Autophagy in Trypanosomatids

Abstract
:1. Introduction
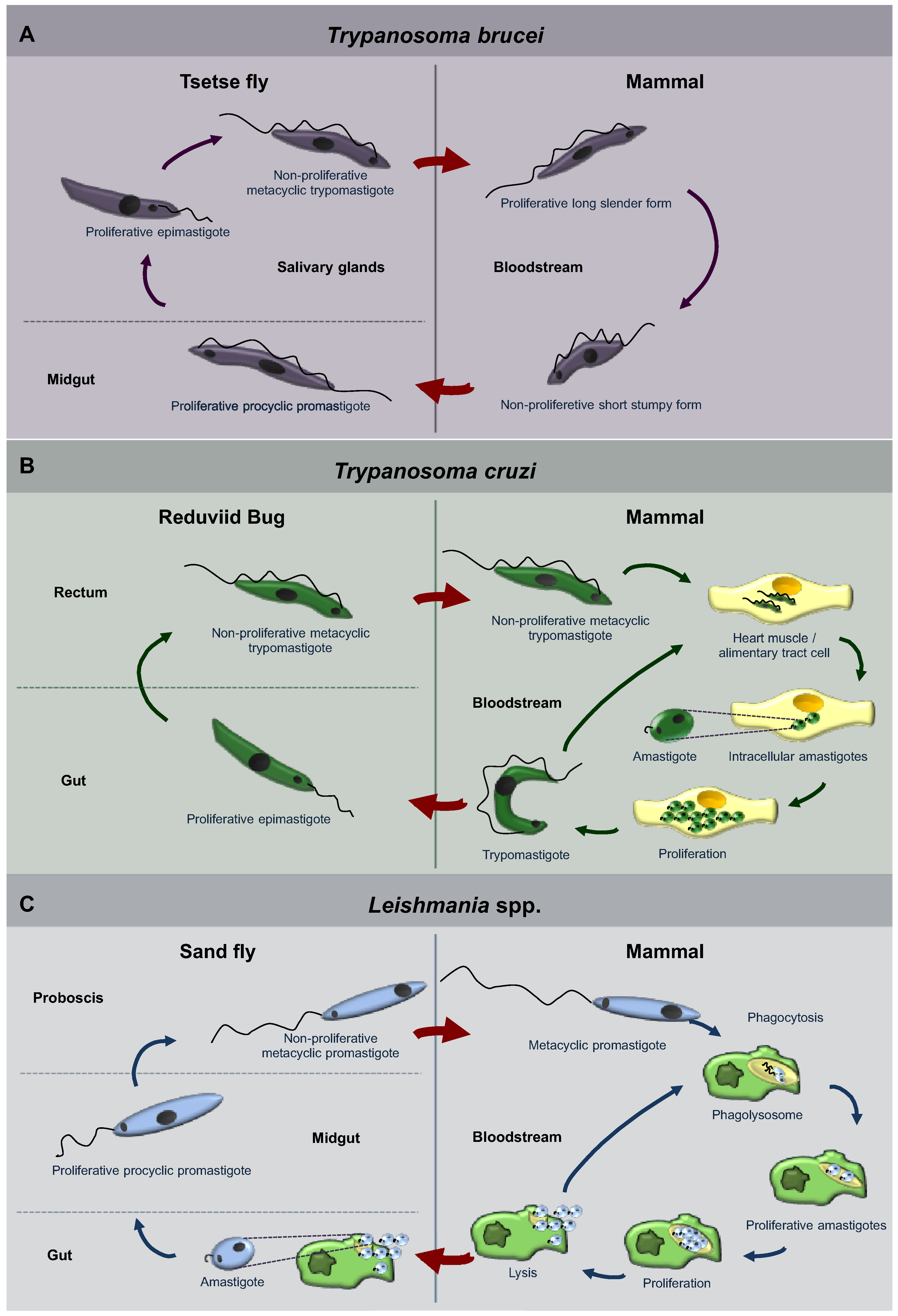
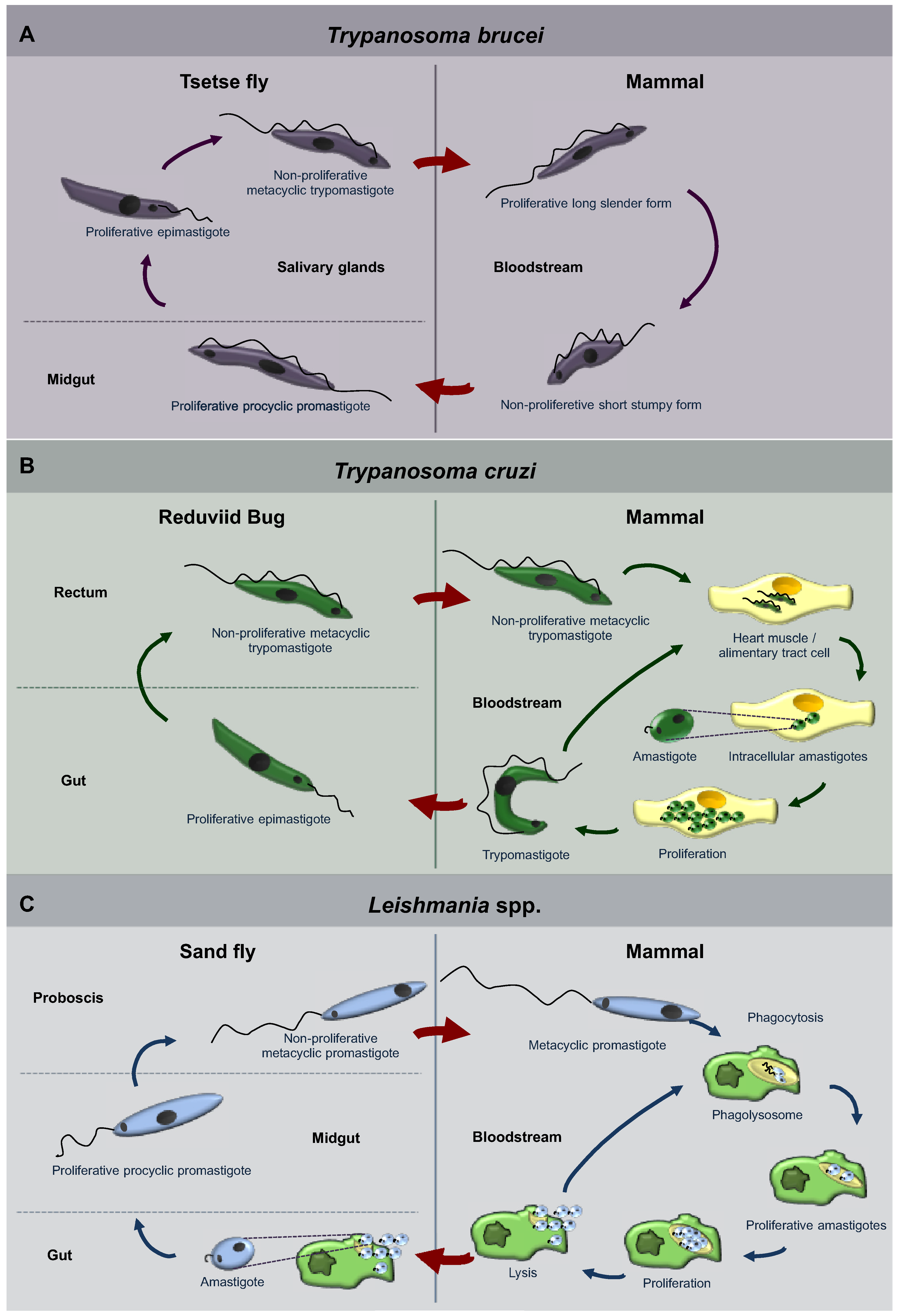
1.1. Trypanosomatidae
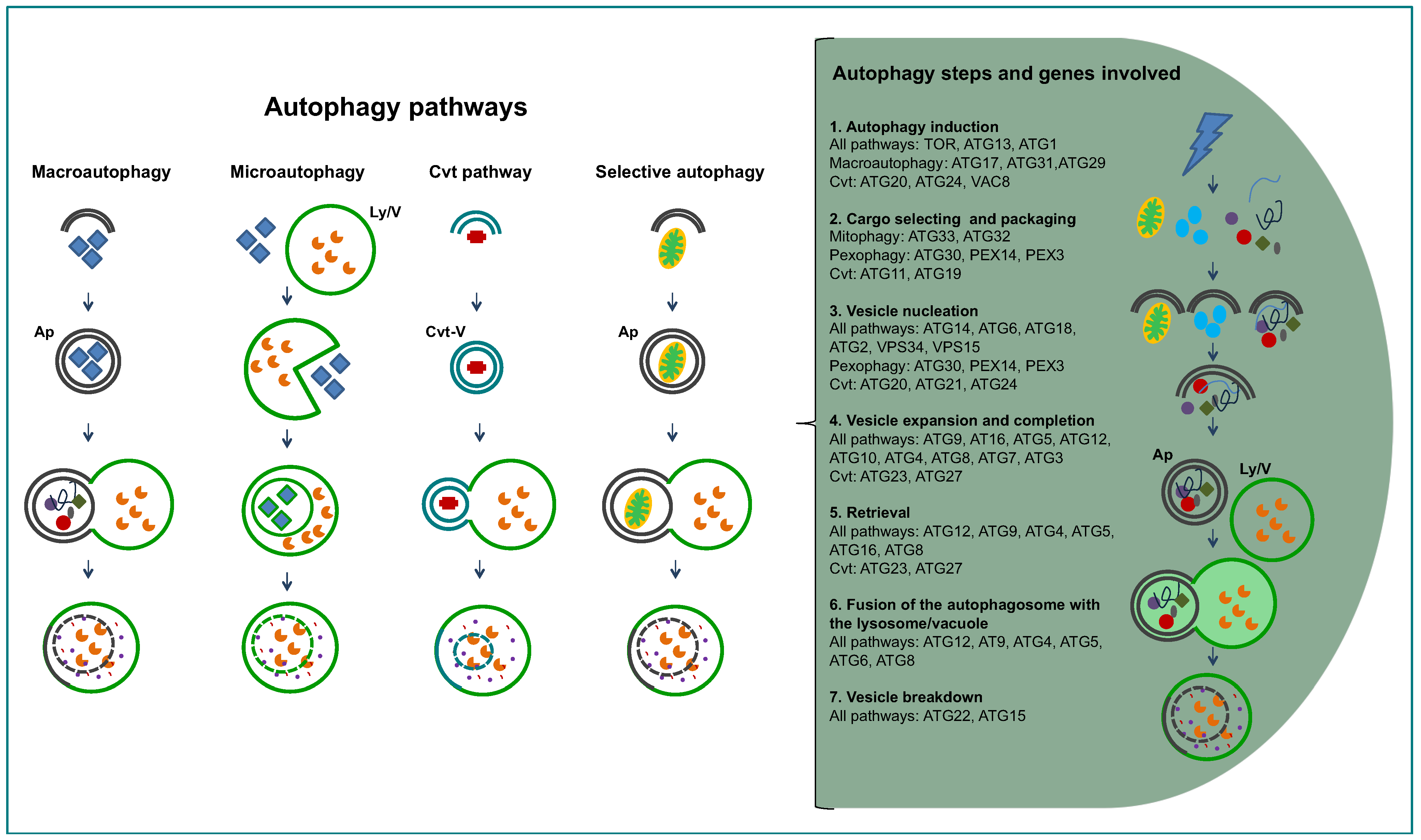
1.2. General Aspects of Autophagy
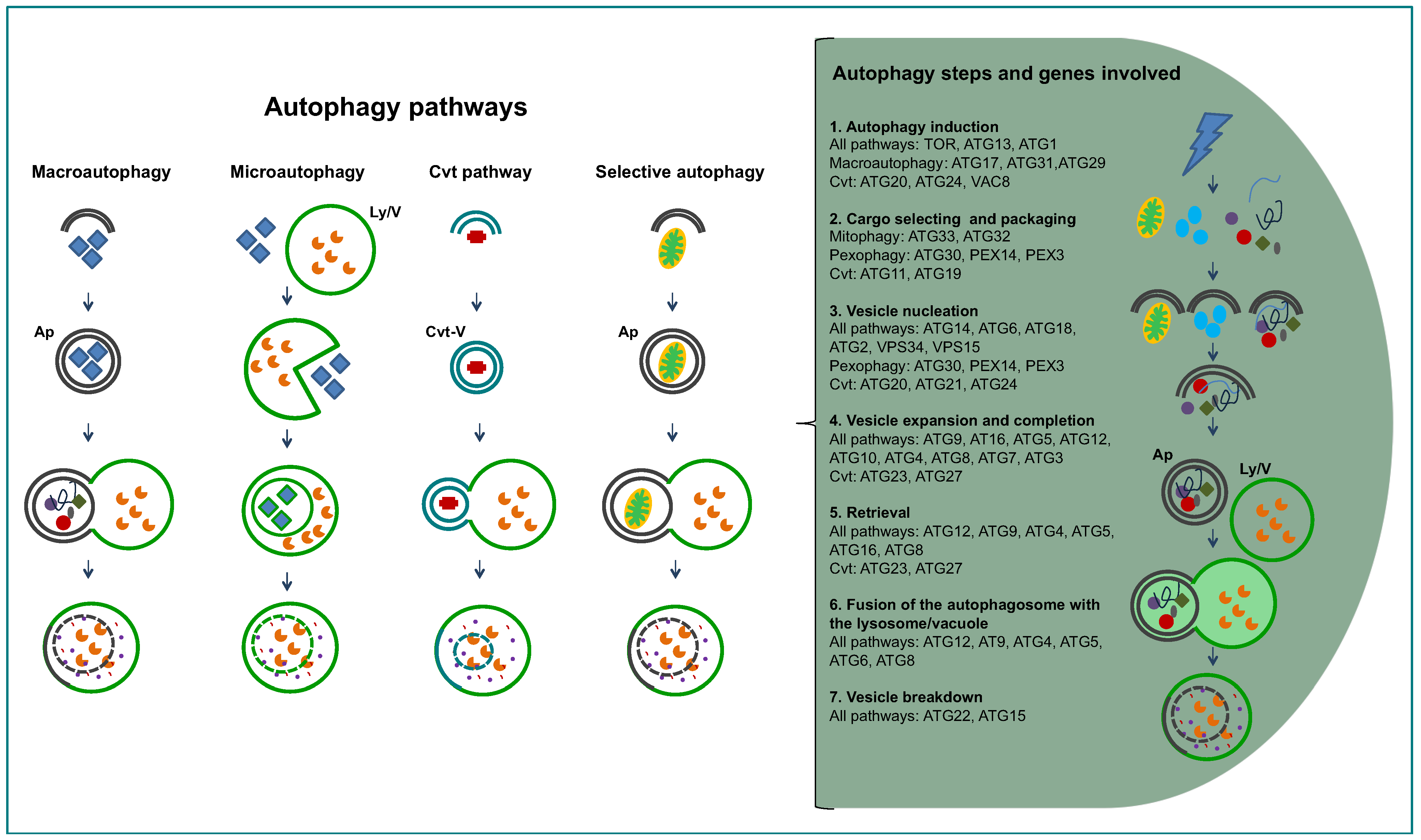
2. Inventory of Genes Encoding Proteins Involved in Autophagy in Trypanosomatids

{kind=link}
{kind=link}
{kind=link}
 |
3. Experimental Studies on Autophagy in Trypanosomatids
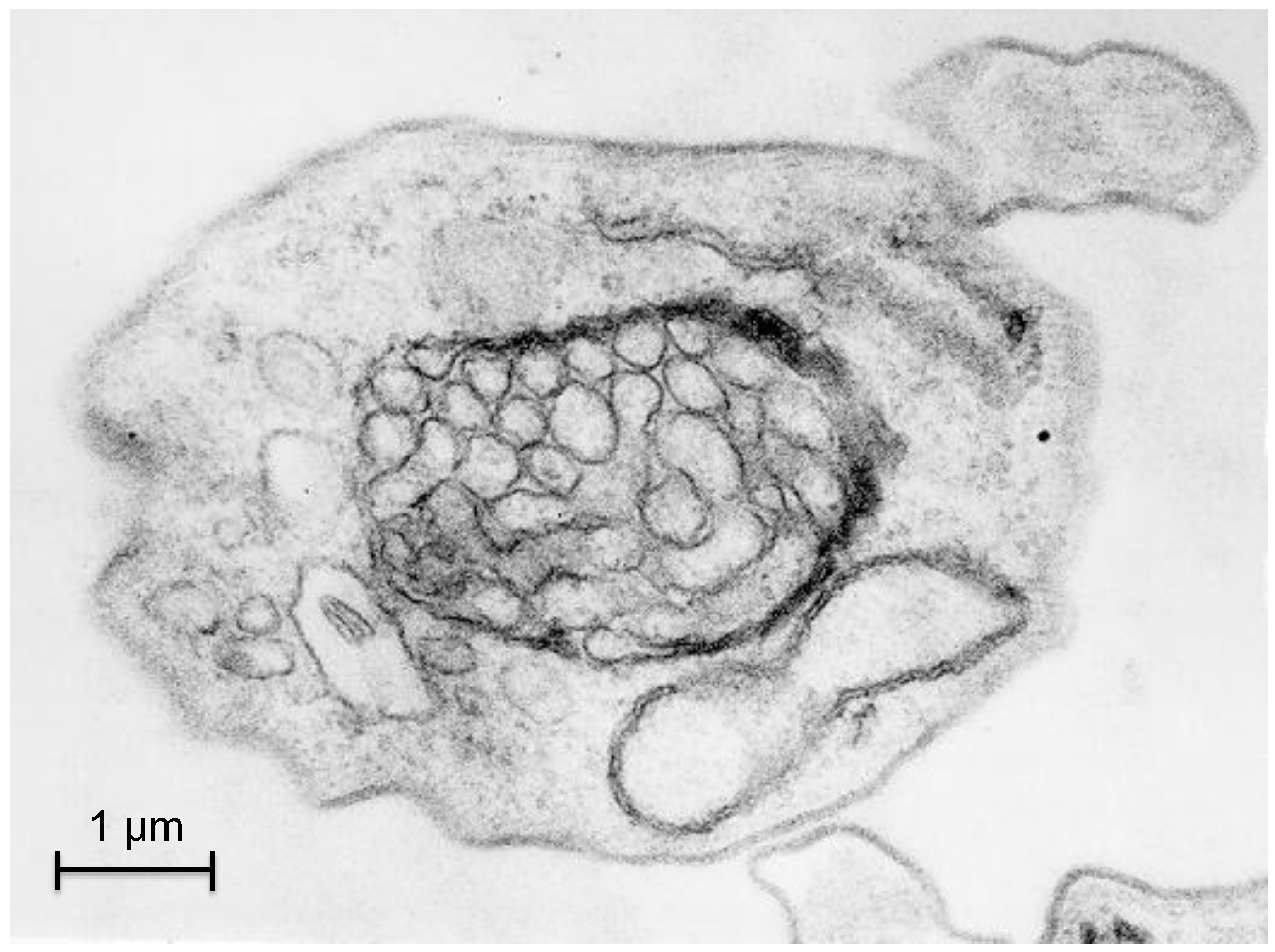
3.1. Autophagy in T. brucei
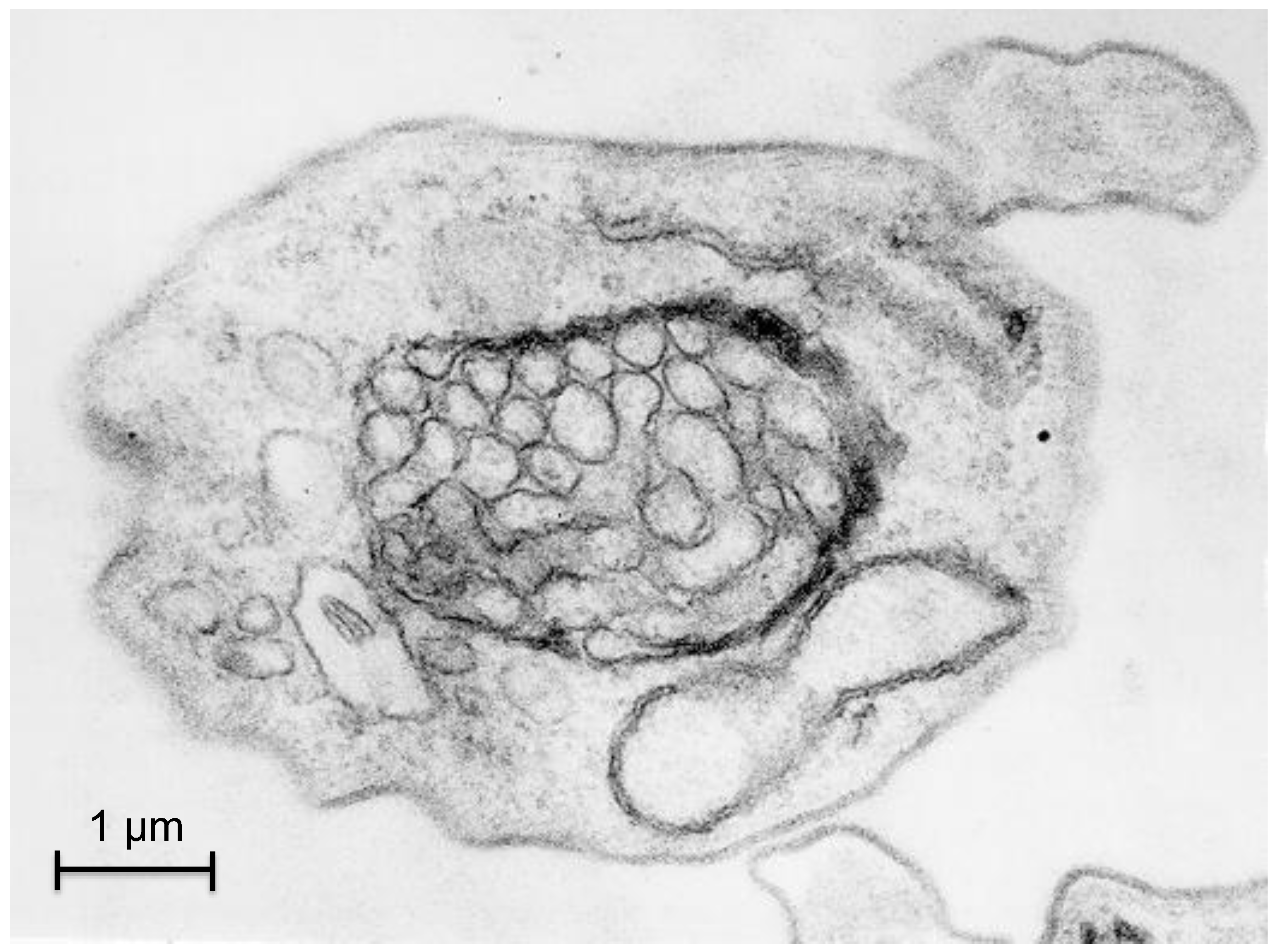
3.2. Autophagy in Trypanosoma cruzi
3.3. Autophagy in Leishmania spp.
3.4. Roles of Autophagy in Trypanosomatids Inferred by Experimental Studies
4. Conclusions and Perspectives
Acknowledgments
Conflict of Interest
References
- Moreira, D.; Lopez-Garcia, P.; Vickerman, K. An updated view of kinetoplastid phylogeny using environmental sequences and a closer outgroup: Proposal for a new classification of the class Kinetoplastea. Int. J. Syst. Evol. Microbiol. 2004, 54, 1861–1875. [Google Scholar] [CrossRef]
- de Souza, W.; Attias, M.; Rodrigues, J.C. Particularities of mitochondrial structure in parasitic protists (Apicomplexa and Kinetoplastida). Int. J. Biochem. Cell Biol. 2009, 41, 2069–2080. [Google Scholar] [CrossRef]
- Dollet, M.; Sturm, N.R.; Campbell, D.A. The internal transcribed spacer of ribosomal RNA genes in plant trypanosomes (Phytomonas spp.) resolves 10 groups. Infect. Genet. Evol. 2012, 12, 299–308. [Google Scholar] [CrossRef]
- Podlipaev, S.A. Insect trypanosomatids: The need to know more. Memorias do Instituto Oswaldo Cruz 2000, 95, 517–522. [Google Scholar] [CrossRef]
- Simpson, A.G.; Stevens, J.R.; Lukes, J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006, 22, 168–174. [Google Scholar] [CrossRef]
- Vickerman, K. The evolutionary expansion of the trypanosomatid flagellates. Int. J. Parasitol. 1994, 24, 1317–1331. [Google Scholar] [CrossRef]
- Zeledon, R.A. Hemoflagellates: Cutaneous and mucocutaneous leishmaniasis. In Barron’s Medical Microbiology; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Gualdron-Lopez, M.; Brennand, A.; Hannaert, V.; Quinones, W.; Caceres, A.J.; Bringaud, F.; Concepcion, J.L.; Michels, P.A. When, how and why glycolysis became compartmentalised in the Kinetoplastea. A new look at an ancient organelle. Int. J. Parasitol. 2012, 42, 1–20. [Google Scholar]
- Michels, P.A.; Bringaud, F.; Herman, M.; Hannaert, V. Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 2006, 1763, 1463–1477. [Google Scholar]
- Furuya, T.; Kessler, P.; Jardim, A.; Schnaufer, A.; Crudder, C.; Parsons, M. Glucose is toxic to glycosome-deficient trypanosomes. Proc. Natl. Acad. Sci. USA 2002, 99, 14177–14182. [Google Scholar]
- Haanstra, J.R.; van Tuijl, A.; Kessler, P.; Reijnders, W.; Michels, P.A.; Westerhoff, H.V.; Parsons, M.; Bakker, B.M. Compartmentation prevents a lethal turbo-explosion of glycolysis in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 17718–17723. [Google Scholar]
- Teixeira, M.M.; Borghesan, T.C.; Ferreira, R.C.; Santos, M.A.; Takata, C.S.; Campaner, M.; Nunes, V.L.; Milder, R.V.; de Souza, W.; Camargo, E.P. Phylogenetic validation of the genera Angomonas and Strigomonas of trypanosomatids harboring bacterial endosymbionts with the description of new species of trypanosomatids and of proteobacterial symbionts. Protist 2011, 162, 503–524. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef]
- Kennedy, P.G. The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol. 2008, 64, 116–126. [Google Scholar] [CrossRef]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef]
- Piscopo, T.V.; Mallia Azzopardi, C. Leishmaniasis. Postgrad. Med. 2007, 83, 649–657. [Google Scholar]
- David, C.V.; Craft, N. Cutaneous and mucocutaneous leishmaniasis. Dermatol. Ther. 2009, 22, 491–502. [Google Scholar] [CrossRef]
- Moore, E.M.; Lockwood, D.N. Treatment of visceral leishmaniasis. J. Global Infect. Dis. 2010, 2, 151–158. [Google Scholar] [CrossRef]
- Castillo, E.; Dea-Ayuela, M.A.; Bolas-Fernandez, F.; Rangel, M.; Gonzalez-Rosende, M.E. The kinetoplastid chemotherapy revisited: Current drugs, recent advances and future perspectives. Curr. Med. Chem. 2010, 17, 4027–4051. [Google Scholar]
- Simarro, P.P.; Franco, J.; Diarra, A.; Postigo, J.A.; Jannin, J. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology 2012, 139, 842–846. [Google Scholar] [CrossRef]
- Hoare, C.A. Rationalization of the terminology for the developmental stages of trypanosomatid flagellates. Meditsinskaia parazitologiia i parazitarnye bolezni 1971, 40, 307–309. [Google Scholar]
- Fenn, K.; Matthews, K.R. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 2007, 10, 539–546. [Google Scholar]
- de Souza, W.; de Carvalho, T.M.; Barrias, E.S. Review on Trypanosoma cruzi: Host cell interaction. Int. J. Cell Biol. 2010, 2010. pii: 295394. [Google Scholar]
- Bates, P.A. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int. J. Parasitol. 2007, 37, 1097–1106. [Google Scholar] [CrossRef]
- de Duve, C. The lysosome turns fifty. Med. Sci. (Paris) 2005, 21, 12–15. [Google Scholar] [CrossRef]
- Armstrong, J. Yeast vacuoles: More than a model lysosome. Trends Cell Biol. 2010, 20, 580–585. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Bio. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Meijer, W.H.; van der Klei, I.J.; Veenhuis, M.; Kiel, J.A. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007, 3, 106–116. [Google Scholar]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef]
- Lynch-Day, M.A.; Klionsky, D.J. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010, 584, 1359–1366. [Google Scholar] [CrossRef]
- Dunn, W.A., Jr.; Cregg, J.M.; Kiel, J.A.; van der Klei, I.J.; Oku, M.; Sakai, Y.; Sibirny, A.A.; Stasyk, O.V.; Veenhuis, M. Pexophagy: The selective autophagy of peroxisomes. Autophagy 2005, 1, 75–83. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Bio. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Roberts, P.; Moshitch-Moshkovitz, S.; Kvam, E.; O'Toole, E.; Winey, M.; Goldfarb, D.S. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 129–141. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Bio. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 4544–4556. [Google Scholar]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Duszenko, M.; Ginger, M.L.; Brennand, A.; Gualdron-Lopez, M.; Colombo, M.I.; Coombs, G.H.; Coppens, I.; Jayabalasingham, B.; Langsley, G.; de Castro, S.L.; et al. Autophagy in protists. Autophagy 2011, 7, 127–158. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Takano, Y.; Sakai, Y. Autophagy in plants and phytopathogens. FEBS Lett. 2010, 584, 1350–1358. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Gene Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Noda, T.; Kim, J.; Huang, W.P.; Baba, M.; Tokunaga, C.; Ohsumi, Y.; Klionsky, D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000, 148, 465–480. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef]
- Herman, M.; Gillies, S.; Michels, P.A.; Rigden, D.J. Autophagy and related processes in trypanosomatids: Insights from genomic and bioinformatic analyses. Autophagy 2006, 2, 107–118. [Google Scholar]
- Rigden, D.J.; Michels, P.A.; Ginger, M.L. Autophagy in protists: Examples of secondary loss, lineage-specific innovations, and the conundrum of remodeling a single mitochondrion. Autophagy 2009, 5, 784–794. [Google Scholar]
- Barquilla, A.; Crespo, J.L.; Navarro, M. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. Proc. Natl. Acad. Sci. USA 2008, 105, 14579–14584. [Google Scholar]
- Madeira da Silva, L.; Beverley, S.M. Expansion of the target of rapamycin (TOR) kinase family and function in Leishmania shows that TOR3 is required for acidocalcisome biogenesis and animal infectivity. Proc. Natl. Acad. Sci. USA 2010, 107, 11965–11970. [Google Scholar] [CrossRef]
- Alvarez, V.E.; Kosec, G.; Sant'Anna, C.; Turk, V.; Cazzulo, J.J.; Turk, B. Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J. Biol. Chem. 2008, 283, 3454–3464. [Google Scholar]
- Brennand, A.; Gualdron-Lopez, M.; Coppens, I.; Rigden, D.J.; Ginger, M.L.; Michels, P.A. Autophagy in parasitic protists: Unique features and drug targets. Mol. Biochem. Parasit. 2011, 177, 83–99. [Google Scholar] [CrossRef]
- Williams, R.A.; Woods, K.L.; Juliano, L.; Mottram, J.C.; Coombs, G.H. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy 2009, 5, 159–172. [Google Scholar] [CrossRef]
- Li, F.J.; Shen, Q.; Wang, C.; Sun, Y.; Yuan, A.Y.; He, C.Y. A role of autophagy in Trypanosoma brucei cell death. Cell Microbiol. 2012, 14, 1242–1256. [Google Scholar] [CrossRef]
- Koopmann, R.; Muhammad, K.; Perbandt, M.; Betzel, C.; Duszenko, M. Trypanosoma brucei ATG8: Structural insights into autophagic-like mechanisms in protozoa. Autophagy 2009, 5, 1085–1091. [Google Scholar] [CrossRef]
- Hall, B.S.; Gabernet-Castello, C.; Voak, A.; Goulding, D.; Natesan, S.K.; Field, M.C. TbVps34, the trypanosome orthologue of Vps34, is required for Golgi complex segregation. J. Biol. Chem. 2006, 281, 27600–27612. [Google Scholar]
- Vickerman, K.; Tetley, L. Recent ultrastructural studies on trypanosomes. Annales de la Societe belge de medecine tropicale 1977, 57, 441–457. [Google Scholar]
- Opperdoes, F.R.; Borst, P. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: The glycosome. FEBS Lett. 1977, 80, 360–364. [Google Scholar] [CrossRef]
- Galland, N.; Michels, P.A. Comparison of the peroxisomal matrix protein import system of different organisms. Exploration of possibilities for developing inhibitors of the import system of trypanosomatids for anti-parasite chemotherapy. Eur. J. Cell Biol. 2010, 89, 621–637. [Google Scholar] [CrossRef]
- Moyersoen, J.; Choe, J.; Fan, E.; Hol, W.G.; Michels, P.A. Biogenesis of peroxisomes and glycosomes: Trypanosomatid glycosome assembly is a promising new drug target. FEMS Microbiol. Rev. 2004, 28, 603–643. [Google Scholar] [CrossRef]
- Bakker, B.M.; Mensonides, F.I.; Teusink, B.; van Hoek, P.; Michels, P.A.; Westerhoff, H.V. Compartmentation protects trypanosomes from the dangerous design of glycolysis. Proc. Natl. Acad. Sci. USA 2000, 97, 2087–2092. [Google Scholar]
- Herman, M.; Perez-Morga, D.; Schtickzelle, N.; Michels, P.A. Turnover of glycosomes during life-cycle differentiation of Trypanosoma brucei. Autophagy 2008, 4, 294–308. [Google Scholar]
- Sakai, Y.; Oku, M.; van der Klei, I.J.; Kiel, J.A. Pexophagy: Autophagic degradation of peroxisomes. Biochim. Biophys. Acta 2006, 1763, 1767–1775. [Google Scholar]
- Besteiro, S.; Williams, R.A.; Morrison, L.S.; Coombs, G.H.; Mottram, J.C. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J. Biol. Chem. 2006, 281, 11384–11396. [Google Scholar]
- Williams, R.A.; Tetley, L.; Mottram, J.C.; Coombs, G.H. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol. Microbiol. 2006, 61, 655–674. [Google Scholar] [CrossRef]
- Goldshmidt, H.; Matas, D.; Kabi, A.; Carmi, S.; Hope, R.; Michaeli, S. Persistent ER stress induces the spliced leader RNA silencing pathway (SLS), leading to programmed cell death in Trypanosoma brucei. PLoS Pathog. 2010, 6, e1000731. [Google Scholar]
- Brennand, A.; Rico, E.; Rigden, D.J.; Van Der Smissen, P.; Courtoy, P.; Michels, P.A.M. Trypanosoma brucei ATG24 is involved in endocytosis and autophagy. de Duve Institute, Université catholique de Louvain: Brussels, Belgium, 2012; Unpublished work. [Google Scholar]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Loewith, R.; Hall, M.N. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics 2011, 189, 1177–1201. [Google Scholar] [CrossRef] [Green Version]
- Barquilla, A.; Navarro, M. Trypanosome TOR as a major regulator of cell growth and autophagy. Autophagy 2009, 5, 256–258. [Google Scholar] [CrossRef]
- Denninger, V.; Koopmann, R.; Muhammad, K.; Barth, T.; Bassarak, B.; Schonfeld, C.; Kilunga, B.K.; Duszenko, M. Kinetoplastida: Model organisms for simple autophagic pathways? Meth. Enzymol. 2008, 451, 373–408. [Google Scholar]
- Diaz-Gonzalez, R.; Kuhlmann, F.M.; Galan-Rodriguez, C.; Madeira da Silva, L.; Saldivia, M.; Karver, C.E.; Rodriguez, A.; Beverley, S.M.; Navarro, M.; Pollastri, M.P. The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Neglect. Trop. D. 2011, 5, e1297. [Google Scholar]
- Uzcategui, N.L.; Carmona-Gutierrez, D.; Denninger, V.; Schoenfeld, C.; Lang, F.; Figarella, K.; Duszenko, M. Antiproliferative effect of dihydroxyacetone on Trypanosoma brucei bloodstream forms: Cell cycle progression, subcellular alterations, and cell death. Antimicrob. Agents Chemother. 2007, 51, 3960–3968. [Google Scholar]
- Uzcategui, N.L.; Denninger, V.; Merkel, P.; Schoenfeld, C.; Figarella, K.; Duszenko, M. Dihydroxyacetone induced autophagy in African trypanosomes. Autophagy 2007, 3, 626–629. [Google Scholar]
- Campos-Salinas, J.; Gonzalez-Rey, E. Autophagy and neuropeptides at the crossroad for parasites: To survive or to die? Autophagy 2009, 5, 551–554. [Google Scholar] [CrossRef]
- Delgado, M.; Anderson, P.; Garcia-Salcedo, J.A.; Caro, M.; Gonzalez-Rey, E. Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell Death Differ. 2009, 16, 406–416. [Google Scholar] [CrossRef]
- Cunha-e-Silva, N.; Sant'Anna, C.; Pereira, M.G.; Porto-Carreiro, I.; Jeovanio, A.L.; de Souza, W. Reservosomes: Multipurpose organelles? Parasitol. Res. 2006, 99, 325–327. [Google Scholar] [CrossRef]
- Alvarez, V.E.; Niemirowicz, G.T.; Cazzulo, J.J. The peptidases of Trypanosoma cruzi: Digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim. Biophys. Acta 2011, 1824, 195–206. [Google Scholar]
- Cazzulo, J.J.; Stoka, V.; Turk, V. Cruzipain, the major cysteine proteinase from the protozoan parasite Trypanosoma cruzi. Biol. Chem. 1997, 378, 1–10. [Google Scholar] [CrossRef]
- Schoijet, A.C.; Miranda, K.; Girard-Dias, W.; de Souza, W.; Flawia, M.M.; Torres, H.N.; Docampo, R.; Alonso, G.D. A Trypanosoma cruzi phosphatidylinositol 3-kinase (TcVps34) is involved in osmoregulation and receptor-mediated endocytosis. J. Biol. Chem. 2008, 283, 31541–31550. [Google Scholar]
- Braga, M.V.; de Souza, W. Effects of protein kinase and phosphatidylinositol-3 kinase inhibitors on growth and ultrastructure of Trypanosoma cruzi. FEMS Microbiol. Lett. 2006, 256, 209–216. [Google Scholar] [CrossRef]
- Santa-Rita, R.M.; Lira, R.; Barbosa, H.S.; Urbina, J.A.; de Castro, S.L. Anti-proliferative synergy of lysophospholipid analogues and ketoconazole against Trypanosoma cruzi (Kinetoplastida: Trypanosomatidae): Cellular and ultrastructural analysis. J. Antimicrob. Chemoth. 2005, 55, 780–784. [Google Scholar] [CrossRef]
- Fernandes, M.C.; Da Silva, E.N.; Pinto, A.V.; De Castro, S.L.; Menna-Barreto, R.F. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology 2012, 139, 26–36. [Google Scholar] [CrossRef]
- Menna-Barreto, R.F.; Correa, J.R.; Cascabulho, C.M.; Fernandes, M.C.; Pinto, A.V.; Soares, M.J.; De Castro, S.L. Naphthoimidazoles promote different death phenotypes in Trypanosoma cruzi. Parasitology 2009, 136, 499–510. [Google Scholar]
- Menna-Barreto, R.F.; Correa, J.R.; Pinto, A.V.; Soares, M.J.; de Castro, S.L. Mitochondrial disruption and DNA fragmentation in Trypanosoma cruzi induced by naphthoimidazoles synthesized from beta-lapachone. Parasitol. Res. 2007, 101, 895–905. [Google Scholar]
- Menna-Barreto, R.F.; Henriques-Pons, A.; Pinto, A.V.; Morgado-Diaz, J.A.; Soares, M.J.; De Castro, S.L. Effect of a beta-lapachone-derived naphthoimidazole on Trypanosoma cruzi: Identification of target organelles. J. Antimicrob. Chemoth. 2005, 56, 1034–1041. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Atg5: More than an autophagy factor. Nat. Cell Biol. 2006, 8, 1045–1047. [Google Scholar] [CrossRef]
- Besteiro, S.; Williams, R.A.; Coombs, G.H.; Mottram, J.C. Protein turnover and differentiation in Leishmania. Int. J. Parasitol. 2007, 37, 1063–1075. [Google Scholar] [CrossRef]
- Cloutier, S.; Laverdiere, M.; Chou, M.N.; Boilard, N.; Chow, C.; Papadopoulou, B. Translational control through eIF2alpha phosphorylation during the leishmania differentiation process. PLoS One 2012, 7, e35085. [Google Scholar]
- McConville, M.J.; Naderer, T. Metabolic pathways required for the intracellular survival of Leishmania. Annu. Rev. Microbiol. 2011, 65, 543–561. [Google Scholar] [CrossRef]
- Saunders, E.C.; DP, D.E.S.; Naderer, T.; Sernee, M.F.; Ralton, J.E.; Doyle, M.A.; Macrae, J.I.; Chambers, J.L.; Heng, J.; Nahid, A.; et al. Central carbon metabolism of Leishmania parasites. Parasitology 2010, 137, 1303–1313. [Google Scholar]
- Coombs, G.H.; Tetley, L.; Moss, V.A.; Vickerman, K. Three dimensional structure of the Leishmania amastigote as revealed by computer-aided reconstruction from serial sections. Parasitology 1986, 92, 13–23. [Google Scholar] [CrossRef]
- Hart, D.T.; Opperdoes, F.R. The occurrence of glycosomes (microbodies) in the promastigote stage of four major Leishmania species. Mol. Biochem. Parasit. 1984, 13, 159–172. [Google Scholar]
- Waller, R.F.; McConville, M.J. Developmental changes in lysosome morphology and function Leishmania parasites. Int. J. Parasitol. 2002, 32, 1435–1445. [Google Scholar] [CrossRef]
- Williams, R.A.; Smith, T.K.; Cull, B.; Mottram, J.C.; Coombs, G.H. ATG5 Is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 2012, 8, e1002695. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Biswas, A.; Das, P.K. Identification of a protein kinase A regulatory subunit from Leishmania having importance in metacyclogenesis through induction of autophagy. Mol. Microbiol. 2012, 83, 548–564. [Google Scholar] [CrossRef]
- Schurigt, U.; Schad, C.; Glowa, C.; Baum, U.; Thomale, K.; Schnitzer, J.K.; Schultheis, M.; Schaschke, N.; Schirmeister, T.; Moll, H. Aziridine-2,3-dicarboxylate-based cysteine cathepsin inhibitors induce cell death in Leishmania major associated with accumulation of debris in autophagy-related lysosome-like vacuoles. Antimicrob. Agents Chemother. 2010, 54, 5028–5041. [Google Scholar] [CrossRef]
- Monte Neto, R.L.; Sousa, L.M.; Dias, C.S.; Barbosa Filho, J.M.; Oliveira, M.R.; Figueiredo, R.C. Morphological and physiological changes in Leishmania promastigotes induced by yangambin, a lignan obtained from Ocotea duckei. Exp. Parasitol. 2011, 127, 215–221. [Google Scholar] [CrossRef]
- de Macedo-Silva, S.T.; de Oliveira Silva, T.L.; Urbina, J.A.; de Souza, W.; Rodrigues, J.C. Antiproliferative, ultrastructural, and physiological effects of amiodarone on promastigote and amastigote forms of leishmania amazonensis. Mol. Biol. Int. 2011, 2011, 876021. [Google Scholar]
- Bera, A.; Singh, S.; Nagaraj, R.; Vaidya, T. Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Mol. Biochem. Parasit. 2003, 127, 23–35. [Google Scholar] [CrossRef]
- Romano, P.S.; Arboit, M.A.; Vazquez, C.L.; Colombo, M.I. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 2009, 5, 6–18. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Letai, A. Who put the “A” in Atg12: Autophagy or apoptosis? Mol. Cell 2011, 44, 844–845. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef]
- Fulda, S. Autophagy and cell death. Autophagy 2012, 8. in press. [Google Scholar]
- Sengupta, S.; Chowdhury, S.; Bosedasgupta, S.; Wright, C.W.; Majumder, H.K. Cryptolepine-induced cell death of leishmania donovani promastigotes is augmented by inhibition of autophagy. Mol. Biol. Int. 2011, 2011, 187850. [Google Scholar]
- Paugam, A.; Bulteau, A.L.; Dupouy-Camet, J.; Creuzet, C.; Friguet, B. Characterization and role of protozoan parasite proteasomes. Trends Parasitol. 2003, 19, 55–59. [Google Scholar] [CrossRef]
- Robertson, C.D. The Leishmania mexicana proteasome. Mol. Biochem. Parasitol. 1999, 103, 49–60. [Google Scholar] [CrossRef]
- Nkemngu, N.J.; Rosenkranz, V.; Wink, M.; Steverding, D. Antitrypanosomal activities of proteasome inhibitors. Antimicrob. Agents Chemother. 2002, 46, 2038–2040. [Google Scholar]
- King, J.S. Autophagy across the eukaryotes: Is S. cerevisiae the odd one out? Autophagy 2012, 8. in press. [Google Scholar]
- Veiga-Santos, P.; Barrias, E.S.; Santos, J.F.; de Barros Moreira, T.L.; de Carvalho, T.M.; Urbina, J.A.; de Souza, W. Effects of amiodarone and posaconazole on the growth and ultrastructure of Trypanosoma cruzi. Int. J. Antimicrob. Agents 2012, 40, 61–71. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brennand, A.; Rico, E.; Michels, P.A.M. Autophagy in Trypanosomatids. Cells 2012, 1, 346-371. https://doi.org/10.3390/cells1030346
Brennand A, Rico E, Michels PAM. Autophagy in Trypanosomatids. Cells. 2012; 1(3):346-371. https://doi.org/10.3390/cells1030346
Chicago/Turabian StyleBrennand, Ana, Eva Rico, and Paul A. M. Michels. 2012. "Autophagy in Trypanosomatids" Cells 1, no. 3: 346-371. https://doi.org/10.3390/cells1030346