Apoptotic Volume Decrease (AVD) Is Independent of Mitochondrial Dysfunction and Initiator Caspase Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| AMC | fluorochrome 7-amino 4-methyl coumarin |
| AVD | apoptotic volume decrease |
| DIDS | 4,4'-diisothiocyanostilbene-2,2'-disulfonic acid |
| DMSO | dimethylsulfoxide |
| FBS | fetal bovine serum |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide |
| NPPB | 5-nitro-2-(3-phenylpropylamino)benzoic acid |
| ROS | reactive oxygen species |
| SITS | 4-acetamido-4'-isothiocyanostilbene |
| STS | staurosporine |
| VDAC | voltage-dependent anion channel |
| VSOR | volume-sensitive outwardly rectifying |
| zD-dcb | Z-Asp-2,6-dichlorobenzoyloxymethylketone |
| zVAD-fmk | benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone |
1. Introduction
2. Results
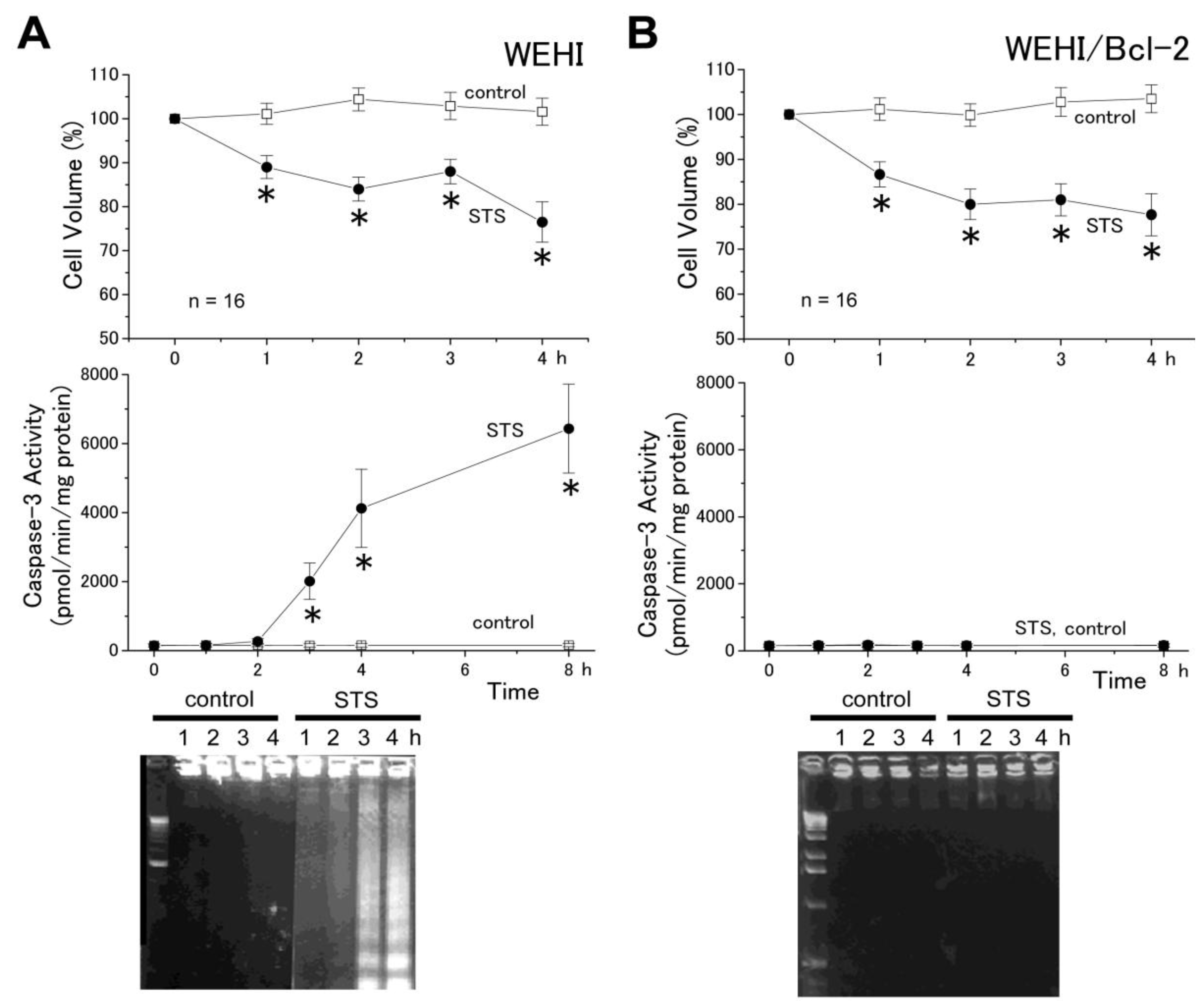
2.1. The AVD Induction Is Independent of the Mitochondrial Apoptosis Pathway


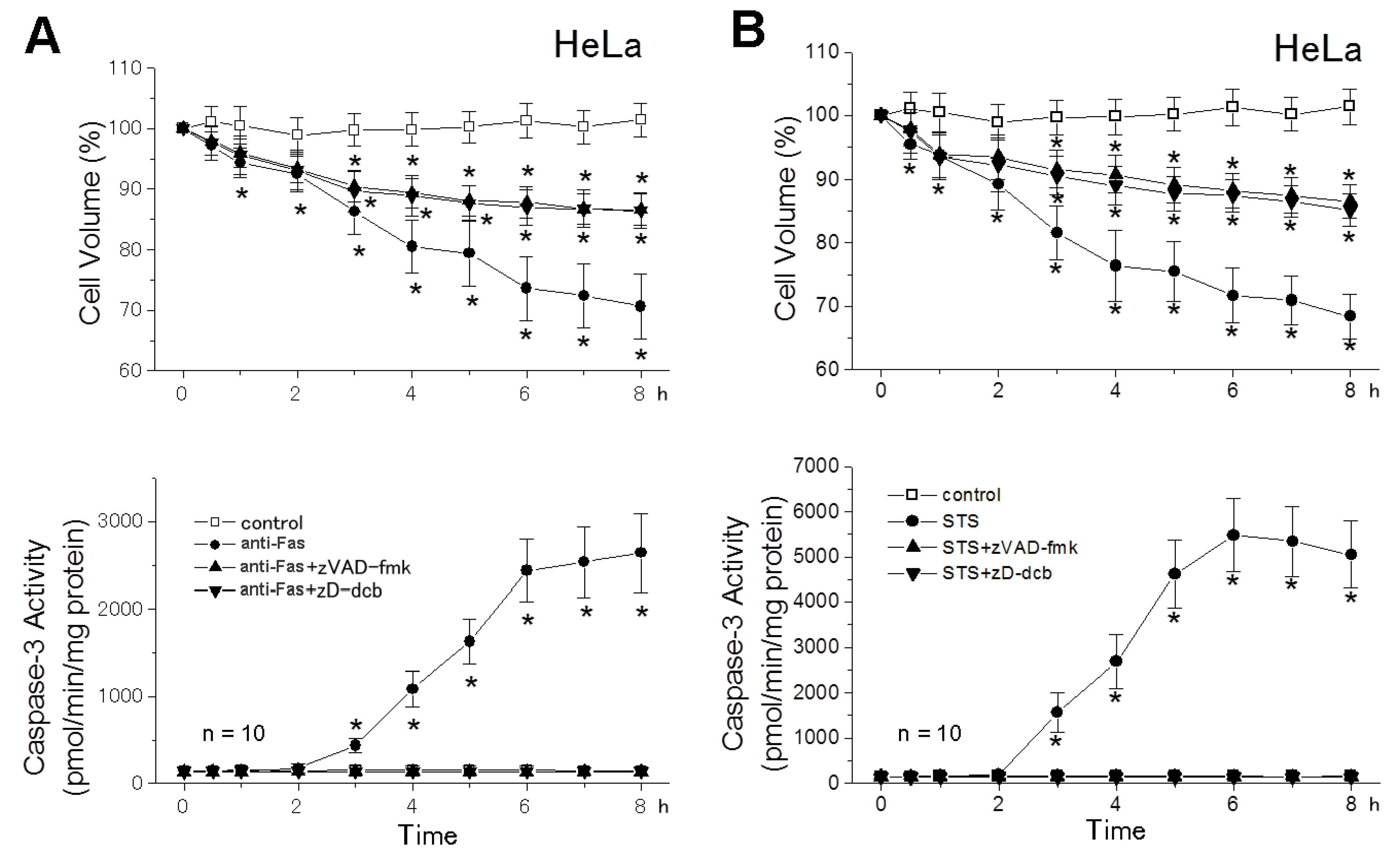
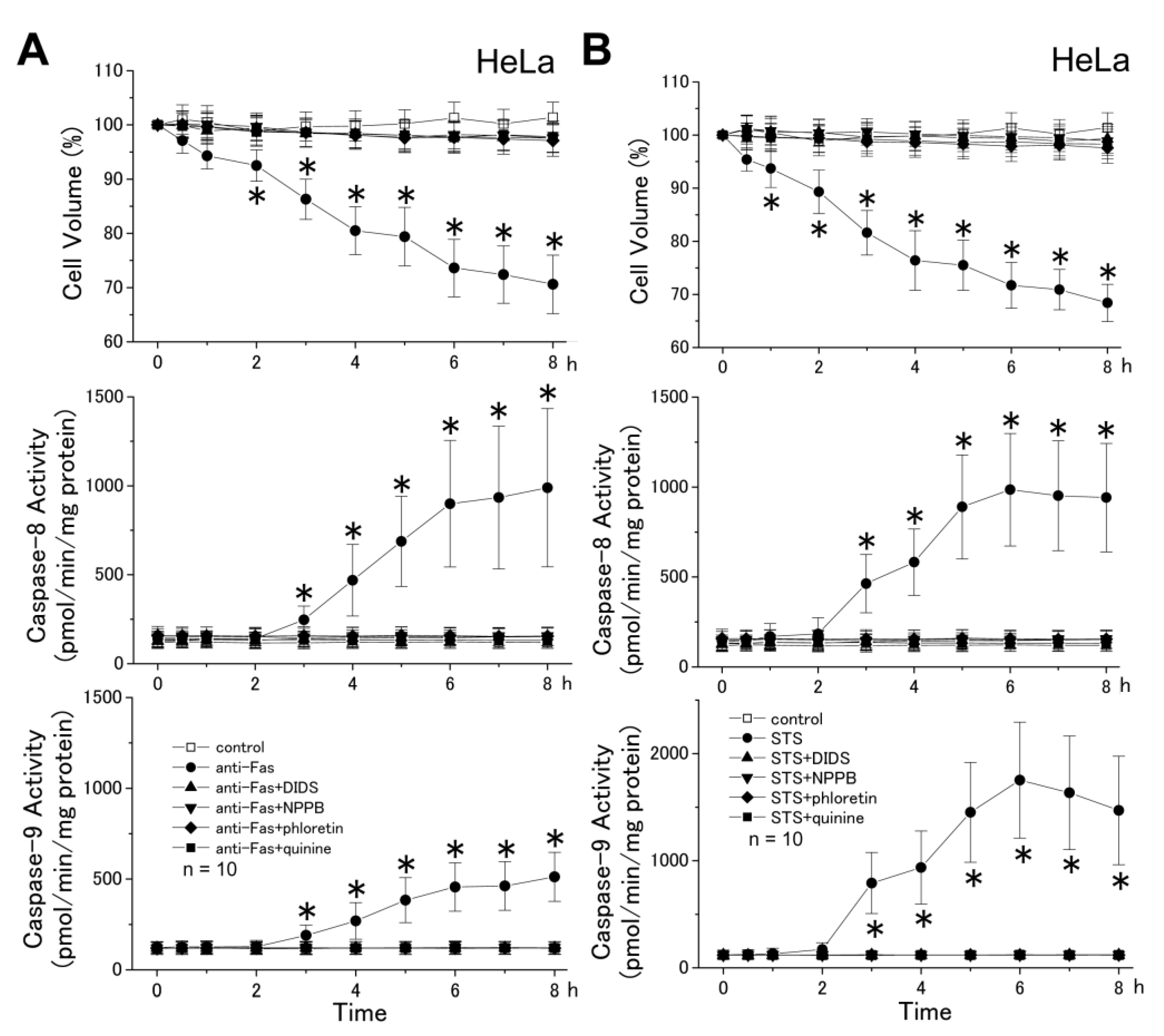
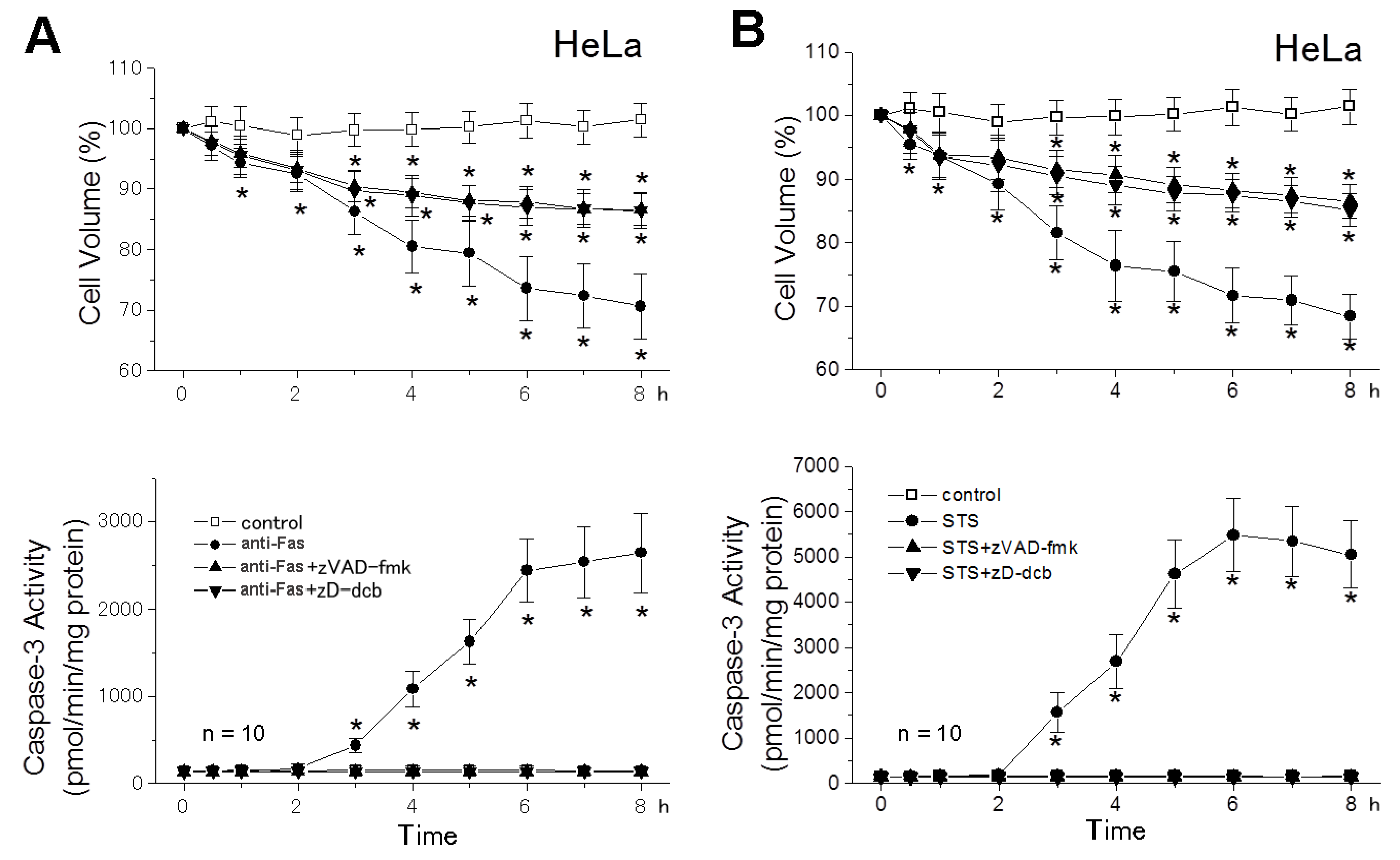
2.2. The AVD Induction Is an Independent Event of Activation of Initiator Caspases
3. Discussion
4. Experimental Section
4.1. Cell Culture and Apoptosis Induction
4.2. Cell Volume Measurements
4.3. Caspase Activity Measurements
4.4. DNA Fragmentation Assay
4.5. Cell Viability Assay
4.6. Chemicals
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage due to disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar]
- Okada, Y.; Maeno, E.; Shimizu, T.; Dezaki, K.; Wang, J.; Morishima, S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol. (London) 2001, 532, 3–16. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Shimizu, T.; Takahashi, N.; Okada, Y. The apoptotic volume decrease is an upstream event of MAP kinase activation during staurosporine-induced apoptosis in HeLa cells. Int. J. Mol. Sci. 2012, 13, 9363–9379. [Google Scholar] [CrossRef]
- Yu, S.P.; Choi, D.W. Ions, cell volume, and apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9360–9362. [Google Scholar]
- Lang, F.; Föller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. K+ channels in apoptosis. J. Membr. Biol. 2006, 209, 3–20. [Google Scholar] [CrossRef]
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J. Membr. Biol. 2006, 209, 21–29. [Google Scholar] [CrossRef]
- Stutzin, A.; Hoffmann, E.K. Swelling-activated ion channels: Functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol. 2006, 187, 27–42. [Google Scholar] [CrossRef]
- Shimizu, T.; Numata, T.; Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar] [CrossRef]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Decaudin, D.; Macho, A.; Hirsch, T.; Susin, S.A.; Petit, P.X.; Mignotte, B.; Kroemer, G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. [Google Scholar] [CrossRef]
- Hortelano, S.; Zeini, M.; Castrillo, A.; Alvarez, A.M.; Boscá, L. Induction of apoptosis by nitric oxide in macrophages is independent of apoptotic volume decrease. Cell Death Differ. 2002, 9, 643–650. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Caspase independent/dependent regulation of K+, cell shrinkage, and mitochondrial membrane potential during lymphocyte apoptosis. J. Biol. Chem. 1999, 274, 21953–21962. [Google Scholar]
- Schrantz, N.; Blanchard, D.A.; Auffredou, M.T.; Sharma, S.; Leca, G.; Vazquez, A. Role of caspases and possible involvement of retinoblastoma protein during TGFβ-mediated apoptosis of human B lymphocytes. Oncogene 19 1999, 18, 3511–3519. [Google Scholar]
- Zhivotovsky, B.; Gahm, A.; Ankarcrona, M.; Nicotera, P.; Orrenius, S. Multiple proteases are involved in thymocyte apoptosis. Exp. Cell Res. 1995, 221, 404–412. [Google Scholar] [CrossRef]
- Wolf, C.M.; Reynolds, J.E.; Morana, S.J.; Eastman, A. The temporal relationship between protein phosphatase, ICE/CED-3 proteases, intracellular acidification, and DNA fragmentation in apoptosis. Exp. Cell Res. 1997, 230, 22–27. [Google Scholar] [CrossRef]
- Hughes, F.M., Jr.; Cidlowski, J.A. Glucocorticoid-induced thymocyte apoptosis: Protease-dependent activation of cell shrinkage and DNA degradation. J. Steroid Biochem. Mol. Biol. 1998, 65, 207–217. [Google Scholar] [CrossRef]
- Lang, F.; Madlung, J.; Siemen, D.; Ellory, C.; Lepple-Wienhues, A.; Gulbins, E. The involvement of caspases in the CD95(Fas/Apo-1)- but not swelling-induced cellular taurine release from Jurkat T-lymphocytes. Pflugers Arch. 2000, 440, 93–99. [Google Scholar]
- Nobel, C.S.; Aronson, J.K.; van den Dobbelsteen, D.J.; Slater, A.F. Inhibition of Na+/K+-ATPase may be one mechanism contributing to potassium efflux and cell shrinkage in CD95-induced apoptosis. Apoptosis 2000, 5, 153–163. [Google Scholar] [CrossRef]
- Vu, C.C.; Bortner, C.D.; Cidlowski, J.A. Differential involvement of initiator caspases in apoptotic volume decrease and potassium efflux during Fas- and UV-induced cell death. J. Biol. Chem. 2001, 276, 37602–37611. [Google Scholar]
- Choi, K.H.; Hama-Inaba, H.; Wang, B.; Haginoya, K.; Odaka, T.; Yamada, T.; Hayata, I.; Ohyama, H. UVC-induced apoptosis in human epithelial tumor A431 cells: Sequence of apoptotic changes and involvement of caspase (-8 and -3) cascade. J. Radiat. Res. 2000, 41, 243–258. [Google Scholar] [CrossRef]
- Scaffidi, C.; Schmitz, I.; Zha, J.; Korsmeyer, S.J.; Krammer, P.H.; Peter, M.E. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem. 1999, 274, 22532–22538. [Google Scholar]
- Eguchi, Y.; Srinivasan, A.; Tomaselli, K.J.; Shimizu, S.; Tsujimoto, Y. ATP-dependent steps in apoptotic signal transduction. Cancer Res. 1999, 59, 2174–2181. [Google Scholar]
- Lindsay, J.; Esposti, M.D.; Gilmore, A.P. Bcl-2 proteins and mitochondria—specificity in membrane targeting for death. Biochim. Biophys. Acta. 2011, 1813, 532–539. [Google Scholar] [CrossRef]
- Benson, R.S.; Heer, S.; Dive, C.; Watson, A.J. Characterization of cell volume loss in CEM-C7A cells during dexamethasone-induced apoptosis. Am. J. Physiol. 1996, 270, C1190–C1203. [Google Scholar]
- Dezaki, K.; Maeno, E.; Sato, K.; Akita, T.; Okada, Y. Early-phase occurrence of K+ and Cl− efflux in addition to Ca2+ mobilization is a prerequisite to apoptosis in HeLa cells. Apoptosis 2012, 17, 821–831. [Google Scholar] [CrossRef]
- Varela, D.; Simon, F.; Riveros, A.; Jorgengen, F.; Stutzin, A. NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J. Biol. Chem. 2004, 279, 13301–13304. [Google Scholar]
- Chung, Y.M.; Kim, J.S.; Yoo, Y.D. A novel protein, Romo1, induces ROS production in the mitochondria. Biochem. Biophys. Res. Commun. 2006, 347, 649–655. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.J.; Shin, J.; Kang, S.T.; Kang, S.; Yoo, Y.D. Bcl-XL prevents serum deprivation-induced oxidative stress mediated by Romo1. Oncology Rep. 2011, 25, 1337–1342. [Google Scholar]
- Okada, Y.; Maeno, E. Apoptosis, cell volume regulation and volume-regulatory chloride channels. Comp. Biochem. Physiol. Part. A 2001, 130, 377–383. [Google Scholar]
- Ishizaki, Y.; Voyvodic, J.T.; Burne, J.F.; Raff, M.C. Control of lens epithelial cell survival. J. Cell Biol. 1993, 121, 899–908. [Google Scholar] [CrossRef]
- Hazama, A.; Okada, Y. Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J. Physiol. (London) 1988, 402, 687–702. [Google Scholar]
- Thornberry, N.A. Interleukin-1 beta converting enzyme. Methods Enzymol. 1994, 244, 615–631. [Google Scholar]
- Shiokawa, D.; Ohyama, H.; Yamada, T.; Takahashi, K.; Tanuma, S. Identification of an endonuclease responsible for apoptosis in rat thymocytes. Eur. J. Biochem. 1994, 226, 23–30. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maeno, E.; Tsubata, T.; Okada, Y. Apoptotic Volume Decrease (AVD) Is Independent of Mitochondrial Dysfunction and Initiator Caspase Activation. Cells 2012, 1, 1156-1167. https://doi.org/10.3390/cells1041156
Maeno E, Tsubata T, Okada Y. Apoptotic Volume Decrease (AVD) Is Independent of Mitochondrial Dysfunction and Initiator Caspase Activation. Cells. 2012; 1(4):1156-1167. https://doi.org/10.3390/cells1041156
Chicago/Turabian StyleMaeno, Emi, Takeshi Tsubata, and Yasunobu Okada. 2012. "Apoptotic Volume Decrease (AVD) Is Independent of Mitochondrial Dysfunction and Initiator Caspase Activation" Cells 1, no. 4: 1156-1167. https://doi.org/10.3390/cells1041156