Heavy Metals and Metalloids as Autophagy Inducing Agents: Focus on Cadmium and Arsenic



Abstract
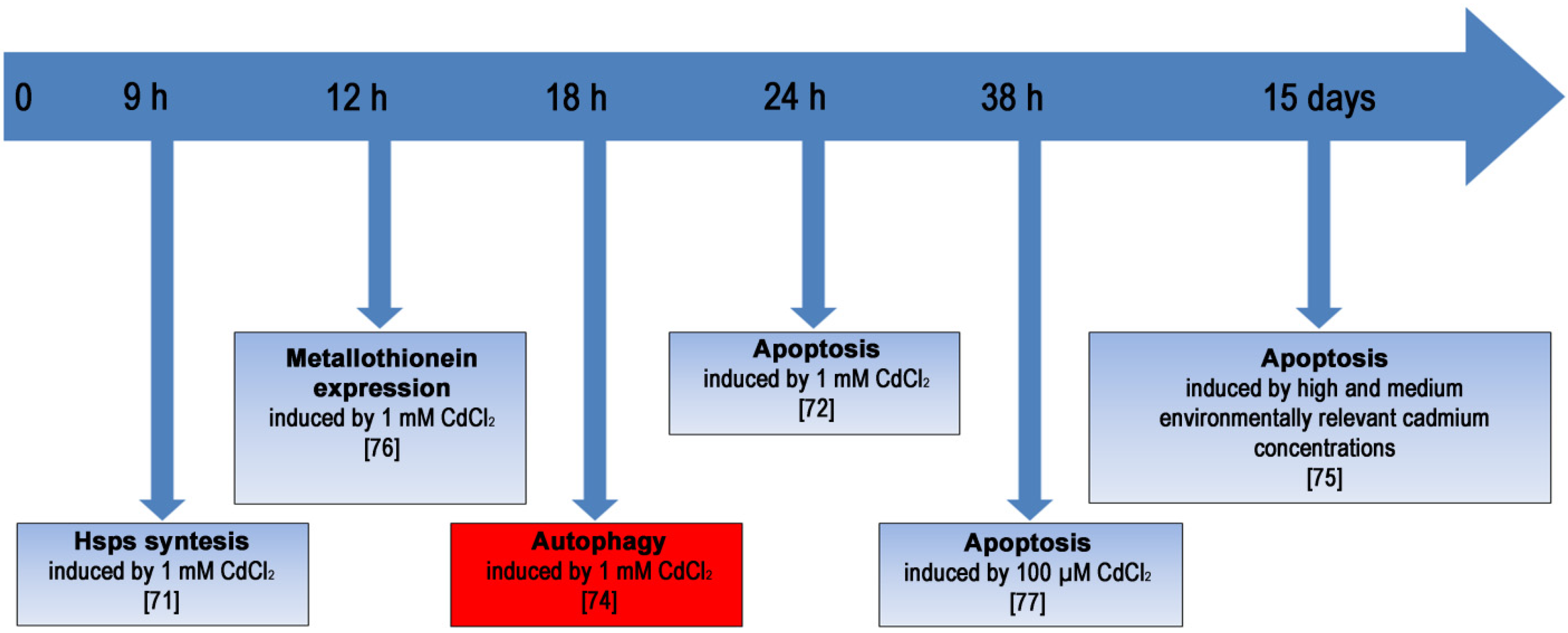
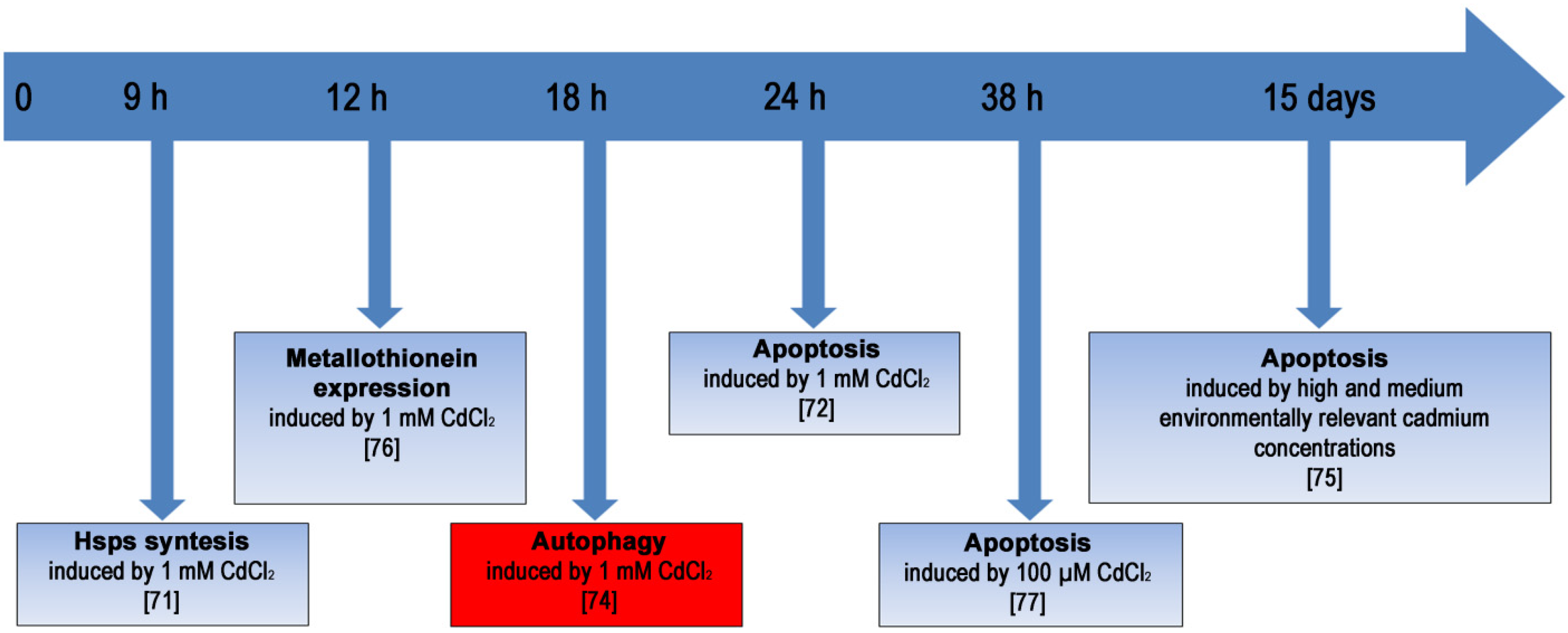
:1. Introduction
1.1. Autophagy
1.2. Heavy Metals and Metalloids
2. The Effects of Heavy Metals and Metalloids on Cells
2.1. Arsenic
2.2. Arsenic in Combination with Other Heavy Metals or Radiation

{kind=link}
{kind=link}
| As compounds (concentrations) | Autophagic effects | Experimental model | References |
|---|---|---|---|
| As2O3 (2 µM) | autophagic cell death, by activation of the MEK/ERK pathway; antileukemic effects | human leukemia cells | [27] |
| As2O3 (0,625–20 μM) | autophagic cell survival, in the earlier period of treatment; apoptosis and/or autophagic cell death, in the later period of treatment | HL60 leukemia cells | [35] |
| As2O3 (1 µM) | autophagy as clearance mechanism of the fusion protein PML/RARA | human leukemia cells | [29] |
| As2O3 (4 µM) | autophagic cell death, by up-regulation of Beclin-1, as well as apoptosis | human leukemia cells | [30] |
| As4O6 (0.5–3 µM) | autophagic cell death, by up-regulation of Beclin-1, as well as apoptosis, by caspase activation | U937 human leukemia cells | [43] |
| As2O3 (1–4 µM) | autophagic cell death, by up-regulation of BNIP3 and ERK 1/2, down-regulation of PI3K/AKT; antitumor effects | human malignant glioma cells | [28,46] |
| NaAsO2 (1–10 µM) | autophagic cell death, including increased levels of LC3B and Beclin-1, as well as apoptosis | human uroepithelial cells | [45] |
| NaAsO2 (6 µM) | autophagic cell death, including increased levels of LC3-II and autophagosomes/autolysosomes, not associated with apoptosis | human lymphoblastoid cells | [41,42] |
2.3. Cadmium
2.3.1. Cadmium in Combination with Chromium
2.3.2. Effects of Cadmium on Aquatic Invertebrates
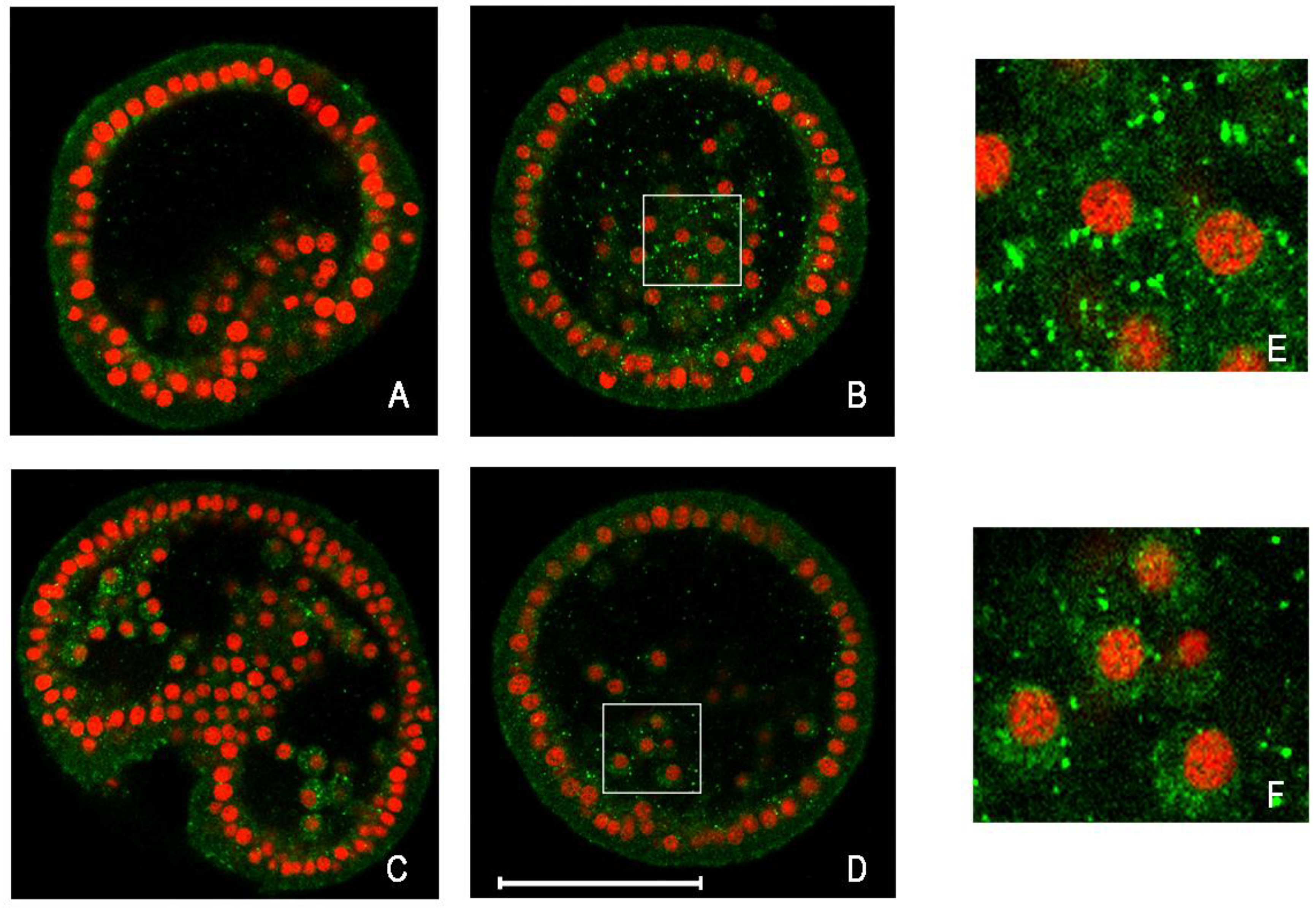
2.3.3. Stress Response in Cd-Exposed Sea Urchin Embryos
2.3.4. Analyzing Autophagy in Cd-Exposed Sea Urchin Embryos

| Cd compounds (concentrations) | Autophagic effects | Experimental model | References |
|---|---|---|---|
| CdCl2 (3–24 µM) | calcium-mediated autophagy and apoptosis, through the ERK-dependent and mitochondria-caspase signaling pathways, respectively | mouse kidney mesangial cells | [54] |
| CdCl2 (1–10 µM) | autophagy that leads to cytotoxicity, as cell death mechanism; detected by an accumulation of autofagosomes and increased levels of LC3-II | mouse epidermal cell line | [57] |
| Cd (NO3)2 (1–10 µM) Cd (NO3)2 (>20 µM) | autophagy, as cell survival mechanism, detected by an accumulation of autolysosomes and increased levels of LC3-II;apoptotic cell death | human vascular endothelial cells | [56] |
| CdCl2 (0.3 mg/kg body mass/1, 3 and 5 days of intoxication ) | autophagy, as cell survival mechanism | rat kidney | [58] |
| CdCl2 (1 mM for 18 hours of exposure) CdCl2 (1 mM for 24 hours of exposure) | autophagy as a survival mechanism detected by an accumulation of autolysosomes and increased levels of LC3-II;Apoptotic cell death | sea urchin embryos | [74] |
3. Conclusions
Acknowledgments
References
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Scarlatti, F.; Granata, R.; Meijer, A.J.; Codogno, P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009, 16, 12–20. [Google Scholar]
- Adastra, K.L.; Chi, M.M.; Riley, J.K.; Moley, K.H. A differential autophagic response to hyperglycemia in the developing murine embryo. Reproduction 2011, 141, 607–615. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar]
- Yu, L.; Lenardo, M.J.; Baehrecke, E.H. Autophagy and caspases: A new cell death program. CellCycle 2004, 3, 1124–1126. [Google Scholar]
- Eisenberg-Lerner, A.; Kimchi, A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 2009, 14, 376–391. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 2009, 4, e6251. [Google Scholar]
- Song, J.; Qu, Z.; Guo, X.; Zhao, Q.; Zhao, X.; Gao, L.; Sun, K.; Shen, F.; Wu, M.; Wei, L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009, 5, 1131–1144. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Invest. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Apel, A.; Zentgraf, H.; Büchler, M.W.; Herr, I. Autophagy-A double-edged sword in oncology. Int. J. Cancer 2009, 125, 991–995. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Sarbassovdos, D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 29, 109–120. [Google Scholar] [CrossRef]
- Scheifler, R.; Coeurdassier, M.; Morilhat, C.; Bernard, N.; Faivre, B.; Flicoteaux, P.; Giraudoux, P.; Noël, M.; Piotte, P.; Rieffel, D.; et al. Lead concentrations in feathers and blood of common blackbirds (Turdus merula) and in earthworms inhabiting unpolluted and moderately polluted urban areas. Sci. Total Environ. 2006, 371, 197–205. [Google Scholar] [CrossRef]
- Jha, A.N.; Noditi, M.; Nilsson, R.; Natarajan, A.T. Genotoxic effects of sodium arsenite on human cells. Mutat. Res. 1992, 284, 215–221. [Google Scholar] [CrossRef]
- Tice, R.R.; Yager, J.W.; Andrews, P.; Crecelius, E. Effect of hepatic methyl donor status on urinary excretion and DNA damage in B6C3F1 mice treated with sodium arsenite. Mutat. Res. 1997, 386, 315–334. [Google Scholar]
- Miller, W.H.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002, 62, 3893–3903. [Google Scholar]
- Miller, W.H., Jr. Molecular targets of arsenic trioxide in malignant cells. Oncologist 2002, 7, 14–19. [Google Scholar] [CrossRef]
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997. [Google Scholar]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef]
- Zhu, X.H.; Shen, Y.L.; Jing, Y.K.; Cai, X.; Jia, P.M.; Huang, Y.; Tang, W.; Shi, G.Y.; Sun, Y.P.; Dai, J.; et al. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J. Natl. CancerInst. 1999, 91, 772–778. [Google Scholar] [CrossRef]
- Sturlan, S.; Baumgartner, M.; Roth, E.; Bachleitner-Hofmann, T. Docosahexaenoic acid enhances arsenic trioxide-mediated apoptosis in arsenic trioxide-resistant HL-60 cells. Blood 2003, 101, 4990–4997. [Google Scholar]
- Glienke, W.; Chow, K.U.; Bauer, N.; Bergmann, L. Down-regulation of wt1 expression in leukemia cell lines as part of apoptotic effect in arsenic treatment using two compounds. Leuk. Lymphoma 2006, 47, 1629–1638. [Google Scholar] [CrossRef]
- Charoensuk, V.; Gati, W.P.; Weinfeld, M.; Le, X.C. Differential cytotoxic effects of arsenic compounds in human acute promyelocytic leukemia cells. Toxicol. Appl. Pharmacol. 2009, 239, 64–70. [Google Scholar] [CrossRef]
- Yang, Y.P.; Liang, Z.Q.; Gao, B.; Jia, Y.L.; Qin, Z.H. Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells. Acta Pharmacol. Sin. 2008, 29, 123–134. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar]
- Gibson, S.B. A matter of balance between life and death: Targeting reactive oxygen species (ROS)-induced autophagy for cancer therapy. Autophagy 2010, 6, 835–837. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Raffoul, F.; Campla, C.; Nanjundan, M. SnoN/SkiL, a TGFβ signaling mediator: A participant in autophagy induced by arsenic trioxide. Autophagy 2010, 6, 955–957. [Google Scholar] [CrossRef]
- Bolt, A.M.; Byrd, R.M.; Klimecki, W.T. Autophagy is the predominant process induced by arsenite in human lymphoblastoid cell lines. Toxicol. Appl. Pharmacol. 2010, 244, 366–373. [Google Scholar] [CrossRef]
- Bolt, A.M.; Douglas, R.M.; Klimecki, W.T. Arsenite exposure in human lymphoblastoid cell lines induces autophagy and coordinated induction of lysosomal genes. Toxicol. Lett. 2010, 199, 153–159. [Google Scholar] [CrossRef]
- Han, M.H.; Lee, W.S.; Lu, J.N.; Yun, J.W.; Kim, G.; Jung, J.M.; Kim, G.Y.; Lee, S.J.; Kim, W.J.; Choi, Y.H. Tetraarsenic Hexoxide Induces Beclin-1-Induced Autophagic Cell Death as well as Caspase-Dependent Apoptosis in U937 Human Leukemic Cells. Evid.-Based Complement Altern. Med. 2012, 2012. [Google Scholar]
- Cheng, T.J.; Wang, Y.J.; Kao, W.W.; Chen, R.J.; Ho, Y.S. Protection against arsenic trioxide-induced autophagic cell death in U118 human glioma cells by use of lipoic acid. Food Chem. Toxicol. 2007, 45, 1027–1038. [Google Scholar] [CrossRef]
- Huang, Y.C.; Hung, W.C.; Chen, W.T.; Yu, H.S.; Chai, C.Y. Sodium arsenite-induced DAPK promoter hypermethylation and autophagy via ERK1/2 phosphorylation in human uroepithelial cells. Chem. Biol. Interact. 2009, 181, 254–262. [Google Scholar]
- Chiu, H.W.; Ho, S.Y.; Guo, H.R.; Wang, Y.J. Combination treatment with arsenic trioxide and irradiation enhances autophagic effects in U118-MG cells through increased mitotic arrest and regulation of PI3K/Akt and ERK1/2 signaling pathways. Autophagy 2009, 5, 472–483. [Google Scholar] [CrossRef]
- Chiu, H.W.; Lin, J.H.; Chen, Y.A.; Ho, S.Y.; Wang, Y.J. Combination treatment with arsenic trioxide and irradiation enhances cell-killing effects in human fibrosarcoma cells in vitro and in vivo through induction of both autophagy and apoptosis. Autophagy 2010, 6, 353–365. [Google Scholar] [CrossRef]
- Larson, J.L.; Somji, S.; Zhou, X.D.; Sens, M.A.; Garrett, S.H.; Sens, D.A.; Dunlevy, J.R. Beclin-1 expression in normal bladder and in Cd2+ and As3+ exposed and transformed human urothelial cells (UROtsa). Toxicol. Lett. 2010, 195, 15–22. [Google Scholar] [CrossRef]
- Roccheri, M.C.; Matranga, V. Cellular, Biochemical and Molecular Effects of Cadmium on Marine Invertebrates: Focus on Paracentrotus lividus Sea Urchin Development. In Cadmium in the Environment; Parvau, R.G., Ed.; Nova Science Publishers: New York, NY, USA, 2010; pp. 337–366. [Google Scholar]
- Achanzar, W.E.; Diwan, B.A.; Liu, J.; Quader, S.M.; Webber, M.M.; Waalkes, M.P. Cadmium-induced malignant transformation of human prostate epithelial cells. Cancer Res. 2001, 61, 455–458. [Google Scholar]
- Joseph, P. Mechanisms of cadmium carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 272–279. [Google Scholar] [CrossRef]
- Templeton, D.M.; Liu, Y. Multiple roles of cadmium in cell death and survival. Chem. Biol. Interact. 2010, 188, 267–275. [Google Scholar] [CrossRef]
- Yang, L.Y.; Wu, K.H.; Chiu, W.T.; Wang, S.H.; Shih, C.M. The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy 2009, 5, 571–572. [Google Scholar] [CrossRef]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Wang, S.H.; Shih, Y.L.; Kuo, T.C.; Ko, W.C.; Shih, C.M. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol. Sci. 2009, 108, 124–131. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, L.; Xu, J.; Li, Y.; Zhang, Y.; Zhang, S.; Miao, J. Promotion of autophagy and inhibition of apoptosis by low concentrations of cadmium in vascular endothelial cells. Toxicol. In Vitro 2009, 23, 105–110. [Google Scholar] [CrossRef]
- Son, Y.O.; Wang, X.; Hitron, J.A.; Zhang, Z.; Cheng, S.; Budhraja, A.; Ding, S.; Lee, J.C.; Shi, X. Cadmium induces autophagy through ROS-dependent activation of the LKB1-AMPK signaling in skin epidermal cells. Toxicol. Appl. Pharmacol. 2011, 255, 287–296. [Google Scholar] [CrossRef]
- Chargui, A.; Zekri, S.; Jacquillet, G.; Rubera, I.; Ilie, M.; Belaid, A.; Duranton, C.; Tauc, M.; Hofman, P.; Poujeol, P.; et al. Cadmium-induced autophagy in rat kidney: An early biomarker of subtoxic exposure. Toxicol. Sci. 2011, 121, 31–42. [Google Scholar] [CrossRef]
- Sansanwal, P.; Li, L.; Hsieh, S.C.; Sarwal, M.M. Insights into novel cellular injury mechanisms by gene expression profiling in nephropathic cystinosis. J. Inherit. Metab. Dis. 2010, 33, 775–786. [Google Scholar] [CrossRef]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Petrarca, C.; Perrone, A.; Farina, M.; Sabbioni, E.; Hartung, T.; Martino, S.; Esposito, D.L.; Lotti, L.V.; Mariani-Costantini, R. Autophagy as an ultrastructural marker of heavy metal toxicity in human cord blood hematopoietic stem cells. Sci. Total Environ. 2008, 392, 50–58. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Petrarca, C.; Perrone, A.; Martino, S.; Esposito, D.L.; Lotti, L.V.; Mariani-Costantini, R. Autophagy in hematopoietic stem/progenitor cells exposed to heavy metals: Biological implications and toxicological relevance. Autophagy 2008, 4, 537–539. [Google Scholar]
- Bagchi, D.; Joshi, S.S.; Bagchi, M.; Balmoori, J.; Benner, E.J.; Kuszynski, C.A.; Stohs, S.J. Cadmium- and chromium-induced oxidative stress, DNA damage, and apoptotic cell death in cultured human chronic myelogenous leukemic K562 cells, promyelocytic leukemic HL-60 cells, and normal human peripheral blood mononuclear cells. J. Biochem. Mol. Toxicol. 2000, 14, 33–41. [Google Scholar] [CrossRef]
- Viarengo, A. Heavy Metal in Marine Invertebrates: Mechanisms of Regulation and Toxicity at the Cellular Level. In Aquatic Sciences; Birkhäuse: Basel, Switzerland, 1989; Volume 1, pp. 295–317. [Google Scholar]
- Rainbow, P.S. Trace metal concentrations in aquatic invertebrates: Why and so what? Environ. Pollut. 2002, 120, 497–507. [Google Scholar] [CrossRef]
- Nigro, M.; Regoli, F.; Rocchi, R.; Orlando, E. Heavy metals in Antarctic molluscs. In Antarctic Communities: Species, Structure and Survival; Battaglia, B., Valencia, J., Walton, D.W.H., Eds.; Cambridge University Press: Cambridge, UK, 1997; pp. 409–412. [Google Scholar]
- Podgurskaya, O.V.; Kavun, V.Y. Cadmium concentration and subcellular distribution in organs of the mussel Crenomytilus grayanus from upwelling regions of Okhotsk Sea and Sea of Japan. Arch. Environ. Contam. Toxicol. 2006, 51, 567–572. [Google Scholar] [CrossRef]
- Rodrigues, E.; da Silva Santos, M.R.; Rodrigues, E., Jr.; Gannabathula, S.V.; Passeri Lavrado, H. Arginine metabolism of the Antarctic Bivalve Laternula elliptica (King & Broderip, 1831): An ecophysiological approach. Polar Biol. 2009, 32, 691–702. [Google Scholar] [CrossRef]
- Chora, S.; Starita-Geribaldi, M.; Guigonis, J.M.; Samson, M.; Roméo, M.; Bebianno, M.J. Effect of cadmium in the clam Ruditapes decussatus assessed by proteomic analysis. Aquat. Toxicol. 2009, 94, 300–308. [Google Scholar] [CrossRef]
- Vacquier, V.D. Laboratory on sea urchin fertilization. Mol. Reprod. Dev. 2011, 78, 553–564. [Google Scholar] [CrossRef]
- Roccheri, M.C.; Agnello, M.; Bonaventura, R.; Matranga, V. Cadmium induces the expression of specific stress proteins in sea urchin embryos. Biochem. Biophys. Res. Commun. 2004, 321, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Agnello, M.; Filosto, S.; Scudiero, R.; Rinaldi, A.M.; Roccheri, M.C. Cadmium induces an apoptotic response in sea urchin embryos. Cell Stress Chaperones 2007, 12, 44–50. [Google Scholar] [CrossRef]
- Hamdoun, A.; Epel, D. Embryo stability and vulnerability in an always changing world. Proc. Natl. Acad. Sci. USA 2007, 104, 1745–1750. [Google Scholar] [CrossRef]
- Chiarelli, R.; Agnello, M.; Roccheri, M.C. Sea urchin embryos as a model system for studying autophagy induced by cadmium stress. Autophagy 2011, 7, 1028–1034. [Google Scholar]
- Filosto, S.; Roccheri, M.C.; Bonaventura, R.; Matranga, V. Environmentally relevant cadmium concentrations affect development and induce apoptosis of Paracentrotus lividus larvae cultured in vitro. CellBiol. Toxicol. 2008, 24, 603–610. [Google Scholar] [CrossRef]
- Russo, R.; Bonaventura, R.; Zito, F.; Schröder, H.C.; Müller, I.; Müller, W.E.; Matranga, V. Stress to cadmium monitored by metallothionein gene induction in Paracentrotus lividus embryos. Cell Stress Chaperones 2003, 8, 232–241. [Google Scholar] [CrossRef]
- Agnello, M.; Filosto, S.; Scudiero, R.; Rinaldi, A.M.; Roccheri, M.C. Cadmium accumulation induces apoptosis in P. lividus embryos. Caryologia 2006, 59, 403–408. [Google Scholar]
- Agnello, M.; Roccheri, M.C. Apoptosis: Focus on sea urchin development. Apoptosis 2010, 15, 322–330. [Google Scholar] [CrossRef]
- Alhama, J.; Romero-Ruiz, A.; Jebali, J.; López-Barea, J. Total Metallothionein Quantification by Reversed-Phase High-Performance Liquid Chromatography Coupled to Fluorescence Detection after Monobromobimane Derivatization. In Cadmium in the Environment; Parvau, R.G., Ed.; Nova Science Publishers: New York, NY, USA, 2010; pp. 389–405. [Google Scholar]
- Samali, A.; Cotter, T.G. Heat shock proteins increase resistance to apoptosis. Exp. Cell Res. 1996, 223, 163–170. [Google Scholar] [CrossRef]
- Hamada, T.; Tanimoto, A.; Sasaguri, Y. Apoptosis induced by cadmium. Apoptosis 1997, 2, 359–367. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Lim, S.C.; Hahm, K.S.; Lee, S.H.; Oh, S.H. Autophagy involvement in cadmium resistance through induction of multidrug resistance-associated protein and counterbalance of endoplasmic reticulum stress WI38 lung epithelial fibroblast cells. Toxicology 2010, 276, 18–26. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiarelli, R.; Roccheri, M.C. Heavy Metals and Metalloids as Autophagy Inducing Agents: Focus on Cadmium and Arsenic. Cells 2012, 1, 597-616. https://doi.org/10.3390/cells1030597
Chiarelli R, Roccheri MC. Heavy Metals and Metalloids as Autophagy Inducing Agents: Focus on Cadmium and Arsenic. Cells. 2012; 1(3):597-616. https://doi.org/10.3390/cells1030597
Chicago/Turabian StyleChiarelli, Roberto, and Maria Carmela Roccheri. 2012. "Heavy Metals and Metalloids as Autophagy Inducing Agents: Focus on Cadmium and Arsenic" Cells 1, no. 3: 597-616. https://doi.org/10.3390/cells1030597