767. [Google Scholar] [CrossRef]
- Moad, G. The synthesis of polyolefin graft copolymers by reactive extrusion. Prog. Polym. Sci. 1999, 24, 81–142. [Google Scholar] [CrossRef]
- Bettini, S.H.P.; de Mello, L.C.; Muñoz, P.A.R.; Ruvolo-Filho, A. Grafting of maleic anhydride onto polypropylene, in the presence and absence of styrene, for compatibilization of poly(ethylene terephthalate)/(ethylene–propylene) blends. J. Appl. Polym. Sci. 2013. [Google Scholar] [CrossRef]
- Webb, R.N.; Shaffer, T.D.; Tsou, A.H. Butyl Rubber. In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 1998; Volume 8. [Google Scholar]
- Baldwin, F.P.; Rae, J.A.; Ver Strate, G. Method for Preparing Moisture Curable Polymers Containing Randomly Distributed Sites of Conjugated Olefinic Unsaturation. U.S. Patent 4,048,258 A, 13 September 1977. [Google Scholar]
- Canter, N.H.; Kennedy, J.P.; Exxon Research Engineering Co. Butyl Rubber Reaction Products. U.S. Patent 3,646,166 A, 29 February 1972. [Google Scholar]
- Abd Rabo Moustafa, M.M.; Gillies, E.R. Rubber functionalization by Diels−Alder chemistry: From cross-linking to multifunctional graft copolymer synthesis. Macromolecules 2013, 46, 6024–6030. [Google Scholar] [CrossRef]
- Ashiura, M.; Hiratsuka, J.P. Production of Maleic Anhydride Modified Butyl Rubber and Use Thereof. U.S. Patent 6,930,153 B, 16 August 2005. [Google Scholar]
- McLean, J.K.; Guillen-Castellanos, S.A.; Parent, J.S.; Whitney, R.A.; Resendes, R. Synthesis of graft copolymer derivatives of brominated poly(isobutylene-co-isoprene). Eur. Polym. J. 2007, 43, 4619–4627. [Google Scholar] [CrossRef]
- Parent, J.S.; Resendes, R.; Whitney, R.A. Liquid Maleated Butyl Rubber. U.S. Patent US20,090,189,118 A1, 30 July 2009. [Google Scholar]
- Haldar, S.K.; Singha, N.K. Grafting of butyl acrylate and methyl methacrylate on butyl rubber using electron beam radiation. J. Appl. Polym. Sci. 2006, 101, 1340–1346. [Google Scholar] [CrossRef]
- Perova, M.S.; Makasheva, A.M.; Khakimullin, Y.N. Influence of maleic anhydrate on properties of incongealable sealants based on butyl rubber. Polym. Sci. Ser. D 2011, 4, 125–128. [Google Scholar] [CrossRef]
- Bonduelle, C.V.; Gillies, E.R. Patterning of a butyl rubber-poly(ethylene oxide) graft copolymer revealed by protein adsorption. Macromolecules 2010, 43, 9230–9233. [Google Scholar] [CrossRef]
- Kaszas, G. Bromination of butyl rubber in the presence of electrophilic solvents and oxidizing agents. Rubber Chem. Technol. 2000, 73, 356–365. [Google Scholar] [CrossRef]
- Scott, M.E.; Parent, J.S.; Dupont, J.; Whitney, R.A. Controlled radical grafting: Nitroxyl-mediated maleation of model hydrocarbons. Ind. Eng. Chem. Res. 2003, 42, 3662–3670. [Google Scholar] [CrossRef]
- Bonilla-Cruz, J.; Saldívar-Guerra, E.; Torres-Lubián, J.R.; Guerrero-Santos, R.; López-Carpy, B.; Luna-Bárcenas, G. Controlled grafting-from of polystyrene on polybutadiene: Mechanism and spectroscopic evidence of the functionalization of polybutadiene with 4-oxo-TEMPO. Macromol. Chem. Phys. 2008, 209, 2268–2283. [Google Scholar] [CrossRef]
- Saldívar-Guerra, E.; Bonilla-Cruz, J.; Hernández-Mireles, B.; Ramírez-Manzanares, G. Progress in controlled grafting-from by nitroxide chemistry. Macromol. Symp. 2009, 283, 110–119. [Google Scholar] [CrossRef]
- Bonilla-Cruz, J.; Guerrero-Sanchez, C.; Schubert, U.; Saldívar-Guerra, E. Controlled “grafting-from” of poly[styrene-co-maleic anhydride] onto polydienes using nitroxide chemistry. Eur. Polym. J. 2010, 46, 298–312. [Google Scholar] [CrossRef]
- Van Dyke, J.D.; Gnatowski, M.; Koutsandreas, A.; Burczyk, A. Effect of butyl rubber type on properties of polyamide and butyl rubber blends. J. Appl. Polym. Sci. 2004, 93, 1423–1435. [Google Scholar] [CrossRef]
- Rajasekar, R.; Das, R.C.K. Development of butyl rubber nanocomposites in presence and absence of compatibiliser. Plast. Rubber Compos. 2011, 40, 407–412. [Google Scholar] [CrossRef]
- Corté, L.; Beaume, F.; Leibler, L. Crystalline organization and toughening: Example of polyamide-12. Polymer 2005, 46, 2748–2757. [Google Scholar] [CrossRef]
- Moad, G.; Solomon, D.H. The Chemistry of Free Radical Polymerization, 1st ed.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1995; p. 105. [Google Scholar]
- Finn, M.; Friedline, R.; Suleman, N.K.; Wohl, C.J.; Tanko, J.M. Chemistry of the t-Butoxyl Radical: Evidence that most hydrogen abstractions from carbon are entropy-controlled. J. Am. Chem. Soc. 2004, 126, 7578–7584. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.L.; Pavelich, W.A.; Clarey, H.D. Homopolymerization of maleic anhydride. I. Preparation of the polymer. J. Polym. Sci. Part A 1963, 1, 1123–1136. [Google Scholar] [CrossRef]
- Harth, E.; Hawker, C.J.; Fan, W.; Waymouth, R.M. Chain end functionalization in nitroxide-mediated “living” free radical polymerizations. Macromolecules 2001, 34, 3856–3862. [Google Scholar] [CrossRef]
- Bonilla-Cruz, J.; Caballero, J.; Albores-Velasco, M.; Saldívar-Guerra, E.; Percino, J.; Chapela, V. Mechanism and kinetics of the induction period in nitroxide mediated thermal autopolymerizations. Application to the spontaneous copolymerization of styrene and maleic anhydride. Macromol. Symp. 2007, 248, 132–140. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- White, J.L.; Shaffer, T.D.; Ruff, C.J.; Cross, J.P. Incorporation of isoprene in isobutylene/isoprene copolymers: NMR identification of branching in butyl rubber. Macromolecules 1995, 28, 3290–3300. [Google Scholar] [CrossRef]
- Huang, J.J.; Keskkula, H.; Paul, D.R. Comparison of the toughening behavior of nylon 6 versus an amorphous polyamide using various maleated elastomers. Polymer 2006, 47, 639–651. [Google Scholar] [CrossRef]
- Majumdar, B.; Keskkula, H.; Paul, D.R. Morphology development in toughened aliphatic polyamides. Polymer 1994, 35, 1386–1398. [Google Scholar] [CrossRef]
- Okada, O.; Keskkula, H.; Paul, D.R. Fracture toughness of nylon 6 blends with maleated ethylene/propylene rubbers. Polymer 2000, 41, 8061–8074. [Google Scholar] [CrossRef]
- Oshinski, A.J.; Keskkula, H.; Paul, D.R. The role of matrix molecular weight in rubber toughened nylon 6 blends: 1. Morphology. Polymer 1996, 37, 4891–4907. [Google Scholar] [CrossRef]
- Oshinski, A.J.; Keskkula, H.; Paul, D.R. Rubber toughening of polyamides with functionalized block copolymers: 1. Nylon-6. Polymer 1992, 33, 268–283. [Google Scholar] [CrossRef]
- Borggreve, R.J.M.; Gaymans, R.J.; Schuijer, J.; Housz, J.F.I. Brittle-tough transition in nylon-rubber blends: Effect of rubber concentration and particle size. Polymer 1987, 28, 1489–1496. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAG | PA-6 (wt %) | IIR (wt %) | IIR-g-MA (wt %) | IIR-g-SMA (wt %) | Residence time, min | Temperature (°C) |
|---|---|---|---|---|---|---|
| B1 | 100 | - | - | - | 15 | 240 |
| B2 | 80 | 20 | - | - | 15 | 240 |
| B3 | 99 | - | 1 | - | 15 | 240 |
| B4 | 90 | - | 10 | - | 15 | 240 |
| B5 | 85 | - | 15 | - | 15 | 240 |
| B6 | 80 | - | 20 | - | 15 | 240 |
| B7 | 99 | - | 1 | 15 | 240 | |
| B8 | 80 | - | - | 20 | 15 | 240 |
| B9 | 80 * | 20 * | - | 1 * | 15 | 240 |
| IIR-g-MA (wt %) | IIR (wt %) | (μm) | (μm) | (μm) | ||
|---|---|---|---|---|---|---|
| 0 | 20 | 0.85 | 1.26 | 2.41 | 1.48 | 2.84 |
| 10 | 0 | 0.20 | 0.21 | 0.23 | 1.06 | 1.19 |
| 20 | 0 | 0.17 | 0.20 | 0.24 | 1.13 | 1.42 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonilla-Cruz, J.; Hernández-Mireles, B.; Mendoza-Carrizales, R.; Ramírez-Leal, L.A.; Torres-Lubián, R.; RamosdeValle, L.F.; Paul, D.R.; Saldívar-Guerra, E. Chemical Modification of Butyl Rubber with Maleic Anhydride via Nitroxide Chemistry and Its Application in Polymer Blends. Polymers 2017, 9, 63. https://doi.org/10.3390/polym9020063
Bonilla-Cruz J, Hernández-Mireles B, Mendoza-Carrizales R, Ramírez-Leal LA, Torres-Lubián R, RamosdeValle LF, Paul DR, Saldívar-Guerra E. Chemical Modification of Butyl Rubber with Maleic Anhydride via Nitroxide Chemistry and Its Application in Polymer Blends. Polymers. 2017; 9(2):63. https://doi.org/10.3390/polym9020063
Chicago/Turabian StyleBonilla-Cruz, José, Brenda Hernández-Mireles, Ricardo Mendoza-Carrizales, Luis A. Ramírez-Leal, Román Torres-Lubián, Luis F. RamosdeValle, Donald R. Paul, and Enrique Saldívar-Guerra. 2017. "Chemical Modification of Butyl Rubber with Maleic Anhydride via Nitroxide Chemistry and Its Application in Polymer Blends" Polymers 9, no. 2: 63. https://doi.org/10.3390/polym9020063