1. Introduction
Soybeans (
Glycine max L.), used for food, livestock feed, and biofuel, is one of the most important agricultural crops worldwide. Soybeans are consumed directly by humans, especially in many Asian countries, in the form of traditional food products such as tofu, soy flour, and soymilk [
1,
2]. Soybean seeds are composed of 40%–42% protein, 18%–22% oil (85% unsaturated and 15% saturated fatty acids), 28% carbohydrates, and abundant quantities of other nutrients, such as phosphorus, calcium, iron, lysine, and vitamins A, B, and D [
3]. In addition, soybeans play an important role in crop diversification and improve other crops through its addition of nitrogen to the soil during crop rotation [
4].
Because the rate of spontaneous mutations in higher plants is quite low (10
−5 to 10
−8) [
5], physical and chemical mutagens can be used to induce mutations in cultivated plants [
6]. Gamma radiation is a very effective tool to induce genetic variation in many plant characters, with the resulting changes dependent on the irradiation dose. Various plant organisms, such as seeds, pollen, whole plants, and embryoid bodies, can be irradiated [
7]. Because gamma rays can also cause various types of DNA damage, including single- or double-strand breaks and substitutions [
8,
9], agronomic traits, such as flowering, maturation date, seed coat color, chloroplast number, and biomass yield, are frequently altered in soybean [
10,
11]. At present, 3200 mutant varieties of more than 210 plant species have been produced for commercial use. Approximately 170 mutant varieties of soybean, the second-most registered species after rice, are found in the FAO/IAEA Mutant Variety Database (
http://mvd.iaea.org).
The use of molecular marker-based techniques in genetic studies, such as estimation of genetic diversity and population structure, has advanced remarkably in recent years. Among the different types of DNA markers, restriction fragment length polymorphisms (RFLPs), random amplified polymorphic DNAs (RAPDs), amplified fragment length polymorphisms (AFLPs), and inter-simple sequence repeats (ISSRs) have been extensively used in soybeans, each with their own advantages and limitations [
12]. In addition, SNPs, which are widely distributed throughout genomes in both non-coding and coding regions, constitute the most abundant molecular markers recently used in plant genetic breeding [
13], but their development is time-consuming and costly. The target region amplification polymorphism (TRAP) is a relatively new, simple, polymerase chain reaction (PCR)-based marker system that takes advantage of the available EST database sequence information to generate polymorphic markers targeting candidate gene [
14]. Essentially, it derives an 18-mer primer from the EST sequence and pairs it with an arbitrary primer that targets the intron and/or exon region (AT- or GC-rich core). Because it can be used to generate markers for specific gene sequences, the TRAP technique is useful for genotyping germplasm and generating markers associated with desirable crop agronomic traits for marker-assisted breeding [
15]. In recent years, the TRAP marker technique has been applied for genetic diversity analyses [
16,
17] and genetic mapping [
18]. In addition, Im et al. [
19] have developed a transposable element-based TRAP (TE-TRAP) marker system that is reportedly suitable for the mutation breeding of sorghum. Although TRAP markers have most commonly been used for genetic mapping and phylogenetic studies, they have also recently been applied to detect DNA mutations [
20].
Rapid advances in the field of molecular biology and its allied sciences have led to the routine use of molecular markers, thereby providing plant breeders with a precise genetic-diversity analysis tool for plant improvement [
21,
22]. A combined molecular and morphological analysis is one of the most widely used approaches for the estimation of genetic distances within a group of genotypes, and molecular markers serve as an excellent tool for obtaining genetic information. Molecular markers are also of great value to plant breeders for assessment of genetic divergence among genotypes for various agronomic traits [
23]. Another recent strategy for analyzing agronomic traits, association analysis based on molecular-marker linkage disequilibrium (LD), can reduce experimental time and costs. Association analysis has therefore been widely applied to study a variety of crops, such as rice [
24], maize [
25], and soybeans [
26].
In this study, we constructed 208 mutant diversity pool (MDP) lines based on agronomic traits and investigated their genetic diversity and relationships using TRAP markers. Finally, we performed an association analysis between agronomic traits and polymorphic TRAP amplicons.
4. Discussion
In this study, we constructed an MDP from populations of 1695 gamma-irradiated mutants in two selection phases over M
1 to M
12 generations; first, in the M
5 generation, we selected 523 mutant lines exhibiting at least 30% superior agricultural characteristics, and, second, we eliminated redundant morphological phenotypes in the M
12 generation (
Figure 1). Finally, we constructed 208 MDP lines and investigated 11 agronomic traits. Our collection strategy for selecting MDP lines differed in some respects from the general core-collection method. With the latter approach, a core collection assembled from an existing collection is chosen to represent the genetic and phenotypic diversity of the larger collection without overlapping phenotypes [
30]. Such an approach has become accepted as an efficient tool for improving the conservation of many crops [
31,
32]. In our study, we similarly eliminated overlapping phenotypes from our collected MDP lines in the second selection phase, but we considered specific changed agronomic characteristics of individual mutants rather than their representation of the original populations.
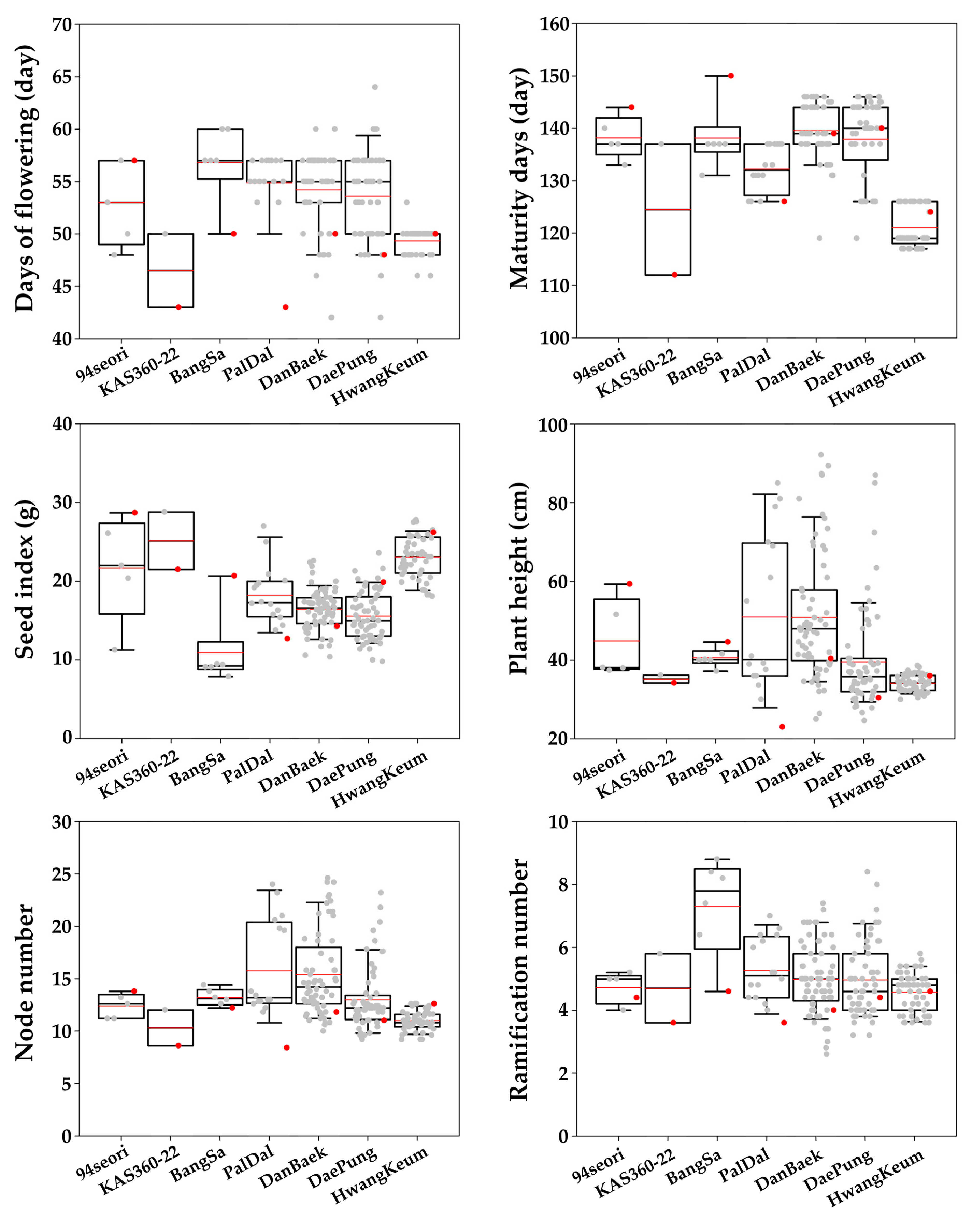
Our examination of agronomic traits in the MDP lines revealed a variety of DF, MD, GT, FC, SA, PH, NN, and RN phenotypes as well as those related to seed traits, such as SCC, SHC, and SI (
Table 2,
Figure 2,
Table S1). We also observed changes in phenotypes between MDP lines and their wild types (
Figure 3,
Table S3, Figure S1). The FAO/IAEA mutant variety database (MVD,
http://mvd.iaea.org) includes 174 publicly released soybean mutants. These mutants have various desirable agronomical and biochemical characteristics, such as an improved maturity date, yield, protein content, fatty acid content, and changed seed/stem color, with approximately 62% of released mutants mainly selected for their altered maturity dates and yields. In our phenotypic evaluation of the 208 MDP lines, we detected a wider variety of changes to the quantitative traits, including SI, PH, NN, and RN (
Figure 3), as well as to the qualitative traits, such as FC, SCC, and SHC (
Figure S1). According to our previous study, in addition, some of DB- and DP-derived mutants in the MDP lines had changed compositions of fatty acids, including linolenic acid and oleic acid [
33]. Given all of these results, our MDP lines may be useful resources as a genetic diversity pool for soybean breeding.
To investigate genetic relationships among the 208 MDP lines, we evaluated DNA polymorphism patterns in these lines using TRAP markers. In the rapid, efficient PCR-based TRAP marker system, expressed sequence tag database information and bioinformatics tools are used to generate polymorphic markers around targeted candidate gene sequences. Previous studies of lettuce (
Lactuca sativa) [
34], sugarcane (
Saccharum officinarum) [
35], spinach (
Spinacia oleracea) [
29], geranium (
Pelargonium inquinans) [
36], sunflower (
Helianthus annuus) [
37], and faba beans (
Vicia faba) [
16] have demonstrated that TRAP markers are useful for assessing genetic diversity. Using this system in the present study, we PCR-amplified 551 fragments with 16 primer combinations and observed considerable variation in the percentage of polymorphic amplicons among primer pairs—from 32.35% to 84.00% (
Table 4). In a study of faba beans, Kwon et al. [
16] obtained 221 amplified fragments with 12 TRAP primer combinations and observed an average polymorphism rate of 55.2%. In the present study, we observed a polymorphism level of 59.7% among 551 amplified fragments. In contrast, a study of sugarcane detected a polymorphism rate of 74% from 925 amplified fragments [
17], a level much higher than in the soybeans (
Glycine max) and the faba beans. Compared with the results of previous studies of soybeans based on ISSR and RAPD techniques [
38,
39], the use of the TRAP system yielded more DNA fragments per primer combination. A previous AFLP analysis generated an average of 40 to 50 DNA fragments per primer pair [
40,
41], similar to the outcome of our TRAP analysis. The present results demonstrate that the TRAP marker system is a simple yet powerful technique for estimating soybean genetic diversity.
To reveal relationships among the 208 MDP lines, we constructed a UPGMA-based dendrogram using the TRAP marker data. On the basis of genetic distances, the 208 MDP lines clustered into four groups. An analysis of the population structure based on an ad-hoc statistic (
ΔK) likewise divided the MDP lines into four groups. These results indicate that four genotype-based sub-populations are present in the 208 MDP lines (
Figure 4) that largely correspond to their wild-type cultivars. As denoted by different colors, the main membership composition of the four groups and their subgroups is as follows: Group I including two wild types (94seori, and KAS360-22) possessed 52% red and 46% blue; Group II including BS and P was 80% blue; Group III-a including DP was 88% yellow; Group III-b including HK was 71% green; and Group IV including DB was 90% red. In a previous genetic diversity analysis based on 20 SSR markers, 91 Korean soybean cultivars were divided into seven groups at a genetic distance of 0.81. In that study, HK and P were clearly separated, but three cultivars (BS, DB, and DP) grouped together [
42]. Using TRAP markers in the present study, we were able to better resolve groups of wild-type cultivars. In addition, we performed an AMOVA to separate the total molecular variance of the mutants into inter- and intra-population components (
Table 5) and assessed their significance using permutational testing procedures. Overall, based on the dendrogram and population structure, 201 mutant lines grouped with their wild type except 29 (14%) mutant lines, including 22 DP- and 7 HK-mutants. Nevertheless, these mutant lines also possessed their genetic membership according to population structure. In AMOVA, all intra-mutant population also showed lower variation than inter-mutant population except for two populations, DB- and DP-, since DB- and DP- had most large mutant lines, 64 and 60, respectively. A similar result was described by Lee et al. [
20]. Each of the ten wild types was clustered with their M
1 generation mutants by gamma radiation in faba bean. However, the genetic variation of the mutants is not much higher than among cultivars or accessions. Although TRAP markers have most commonly been used for genetic mapping and dendrogram studies, they have also recently been applied to detect DNA mutations. Because of their many advantages, including simplicity, reliability, moderate throughput, and ease of sequencing of selected bands, TRAP markers have been used widely in plants. For example, the TRAP system has been used to study genetic variability induced by gamma ray treatments in sugarcane [
43] and sorghum (
Sorghum bicolor) [
19]. Lee et al. [
20] recently exploited a TRAP marker to estimate the frequency of mutations induced by gamma rays in an M
1 generation of faba bean. The 242 amplified fragments obtained using eight primer combinations had an average polymorphism rate of 66.7%, which is higher than the percentage in our study because they used early generation. TRAP markers have several advantages over other types of markers: they are easy to use (like RAPDs), high in polymorphisms (like AFLPs), and their primers can be readily designed from known sequences of putative genes [
44].
In association mapping, false discoveries are a major concern and can be partially attributed to spurious associations caused by population structure and unequal relatedness among individuals. Two major approaches, namely, GLM and MLM, are used to study marker–trait associations. The number of SMTAs detected by GLM is generally much higher than that revealed by MLM [
45]. GLM-based studies of marker–trait associations consider only the Q matrix generated during the study of population structure. In contrast, MLM simultaneously accounts for both population structure and kinship (genetic relatedness among individuals) and is hence more reliable. In the present study, the GLM method (Q) uncovered 178 SMTAs between the 11 phenotypic traits and 27 TRAP markers. Using the MLM method (Q + K), 143 SMTAs involving 27 TRAP markers were identified (
Table 6,
Table S2). These results confirm a previous observation that the number of SMTAs estimated with GLM is higher than that uncovered with MLM [
46,
47]. Most interestingly, the three approaches considering kinship and/or population structure in the MDP collection in this study revealed six SMTAs at
p < 0.0001 in all approach methods. These six SMTAs involved five agronomic traits: GT (2), FC (1), SCC (1), PH (1), and NN (1).

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}