Optimizing ddRADseq in Non-Model Species: A Case Study in Eucalyptus dunnii Maiden

 , , , , ,
, , , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Evaluation of Enzymes and Size Selection Range
2.3. Protocol 1 (P1): Optimized ddRADseq
2.4. Protocol 2 (P2): Optimized ddRADseq (Scaling Up to 24 Samples)
2.5. ddRADseq Data Analyses
2.6. SSR Identification
2.7. Evaluation of Robustness—Sequencing Platform Comparison
3. Results
3.1. Evaluation of Enzymes and Size Selection Range
3.2. Protocol 1: Analysis in Samples A and B
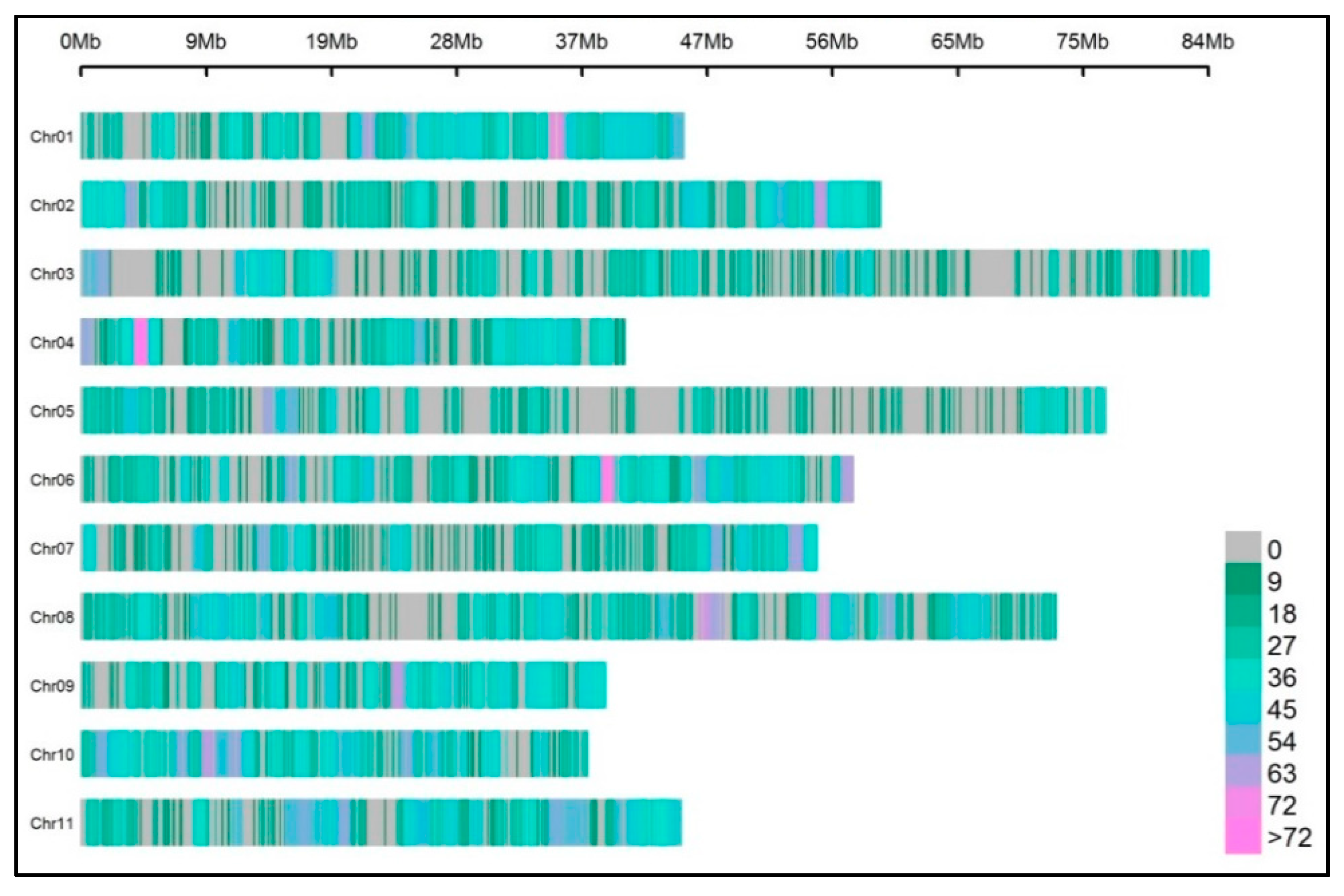
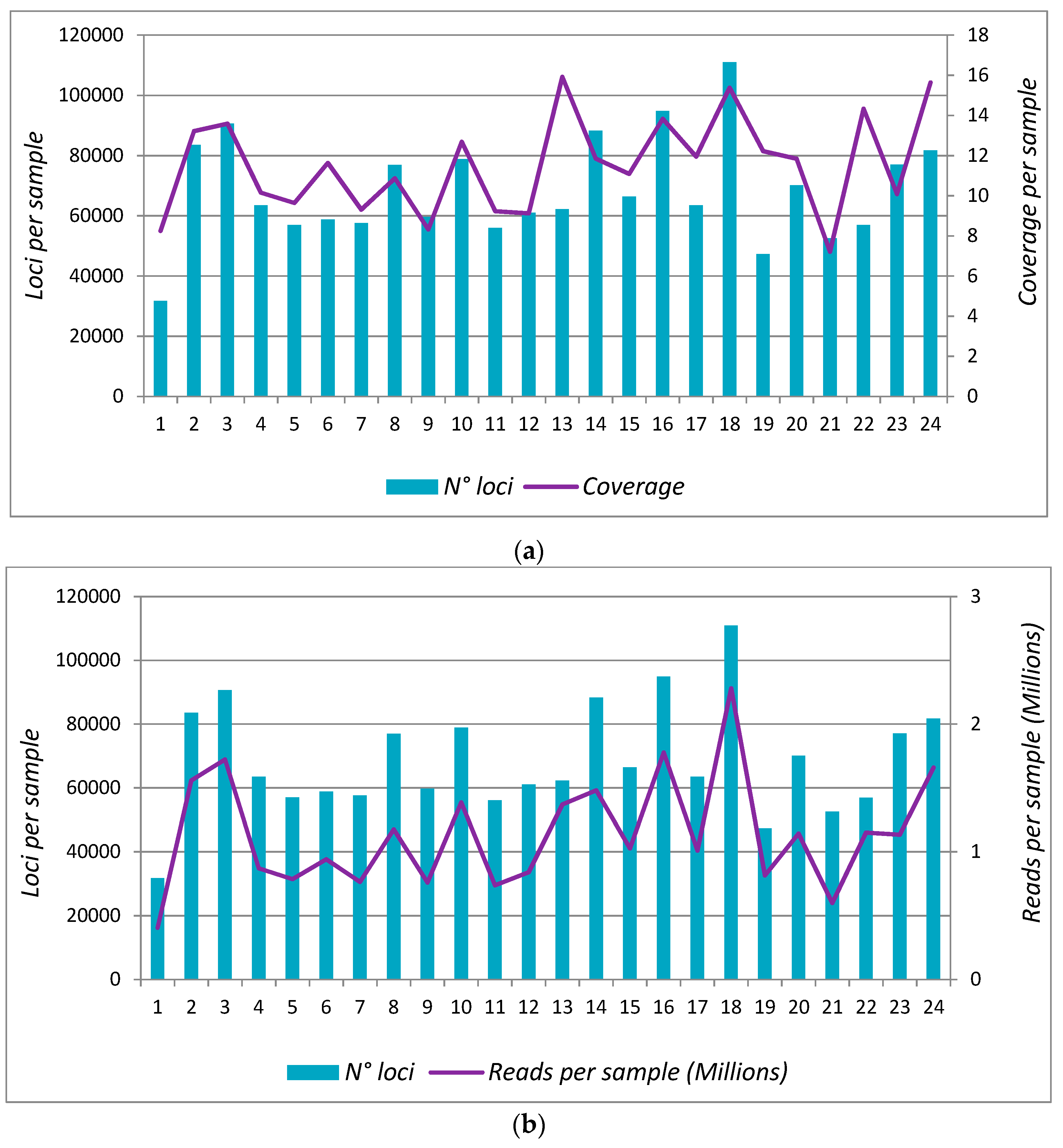
3.3. Protocol 2: Analysis in 24 Samples (Scaling-Up)
3.4. Evaluation of Robustness—Sequencing Platform Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torkamaneh, D.; Boyle, B.; Belzile, F. Efficient Genome wide genotyping strategies and data integration in crop plants. Theor. Appl. Genet. 2018, 131, 499. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, H.; Weigand, H.; Weiss, M.; Leese, F.; Rahmann, S. DDRAGE: A data set generator to evaluate ddRADseq analysis software. Mol. Ecol. Resour. 2018, 18, 681–690. [Google Scholar] [CrossRef]
- Wang, S.; Meyer, E.; Mckay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef]
- Toonen, R.J.; Puritz, J.B.; Forsman, Z.H.; Whitney, J.L.; Fernandez-Silva, I.; Andrews, K.R.; Bird, C.E. ezRAD: A simplified method for genomic genotyping in non-model organisms. PeerJ 2013, 1, e203. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimun sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Pyne, R.; Honig, J.; Vaiciunas, J.; Koroch, A.; Wyenandt, C.; Bonos, S.; Simon, J. A first linkage map and downy mildew resistance QTL discovery for sweet basil (Ocimum basilicum) facilitated by double digestion restriction site associated DNA sequencing (ddRADseq). PLoS ONE 2017, 12, e0184319. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.C.; Moitra, K.; De Sarker, D. Assessment of genetic diversity among four orchids based on ddRAD sequencing data for conservation purposes. Physiol. Mol. Biol. Plants 2017, 23, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Vargas, O.M.; Ortiz, E.M.; Simpson, B.B. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 2017, 214, 1736–1750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xia, Y.; Ren, X.; Chen, Y.; Huang, L.; Huang, S.; Liao, B.; Lei, Y.; Yan, L.; Jiang, H. Construction of SNP-based genetic linkage map in cultivated peanut basedon large scale marker development using next-generation double-digest restriction associated DNA sequencing (ddRADseq). BMC Genom. 2014, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Jahner, J.P.; Uckele, K.A.; Galland, L.M.; Eckert, A.J. RADseq approaches and applications for forest tree genetics. Tree Genet. Genomes 2018, 14. [Google Scholar] [CrossRef]
- Peterson, G.W.; Dong, Y.; Horbach, C.; Fu, Y.B. Genotyping-by-sequencing for plant genetic diversity analysis: A lab guide for SNP genotyping. Diversity 2014, 6, 665–680. [Google Scholar] [CrossRef]
- Yang, G.Q.; Chen, Y.M.; Wang, J.P.; Guo, C.; Zhao, L.; Wang, X.Y.; Guo, Y.; Li, L.; Li, D.Z.; Guo, Z.H. Development of a universal and simplified ddRAD library preparation approach for SNP discovery and genotyping in angiosperm plants. Plant Methods 2016, 12, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchi, L.; Lanteri, S.; Portis, E.; Acquadro, A.; Valè, G.; Toppino, L.; Rotino, G.L. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genom. 2011, 12, 304. [Google Scholar] [CrossRef]
- Torales, S.L.; Rivarola, M.; Gonzalez, S.; Inza, M.V.; Pomponio, M.F.; Fernández, P.; Acuña, C.V.; Zelener, N.; Fornés, L.; Hopp, H.E.; et al. De novo transcriptome sequencing and SSR markers development for Cedrela balansae C.DC., a native tree species of northwest Argentina. PLoS ONE 2018, 13, e0203768. [Google Scholar] [CrossRef]
- Hodel, R.G.J.; Segovia-Salcedo, M.C.; Landis, J.B.; Crowl, A.A.; Sun, M.; Liu, X.; Gitzendanner, M.A.; Douglas, N.A.; Germain-Aubrey, C.C.; Chen, S.; et al. The report of my death was an exaggeration: A review for researchers using microsatellites in the 21st century. Appl. Plant Sci. 2016, 4, 1600025. [Google Scholar] [CrossRef]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D. A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Sansaloni, C.P.; Petroli, C.D.; Carling, J.; Hudson, C.J.; Steane, D.A.; Myburg, A.A.; Grattapaglia, D.; Vaillancourt, R.E.; Kilian, A. A high-density Diversity Arrays Technology (DArT) microarray for genome-wide genotyping in Eucalyptus. Plant Methods 2010, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Grattapaglia, D.; de Alencar, S.; Pappas, G. Genome-wide genotyping and SNP discovery by ultra-deep Restriction-Associated DNA (RAD) tag sequencing of pooled samples of E. grandis and E. globulus. BMC Proc. 2011, 5 (Suppl. 7), P45. [Google Scholar] [CrossRef]
- Hoisington, D.; Khairallah, M.; González-de-león, D. Laboratory Protocols: CIMMYT Applied Molecular Genetics Laboratory, 2nd ed.; CIMMYT: México, DF, Mexico, 1994. [Google Scholar]
- Marcucci Poltri, S.N.; Zelener, N.; Rodriguez Traverso, J.; Gelid, P.; Hopp, H.E. Selection of a seed orchard of Eucalyptus dunnii based on genetic diversity criteria calculated using molecular markers. Tree Physiol. 2003, 23, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepais, O.; Weir, J.T. SimRAD: An R package for simulation-based prediction of the number of loci expected in RADseq and similar genotyping by sequencing approach. Mol. Ecol. Resour. 2014. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, D.; Fornasiero, A.; Pinto, C.; Cattonaro, F.; Spadotto, A.; Infante, R.; Meneses, C.; Messina, R.; Lain, O.; Cipriani, G.; et al. A RAD-based linkage map of kiwifruit (Actinidia chinensis Pl.) as a tool to improve the genome assembly and to scan the genomic region of the gender determinant for the marker-assisted breeding. Tree Genet. Genomes 2015, 11. [Google Scholar] [CrossRef]
- Lange, V.; Böhme, I.; Hofmann, J.; Lang, K.; Sauter, J.; Schöne, B.; Paul, P.; Albrecht, V.; Andreas, J.M.; Baier, D.M. Cost-efficient high-throughput HLA typing by MiSeq amplicon sequencing. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC: A Quality Control Tool for High Throuput Sequencing Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 November 2015).
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 (Bethesda) 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Torkamaneh, D.; Laroche, J.; Belzile, F. Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies. PLoS ONE 2016, 11, e0161333. [Google Scholar] [CrossRef] [PubMed]
- Wickland, D.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 2017, 18, 586. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Qin, H.; Yang, G.; Provan, J.; Liu, J.; Gao, L. Using MiddRADseq data to develop polymorphic microsatellite markers for an endangered yew species. Plant Divers. 2017, 39, 294–299. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A High-performance Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. Bioinformatics 2012. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Durán, R.; Zapata-Valenzuela, J.; Balocchi, C.; Valenzuela, S. Efficiency of EUChip60K pipeline in fingerprinting clonal population of Eucalyptus globulus. Trees 2018, 32, 663. [Google Scholar] [CrossRef]
- Cappa, E.P.; de Lima, B.M.; da Silva-Junior, O.B.; Garcia, C.C.; Mansfield, S.D.; Grattapaglia, D. Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci. 2019. [Google Scholar] [CrossRef]
- Müller, B.S.F.; de Almeida Filho, J.E.; Lima, B.M.; Garcia, C.C.; Missiaggia, A.; Aguiar, A.M.; Takahashi, E.; Kirst, M.; Gezan, S.A.; Silva-Junior, O.B.; et al. Independent and Joint-GWAS for growth traits in Eucalyptus by assembling genome-wide data for 3373 individuals across four breeding populations. New Phytol. 2019, 221, 818–833. [Google Scholar] [CrossRef]
- Suontama, M.; Klápště, J.; Telfer, E.; Graham, N.; Stovold, T.; Low, C.; McKinley, R.; Dungey, H. Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity 2019, 122, 370–379. [Google Scholar] [CrossRef]
- Albrechtsen, A.; Nielsen, F.C.; Nielsen, R. Ascertainment biases in SNP chips affect measures of population divergence. Mol. Biol. Evol. 2010, 27, 2534–2547. [Google Scholar] [CrossRef]
- Bajgain, P.; Rouse, M.N.; Anderson, J.A. Comparing genotyping-by-sequencing and single nucleotide polymorphism chip genotyping for quantitative trait loci mapping in wheat. Crop Sci. 2016, 56, 232–248. [Google Scholar] [CrossRef]
- Li, B.; Kimmel, M. Factors influencing ascertainment bias of microsatellite allele sizes: Impact on estimates of mutation rates. Genetics 2013, 195, 563–572. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: choosing the right tool for the right application. Plant Biotech. J. 2017, 15, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Inglis, P.W.; Pappas, M.C.R.; Resende, L.V.; Grattapaglia, D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS ONE 2018, 13, e0206085. [Google Scholar] [CrossRef]
- Kess, T.; Gross, J.; Harper, F.; Boulding, E.G. Low-cost ddRAD method of SNP discovery and genotyping applied to the periwinkleLittorina saxatilis. J. Molluscan Stud. 2016, 82, 104–109. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, X.; Zhao, Y.; Fei, J.; Hu, X.; Li, N. Optimized double-digest genotyping by sequencing (ddGBS) method with high-density SNP markers and high genotyping accuracy for chickens. PLoS ONE 2017, 12, e0179073. [Google Scholar] [CrossRef]
- Tan, G.; Opitz, L.; Schlapbach, R.; Rehrauer, H. Long fragments achieve lower base quality in Illumina paired-end sequencing. Sci. Rep. 2019, 9, 2856. [Google Scholar] [CrossRef]
- DaCosta, J.M.; Sorenson, M.D. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLoS ONE 2014, 9, e106713. [Google Scholar] [CrossRef]
- Quail, M.A.; Kozarewa, I.; Smith, F.; Scally, A.; Stephens, P.J.; Durbin, R.; Swerdlow, H.; Turner, D.J. A large genome center’s improvements to the Illumina sequencing system. Nat. Methods 2008, 5, 1005–1010. [Google Scholar] [CrossRef]
- Heavens, D.; Garcia Accinelli, G.; Clavijo, B.; Clark, M.D. A method to simultaneously construct up to 12 differently sized Illumina Nextera long mate pair libraries with reduced DNA input, time, and cost. BioTechniques 2015, 59, 42–45. [Google Scholar] [CrossRef] [Green Version]
- Rochette, N.C.; Catchen, J.M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc. 2017, 12, 2640–2659. [Google Scholar] [CrossRef]
- Merino, G. Imputación de Genotipos Faltantes en Datos de Secuencación Masiva. Master’s Thesis, Facultada de Ciencias Agrarias, Universidad Nacional de Córdoba, Córdoba, Argentina, 2018. [Google Scholar]
- Hendre, P.S.; Kamalakannan, R.; Rajkumar, R.; Varghese, M. High-throughput targeted SNP discovery using Next Generation Sequencing (NGS) in few selected candidate genes in Eucalyptus camaldulensis. BMC Proc. 2011, 5, O17. [Google Scholar] [CrossRef]
- Külheim, C.; Hui Yeoh, S.; Maintz, J.; Foley, W.; Moran, G. Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genom. 2009, 10, 452. [Google Scholar] [CrossRef]
- Campbell, E.O.; Davis, C.S.; Dupuis, J.R.; Muirhead, K.; Sperling, F.A.H. Cross-platform compatibility of de novo-aligned SNPs in a nonmodel butterfly genus. Mol. Ecol. Resour. 2017, 17, e84–e93. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Window | Manual Size Selection | Automated Size Selection | ||||
|---|---|---|---|---|---|---|---|
| Insert Size 100 or 150 bp Window (mean) | Fragment Size in Gel (Protocol 1) | Hypothetical Fragment Size in Gel (Protocol 2; mean) | N° of Predicted Fragments (100 or 150 bp window) | Insert Size 70 bp (1 or 2 wells) | N° of Predicted Fragments (100 or 150 bp window) | ||
| SphI-MboI | 100 or 70 bp | 270–370 (320) | 400–500 | 350–450 (400) | 28,107 | 285–355 (1) | 19,184 |
| 320–420 (370) | 450–550 | 400–500 (450) | 24,508 | 335–405 (1) | 17,317 | ||
| 370–470 (420) | 500–600 | 450–550 | 19,347 | 385–455 (1) | 12,906 | ||
| 150 bp | 220–370 (295) | 350–500 | 300–450 | 45,655 | 225–365 (2) | ~ manual selection a | |
| 270–420 (345) | 400–550 | 350–500 | 39,122 | 265–415 (2) | ~ manual selection a | ||
| PstI-MspI | 100 or 70 bp | 270–370 (320) | 400–500 | 350–450 | 13,102 | 285–355 (1) | 9137 |
| 320–420 (370) | 450–550 | 400–500 (450) | 12,026 | 335–405 (1) | 8359 | ||
| 370–470 (420) | 500–600 | 450–550 | 10,940 | 385–455 (1) | 7595 | ||
| 150 bp | 220–370 (295) | 350–500 | 300–450 | 20,749 | 225–365 (2) | ~ manual selection a | |
| 270–420 (345) | 400–550 | 350–500 | 18,826 | 265–415 (2) | ~ manual selection a | ||
| Analysis | Total | Shared | |||||
|---|---|---|---|---|---|---|---|
| SNPs | loci | SSRs | SNPs | loci | SSRs | SSRs Polym. | |
| with reference | 19,525 | 9299 | 4246 | 15,792 | 7346 | 420 | 16 |
| de novo | 33,313 | 18,951 | 7717 | 25,778 | 14,423 | 1366 | 55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre, N.C.; Filippi, C.V.; Zaina, G.; Rivas, J.G.; Acuña, C.V.; Villalba, P.V.; García, M.N.; González, S.; Rivarola, M.; Martínez, M.C.; et al. Optimizing ddRADseq in Non-Model Species: A Case Study in Eucalyptus dunnii Maiden. Agronomy 2019, 9, 484. https://doi.org/10.3390/agronomy9090484
Aguirre NC, Filippi CV, Zaina G, Rivas JG, Acuña CV, Villalba PV, García MN, González S, Rivarola M, Martínez MC, et al. Optimizing ddRADseq in Non-Model Species: A Case Study in Eucalyptus dunnii Maiden. Agronomy. 2019; 9(9):484. https://doi.org/10.3390/agronomy9090484
Chicago/Turabian StyleAguirre, Natalia Cristina, Carla Valeria Filippi, Giusi Zaina, Juan Gabriel Rivas, Cintia Vanesa Acuña, Pamela Victoria Villalba, Martín Nahuel García, Sergio González, Máximo Rivarola, María Carolina Martínez, and et al. 2019. "Optimizing ddRADseq in Non-Model Species: A Case Study in Eucalyptus dunnii Maiden" Agronomy 9, no. 9: 484. https://doi.org/10.3390/agronomy9090484