Characterization of Genetic Diversity in Accessions of Prunus salicina Lindl: Keeping Fruit Flesh Color Ideotype While Adapting to Water Stressed Environments

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction, PCR, and Gel Electrophoresis Conditions
2.3. Genetic Analyses
2.4. Mapping of SSR Markers on the Prunus persica Genome and Identification of Their Flanking Genes
3. Results
3.1. Diversity Study
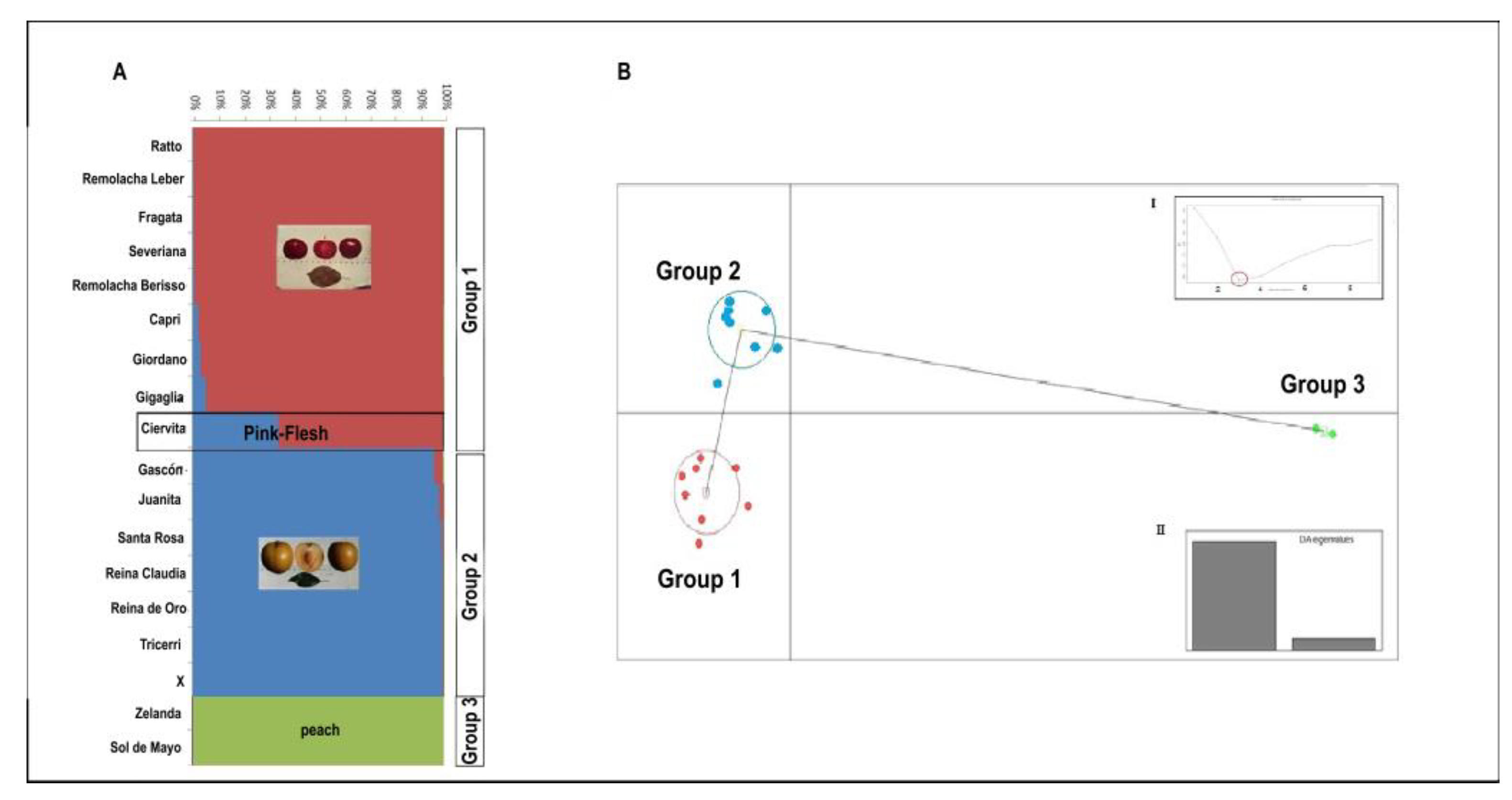
3.2. Cluster and Genetic Structure Analyses
3.3. Mapping of SSR Markers on the Prunus persica Genome and Identification of Their Flanking Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Das, B. Prunus diversity—Early and present development: A review. Int. J. Biodivers. Conserv. 2012, 3, 14. [Google Scholar]
- Mnejja, M.; Garcia-Mas, J.; Howad, W.; Badenes, M.L.; Arús, P. Simple-sequence repeat (SSR) markers of Japanese plum (Prunus salicina Lindl.) are highly polymorphic and transferable to peach and almond. Mol. Ecol. Notes 2004, 4, 163–166. [Google Scholar] [CrossRef]
- Kandus, P.; Quintana, R.D. The Paraná River Delta. In The Wetland Book; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–9. [Google Scholar]
- Caffera, R.; Berbery, H. Climatologia de la Cuenca del Plata. Clim. La Cuenca Del Plata 2006, 20, 19–38. [Google Scholar]
- Zagare, V.M.E. Natural territory, urban growth and climate change in the Parana River Delta and Rio de la Plata estuarine system. An Overv. 2016, 1–16. [Google Scholar]
- Kandus, P.; Malvárez, A.I. Vegetation patterns and change analysis in the lower delta islands of the Parana River (Argentina). Wetlands 2006, 24, 620–632. [Google Scholar] [CrossRef]
- Meis, M.; Llano, M.P. Modelado estadístico del caudal mensual en la baja Cuenca del Plata. Meteorologica 2018, 43, 63–77. [Google Scholar]
- Campoy, J.A.; Lerigoleur-Balsemin, E.; Christmann, H.; Beauvieux, R.; Girollet, N.; Quero-García, J.; Dirlewanger, E.; Barreneche, T. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol. 2016, 16, 49. [Google Scholar] [CrossRef]
- Acuña, C.V.; Fernandez, P.; Villalba, P.V.; García, M.N.; Hopp, H.E.; Marcucci Poltri, S.N. Discovery, validation, and in silico functional characterization of EST-SSR markers in Eucalyptus globulus. Tree Genet. Genom. 2012, 8, 289–301. [Google Scholar] [CrossRef]
- Wünsch, A.; Carrera, M.; Hormaza, J.I. Molecular Characterization of Local Spanish Peach [Prunus persica (L.) Batsch] Germplasm. Genet. Resour. Crop Evol. 2006, 53, 925. [Google Scholar] [CrossRef]
- Arif, I.A.; Khan, H.A.; Bahkali, A.H.; Al Homaidan, A.A.; Al Farhan, A.H.; Al Sadoon, M.; Shobrak, M. DNA marker technology for wildlife conservation. Saudi J. Biol. Sci. 2011, 18, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Aranzana, M.J.; Decroocq, V.; Dirlewanger, E.; Eduardo, I.; Gao, Z.S.; Gasic, K.; Iezzoni, A.; Jung, S.; Peace, C.; Prieto, H.; et al. Prunus genetics and applications after de novo genome sequencing: Achievements and prospects. Hortic. Res. 2019, 6, 58. [Google Scholar] [CrossRef]
- Carrasco, B.; Díaz, C.; Moya, M.; Gebauer, M.; García-González, R. Genetic characterization of Japanese plum cultivars (Prunus salicina) using SSR and ISSR molecular markers. Cienc. E Investig. Agrar. 2012, 39, 533–543. [Google Scholar] [CrossRef]
- Klabunde, G.H.F.; Dalbó, M.A.; Nodari, R.O. DNA fingerprinting of Japanese plum (Prunus salicina) cultivars based on microsatellite markers. Crop. Breed. Appl. Biotechnol. 2014, 14, 139–145. [Google Scholar] [CrossRef]
- Vieira, E.A.; Onofre Nodari, R.; Cibele De Mesquita Dantas, A.; Henri, J.-P.; Ducroquet, J.; Dalbó, M.; Borges, C.V. Genetic Mapping of Japanese Plum. Crop Breeding and Applied Biotechnology: Vicosa, Brazil, 2005; p. 5. [Google Scholar]
- Gonz#xE1;lez, M.; Salazar, E.; Castillo, J.; Morales, P.; Mura-Jornet, I.; Maldonado, J.; Silva, H.; Carrasco, B. Genetic structure based on EST–SSR: a putative tool for fruit color selection in Japanese plum (Prunus salicina L.) breeding programs. Mol. Breed. 2016, 36. [Google Scholar]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar]
- Chakraborty, R.; Jin, L. A unified approach to study hypervariable polymorphisms: Statistical considerations of determining relatedness and population distances. In DNA Fingerprinting: State of the Science; Birkhäuser Basel: Basel, Switzerland, 1993; pp. 153–175. [Google Scholar]
- Langela, O. Populations: A free Population Genetic Software; UCLA: Los Angeles, CA, USA, 2002. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Liu, N.; Zhao, H. A non-parametric approach to population structure inference using multilocus genotypes. Hum. Genom. 2006, 2, 353. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Arús, P.; Verde, I.; Sosinski, B.; Zhebentyayeva, T.; Abbott, A.G. The peach genome. Tree Genet. Genomes 2012, 8, 531–547. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Olmstead, J.W.; Sebolt, A.M.; Cabrera, A.; Sooriyapathirana, S.S.; Hammar, S.; Iriarte, G.; Wang, D.; Chen, C.Y.; Van Der Knaap, E.; Iezzoni, A.F. Construction of an intra-specific sweet cherry (Prunus avium L.) genetic linkage map and synteny analysis with the Prunus reference map. Tree Genet. Genomes 2008, 4, 897–910. [Google Scholar] [CrossRef]
- Yan, A.; Chen, Z. The pivotal role of abscisic acid signaling during transition from seed maturation to germination. Plant Cell Rep. 2017, 36, 689–703. [Google Scholar] [CrossRef]
- Li-hui, Z.; Zhi-xiao, H.; Hai-yong, L.; YANG Min-sheng, A. Analysis of Genetic Diversity of Prunus salicina from Different Producing Areas by SSR Markers. Acta Hortic. Sin. 2015, 42, 11–118. [Google Scholar]
- Rojas, G.; Méndez, M.A.; Muñoz, C.; Lemus, G.; Hinrichsen, P. Identification of a minimal microsatellite marker panel for the fingerprinting of peach and nectarine cultivars. Electron. J. Biotechnol. 2008, 11, 4–5. [Google Scholar] [CrossRef]
- Tani, E.; Polidoros, A.N.; Tsaftaris, A.S. Characterization and expression analysis of FRUITFULL- and SHATTERPROOF-like genes from peach (Prunus persica) and their role in split-pit formation. Tree Physiol. 2007, 27, 649–659. [Google Scholar] [CrossRef]
- Schueler, S.; Tusch, A.; Schuster, M.; Ziegenhagen, B. Characterization of microsatellites in wild and sweet cherry Prunus avium—Markers for individual identification and reproductive processes. Genome 2003, 46, 95–102. [Google Scholar] [CrossRef]
- Mnejja, M.; Garcia-Mas, J.; Audergon, J.M.; Arús, P. Prunus microsatellite marker transferability across rosaceous crops. Tree Genet. Genomes 2010, 6, 689–700. [Google Scholar] [CrossRef]
- González-Agüero, M.; Troncoso, S.; Gudenschwager, O.; Campos-Vargas, R.; Moya-León, M.A.; Defilippi, B.G. Differential expression levels of aroma-related genes during ripening of apricot (Prunus armeniaca L.). Plant Physiol. Biochem. 2009, 47, 435–440. [Google Scholar]
- Zhang, B.; Shen, J.; Wei, W.; Xi, W.; Xu, C.-J.; Ferguson, I.; Chen, K. Expression of Genes Associated with Aroma Formation Derived from the Fatty Acid Pathway during Peach Fruit Ripening. J. Agric. Food Chem. 2010, 58, 6157–6165. [Google Scholar] [CrossRef]
- Xu, Z.; Sun, L.; Zhou, Y.; Yang, W.; Cheng, T.; Wang, J.; Zhang, Q. Identification and expression analysis of the SQUAMOSA promoter-binding protein (SBP)-box gene family in Prunus mume. Mol. Genet. Genom. 2015, 290, 1701–1715. [Google Scholar] [CrossRef]
- Tani, E.; Polidoros, A.N.; Flemetakis, E.; Stedel, C.; Kalloniati, C.; Demetriou, K.; Katinakis, P.; Tsaftaris, A.S. Characterization and expression analysis of AGAMOUS-like, SEEDSTICK-like, and SEPALLATA-like MADS-box genes in peach Prunus persica fruit. Plant Physiol. Biochem. 2009, 47, 690–700. [Google Scholar] [CrossRef]
- Roy Choudhury, S.; Roy, S.; Nag, A.; Singh, S.K.; Sengupta, D.N. Characterization of an AGAMOUS-like MADS Box Protein, a Probable Constituent of Flowering and Fruit Ripening Regulatory System in Banana. PLoS ONE 2012, 7, e44361. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, G.; Yang, Y.-N.; Le, W.-Q.; Khan, M.A.; Zhang, S.-L.; Gu, C.; Huang, W.-J. Identification of differentially expressed genes related to coloration in red/green mutant pear Pyrus communis. Tree Genet. Genom. 2013, 9, 75–83. [Google Scholar] [CrossRef]
- Gou, J.-Y.; Felippes, F.F.; Liu, C.-J.; Weigel, D.; Wang, J.-W. Negative Regulation of Anthocyanin Biosynthesis in Arabidopsis by a miR156-Targeted SPL Transcription Factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef]
- Jaakola, L.; Poole, M.; Jones, M.O.; Kämäräinen-Karppinen, T.; Koskimäki, J.J.; Hohtola, A.; Häggman, H.; Fraser, P.D.; Manning, K.; King, G.J.; et al. A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiol. 2010, 153, 1619–1629. [Google Scholar] [CrossRef]
- González, M.; Salazar, E.; Cabrera, S.; Olea, P.; Carrasco, B. Analysis of anthocyanin biosynthesis genes expression profiles in contrasting cultivars of Japanese plum Prunus salicina during fruit development. Gene Expr. Patterns 2016, 21, 54–62. [Google Scholar] [CrossRef]
- Xu, X.; Ma, X.; Lei, H.; Yin, L.; Shi, X.; Song, H. MicroRNAs play an important role in the regulation of strawberry fruit senescence in low temperature. Postharvest Biol. Technol. 2015, 108, 39–47. [Google Scholar] [CrossRef]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC proteins: Regulation and role in stress tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef]
- Lan, T.; Yang, Z.L.; Yang, X.; Liu, Y.J.; Wang, X.R.; Zeng, Q.Y. Extensive functional diversification of the Populus glutathione S-transferase supergene family. Plant Cell Online 2009, 21, 3749. [Google Scholar] [CrossRef]
- Zavaliev, R.; Sagi, G.; Gera, A.; Epel, B.L. The constitutive expression of Arabidopsis plasmodesmal-associated class 1 reversibly glycosylated polypeptide impairs plant development and virus spread. J. Exp. Bot. 2010, 61, 131–142. [Google Scholar] [CrossRef]
- Janiak, A.; Mirosłw., K.; Iwonas, S. Gene expression regulation in roots under drought. J. Exp. Botany. 2016, 67, 1003–1014. [Google Scholar] [CrossRef]
- Xu, B.; Ohtani, M.; Yamaguchi, M.; Toyooka, K.; Wakazaki, M.; Sato, M.; Kubo, M.; Nakano, Y.; Sano, R.; Hiwatashi, Y.; et al. Contribution of NAC Transcription Factors to Plant Adaptation to Land. Science 2014, 343, 1505–1508. [Google Scholar] [CrossRef]
- Zhuo, X.; Zheng, T.; Zhang, Z.; Zhang, Y.; Jiang, L.; Ahmad, S.; Sun, L.; Wang, J.; Cheng, T.; Zhang, Q. Genome-Wide Analysis of the NAC Transcription Factor Gene Family Reveals Differential Expression Patterns and Cold-Stress Responses in the Woody Plant Prunus mume. Genes 2018, 9, 494. [Google Scholar] [CrossRef]
- Zhou, H.; Lin-Wang, K.; Wang, H.; Gu, C.; Dare, A.P.; Espley, R.V.; He, H.; Allan, A.C.; Han, Y. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015, 82, 105–121. [Google Scholar] [CrossRef]
- Fang, Z.-Z.; Zhou, D.-R.; Ye, X.-F.; Jiang, C.-C.; Pan, S.-L. Identification of Candidate Anthocyanin-Related Genes by Transcriptomic Analysis of ‘Furongli’ Plum Prunus salicina Lindl. during Fruit Ripening Using RNA-Seq. Front. Plant Sci. 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Kojima, Y.; Maruta, T.; Nishizawa-Yokoi, A.; Yabuta, Y.; Shigeoka, S. Arabidopsis NAC transcription factor, ANAC078, regulates flavonoid biosynthesis under high-light. Plant Cell Physiol. 2009, 50, 2210–2222. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, Y.-H.; Yang, Y.; Chi, Y.-J.; Zhou, J.; Chen, J.-Y.; Wang, F.; Fan, B.; Shi, K.; Zhou, Y.-H.; et al. Structural and Functional Analysis of VQ Motif-Containing Proteins in Arabidopsis as Interacting Proteins of WRKY Transcription Factors. Plant Physiol. 2012, 159, 810–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custers, J.B.M.; Oldenhof, M.T.; Schrauwen, J.A.M.; Cordewener, J.H.G.; Wullems, G.J.; van Lookeren Campagne, M.M. Analysis of microspore-specific promoters in transgenic tobacco. Plant Mol. Biol. 1997, 35, 689–699. [Google Scholar] [CrossRef]
- Cao, K.; Wang, L.; Zhu, G.; Fang, W.; Chen, C.; Luo, J. Genetic diversity, linkage disequilibrium, and association mapping analyses of peach Prunus persica landraces in China. Tree Genet. Genomes 2012, 8, 975–990. [Google Scholar] [CrossRef]
- Barnaud, A.; Lacombe, T.; Doligez, A. Linkage disequilibrium in cultivated grapevine, Vitis vinifera. L. Theor. Appl. Genet. 2006, 112, 708–716. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Landrace | Origin | Flesh Color | Harvest |
|---|---|---|---|
| Ratto | Seedling Delta | red | Very late season—End of January |
| Severiana | Seedling Delta | red | Very late season—End of January to mid-February |
| Remolacha de Berisso | Seedling Delta | red | Very late season—End of January to early February |
| Fragata | Seedling Delta | red | Very late season—End of January |
| Remolacha de Leber | Seedling Delta | red | Middle season—Early/middle December |
| Gigaglia | Seedling Delta | red | Middle season—Mid-November to early December |
| Capri | Seedling Delta | red | Late season—Late December |
| Giordano | Seedling Delta | red | Middle season—Late November to early December |
| Ciervita | Seedling Delta | pink | Early season—Mid-to late November |
| Reina de oro | Seedling Delta | yellow | Late season—Early January |
| Juanita | Seedling Delta | yellow | Early season—Mid-November to early December |
| Gascón | Seedling Delta | yellow | Middle season—Late November to early December |
| Tricerri | Seedling Delta | yellow | Mid-November |
| “X” | Seedling Delta | yellow | Mid-November |
| SSR Marker | Location | AR (pb) | N | Na | Ne | Ho | uHe | PIC |
|---|---|---|---|---|---|---|---|---|
| CPSCT011 (AY426199.1) | C 5 (4163117–4164052) | 171–189 | 13 | 6 | 3.04 | 0.38 | 0.70 | 0.63 |
| CPSCT018 (AY426204.1) | C 8 (123199–124139) 3e- | 151–172 | 13 | 6 | 2.18 | 0.23 | 0.56 | 0.51 |
| CPSCT021 (AY426206.1) | C 2 (27308766–27309699) | 132–157 | 11 | 6 | 3.66 | 0.36 | 0.76 | 0.69 |
| CPSCT022 (AY426207.1) | C 5 (16620819–16621314) | 159–179 | 13 | 7 | 2.91 | 0.77 | 0.68 | 0.62 |
| CPSCT024 (AY426209.1) | C 1 (28058204–28058903) | 157–179 | 13 | 4 | 2.89 | 0.61 | 0.68 | 0.60 |
| CPSCT025 (AY426210.1) | C 3 (6709070–6709740) | 181–223 | 12 | 5 | 2.51 | 0.66 | 0.64 | 0.53 |
| CPSCT026 (AY426211.1) | C 7 (11365276–11365753) | 176–195 | 14 | 4 | 2.78 | 0.43 | 0.66 | 0.60 |
| CPSCT027 (AY426212.1) | C 1 (23010058–23010528) | 137–156 | 13 | 5 | 2.66 | 0.31 | 0.65 | 0.57 |
| CPSCT030 (AY426215.1) | C 5 (15121388–15122031) | 179–200 | 14 | 7 | 4.21 | 0.86 | 0.79 | 0.73 |
| CPSCT034 (AY426219.1) | C 2 (29946149–29946699) | 177–224 | 14 | 6 | 3.70 | 0.64 | 0.76 | 0.68 |
| CPSCT042 (AY426226.1) | C 7 (16682143–16682742) | 167–185 | 14 | 5 | 3.53 | 0.71 | 0.74 | 0.67 |
| CPSCT044 (AY426228.1) | C 2 (20793617–20794126) | 218–241 | 13 | 5 | 3.35 | 0.15 | 0.73 | 0.65 |
| Mean | 5.50 | 3.12 | 0.51 | 0.70 | 0.62 | |||
| SD | 1.00 | 0.58 | 0.23 | 0.06 | 0.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acuña, C.V.; Rivas, J.G.; Brambilla, S.M.; Cerrillo, T.; Frusso, E.A.; García, M.N.; Villalba, P.V.; Aguirre, N.C.; Sabio y García, J.V.; Martínez, M.C.; et al. Characterization of Genetic Diversity in Accessions of Prunus salicina Lindl: Keeping Fruit Flesh Color Ideotype While Adapting to Water Stressed Environments. Agronomy 2019, 9, 487. https://doi.org/10.3390/agronomy9090487
Acuña CV, Rivas JG, Brambilla SM, Cerrillo T, Frusso EA, García MN, Villalba PV, Aguirre NC, Sabio y García JV, Martínez MC, et al. Characterization of Genetic Diversity in Accessions of Prunus salicina Lindl: Keeping Fruit Flesh Color Ideotype While Adapting to Water Stressed Environments. Agronomy. 2019; 9(9):487. https://doi.org/10.3390/agronomy9090487
Chicago/Turabian StyleAcuña, Cintia V., Juan G. Rivas, Silvina M. Brambilla, Teresa Cerrillo, Enrique A. Frusso, Martín N. García, Pamela V. Villalba, Natalia C. Aguirre, Julia V. Sabio y García, María C. Martínez, and et al. 2019. "Characterization of Genetic Diversity in Accessions of Prunus salicina Lindl: Keeping Fruit Flesh Color Ideotype While Adapting to Water Stressed Environments" Agronomy 9, no. 9: 487. https://doi.org/10.3390/agronomy9090487