
Lineage-Selective Disturbance of Early Human Hematopoietic Progenitor Cell Differentiation by the Commonly Used Plasticizer Di-2-ethylhexyl Phthalate via Reactive Oxygen Species: Fatty Acid Oxidation Makes the Difference



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human CD34+ Cell Culture
2.2. HUVEC and HepG2/C3A Cell Culture
2.3. Flow Cytometry
2.4. Caspase 3/7 Activation
2.5. ATP Quantification
2.6. NADPH Quantification
2.7. H2DCFDC-Assay for Detection of ROS
2.8. Isolation of Total RNA and qPCR
2.9. LC-MS-Based Metabolomics
2.10. Western Blot Analysis
2.11. Measurement of Superoxide Dismutase (SOD) Activity
2.12. GC-MS-Based Quantification of DEHP and MEHP in Culture Medium
2.13. Quantification and Statistical Analysis
3. Results
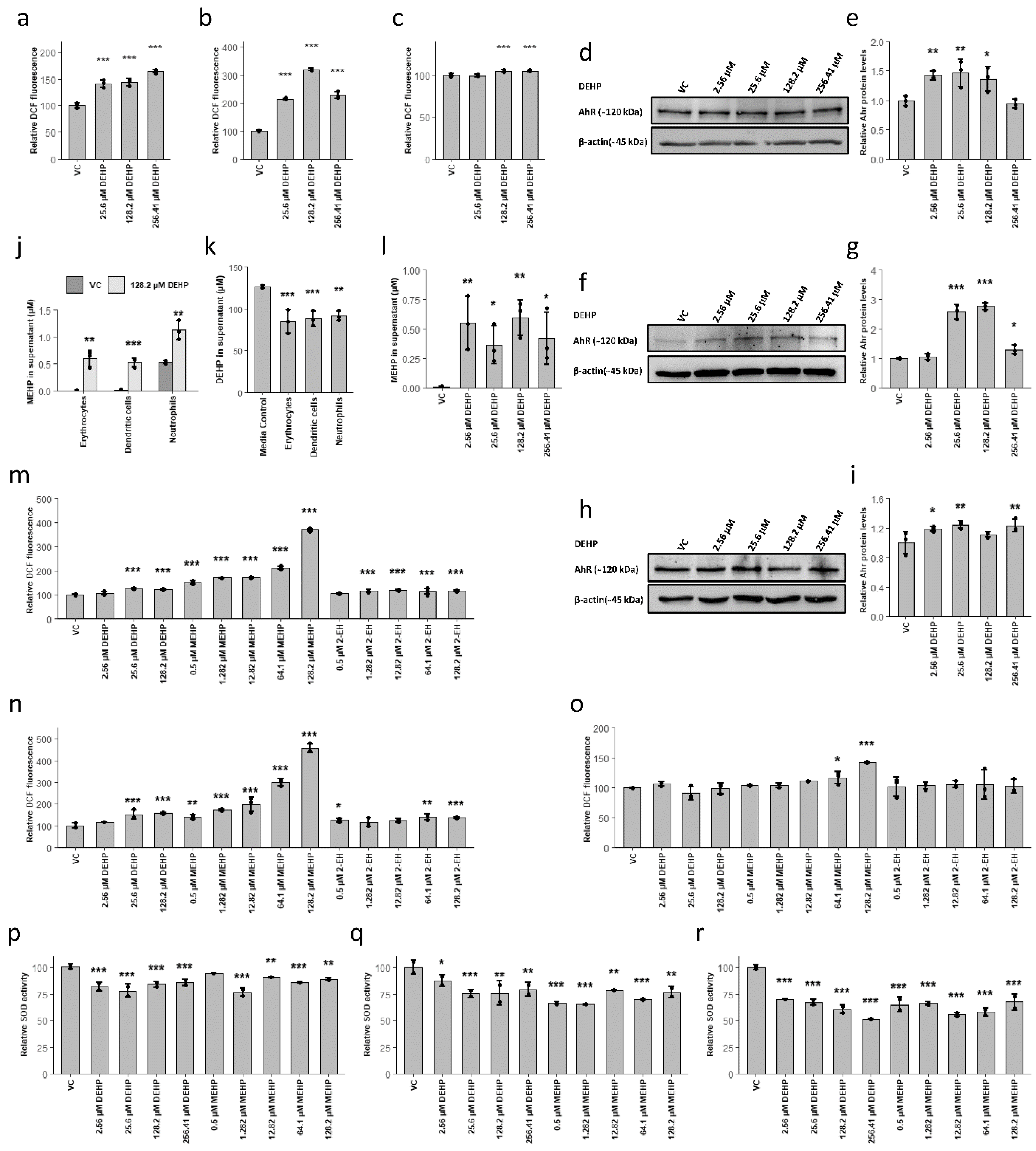
3.1. DEHP Induces Erythrocyte and Dendritic Cell Apoptosis While Enhancing Neutrophil Maturation
3.2. DEHP Alters the Lipidome Composition and Reduces the Rates of Glycolysis, Glutaminolysis, and Polyamine Synthesis in Erythrocytes and Dendritic Cells
3.3. MEHP, the First Metabolite of DEHP, Was Detected at Environmentally Relevant Concentrations and Induced the Accumulation of ROS via the Reduction of SOD Activity
3.4. MEHP Induces a ROS-Mediated Shift from Glycolysis to the Pentose Phosphate Pathway, Resulting in Cell Death in Lineages without Active FAO
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doulatov, S.; Notta, F.; Laurenti, E.; Dick, J.E. Hematopoiesis: A Human Perspective. Cell Stem Cell 2012, 10, 120–136. [Google Scholar] [CrossRef] [Green Version]
- Laiosa, M.D.; Tate, E.R. Fetal Hematopoietic Stem Cells Are the Canaries in the Coal Mine That Portend Later Life Immune Deficiency. Endocrinology 2015, 156, 3458–3465. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M.; Woodruff, T.J.; Vom Saal, F.S. Endocrine-disrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef]
- Lauretta, R.; Sansone, A.; Sansone, M.; Romanelli, F.; Appetecchia, M. Endocrine Disrupting Chemicals: Effects on Endocrine Glands. Front. Endocrinol. 2019, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Schaedlich, K.; Gebauer, S.; Hunger, L.; Beier, L.S.; Koch, H.M.; Wabitsch, M.; Fischer, B.; Ernst, J. DEHP deregulates adipokine levels and impairs fatty acid storage in human SGBS-adipocytes. Sci. Rep. 2018, 8, 3447. [Google Scholar] [CrossRef]
- Den Braver-Sewradj, S.P.; Piersma, A.; Hessel, E.V.S.S. An update on the hazard of and exposure to diethyl hexyl phthalate (DEHP) alternatives used in medical devices. Crit. Rev. Toxicol. 2020, 1–23. [Google Scholar] [CrossRef]
- Ernst, J.; Jann, J.C.J.-C.; Biemann, R.; Koch, H.M.; Fischer, B. Effects of the environmental contaminants DEHP and TCDD on estradiol synthesis and aryl hydrocarbon receptor and peroxisome proliferator-activated receptor signalling in the human granulosa cell line KGN. Mol. Hum. Reprod. 2014, 20, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erythropel, H.C.; Maric, M.; Nicell, J.A.; Leask, R.L.; Yargeau, V. Leaching of the plasticizer di(2-ethylhexyl)phthalate (DEHP) from plastic containers and the question of human exposure. Appl. Microbiol. Biotechnol. 2014, 98, 9967–9981. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.M.; Preuss, R.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP): Human metabolism and internal exposure—An update and latest results1. Int. J. Androl. 2006, 29, 155–165. [Google Scholar] [CrossRef]
- Koch, H.M.; Bolt, H.M.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP) metabolites in human urine and serum after a single oral dose of deuterium-labelled DEHP. Arch. Toxicol. 2004, 78, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Kavlock, R.; Barr, D.; Boekelheide, K.; Breslin, W.; Breysse, P.; Chapin, R.; Gaido, K.; Hodgson, E.; Marcus, M.; Shea, K.; et al. NTP-CERHR Expert Panel Update on the Reproductive and Developmental Toxicity of Di(2-ethylhexyl) phthalate. Reprod. Toxicol. 2006, 22, 291–399. [Google Scholar] [CrossRef]
- Kardas, F.; Bayram, A.K.; Demirci, E.; Akin, L.; Ozmen, S.; Kendirci, M.; Canpolat, M.; Oztop, D.B.; Narin, F.; Gumus, H.; et al. Increased Serum Phthalates (MEHP, DEHP) and Bisphenol A Concentrations in Children With Autism Spectrum Disorder. J. Child Neurol. 2016, 31, 629–635. [Google Scholar] [CrossRef]
- Latini, G.; de Felice, C.; Presta, G.; Del Vecchio, A.; Paris, I.; Ruggieri, F.; Mzzeo, P. In utero exposure to di-(2-ethylhexyl)phthalate and duration of human pregnancy. Environ. Health Perspect. 2003, 111, 1783–1785. [Google Scholar] [CrossRef] [Green Version]
- Cobellis, L.; Latini, G.; DeFelice, C.; Razzi, S.; Paris, I.; Ruggieri, F.; Mazzeo, P.; Petraglia, F. High plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis. Hum. Reprod. 2003, 18, 1512–1515. [Google Scholar] [CrossRef]
- Latini, G.; de Felice, C.; Presta, G.; Del Vecchio, A.; Paris, I.; Ruggieri, F.; Mazzeo, P. Exposure to Di(2-ethylhexyl)phthalate in Humans during Pregnancy. Neonatology 2003, 83, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Hyun Kim, D.; Min Choi, S.; Soo Lim, D.; Roh, T.; Jun Kwack, S.; Yoon, S.; Kook Kim, M.; Sil Yoon, K.; Sik Kim, H.; Wook Kim, D.; et al. Risk assessment of endocrine disrupting phthalates and hormonal alterations in children and adolescents. J. Toxicol. Environ. Heal. Part A 2018, 81, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Couleau, N.; Falla, J.; Beillerot, A.; Battaglia, E.; D’Innocenzo, M.; Plançon, S.; Laval-Gilly, P.; Bennasroune, A. Effects of Endocrine Disruptor Compounds, Alone or in Combination, on Human Macrophage-Like THP-1 Cell Response. PLoS ONE 2015, 10, e0131428. [Google Scholar] [CrossRef] [PubMed]
- Amara, I.; Timoumi, R.; Annabi, E.; Salem, I.B.; Abid-Essefi, S. Di(2-ethylhexyl) phthalate inhibits glutathione regeneration and dehydrogenases of the pentose phosphate pathway on human colon carcinoma cells. Cell Stress Chaperones 2020, 25, 151–162. [Google Scholar] [CrossRef]
- Gaitantzi, H.; Hakenberg, P.; Theobald, J.; Heinlein, H.; Cai, C.; Loff, S.; Wölfl, S.; Ebert, M.P.; Breitkopf-Heinlein, K.; Subotic, U. Di (2-Ethylhexyl) Phthalate and Its Role in Developing Cholestasis. J. Pediatr. Gastroenterol. Nutr. 2018, 66, e28–e35. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, C.; Han, C.; An, Q.; Cheng, Y.; Chen, Y.; Meng, R.; Zhang, Y.; Su, J. Plasticizer Bis(2-ethylhexyl) Phthalate Causes Meiosis Defects and Decreases Fertilization Ability of Mouse Oocytes in Vivo. J. Agric. Food Chem. 2019, 67, 3459–3468. [Google Scholar] [CrossRef]
- She, Y.; Jiang, L.L.; Zheng, L.; Zuo, H.; Chen, M.; Sun, X.; Li, Q.; Geng, C.; Yang, G.; Jiang, L.L.; et al. The role of oxidative stress in DNA damage in pancreatic β cells induced by di-(2-ethylhexyl) phthalate. Chem. Biol. Interact. 2017, 265, 8–15. [Google Scholar] [CrossRef]
- Tripathi, A.; Pandey, V.; Sahu, A.N.; Singh, A.; Dubey, P.K. Di-(2-ethylhexyl) phthalate (DEHP) inhibits steroidogenesis and induces mitochondria-ROS mediated apoptosis in rat ovarian granulosa cells. Toxicol. Res. 2019, 8, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Fang, W.; Cao, H.; Luo, S.; Cong, J.; Liu, S.; Pan, F.; Jia, X. Di-(2-ethylhexyl)-phthalate induces apoptosis via the PPARγ/PTEN/AKT pathway in differentiated human embryonic stem cells. Food Chem. Toxicol. 2019, 131, 110552. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Buhrke, T.; Imber, F.; Jessel, S.; Seidel, A.; Völkel, W.; Lampen, A. Agonistic and antagonistic effects of phthalates and their urinary metabolites on the steroid hormone receptors ERα, ERβ, and AR. Toxicol. Lett. 2017, 277, 54–63. [Google Scholar] [CrossRef]
- You, H.; Chen, S.; Mao, L.; Li, B.; Yuan, Y.; Li, R.; Yang, X. The adjuvant effect induced by di-(2-ethylhexyl) phthalate (DEHP) is mediated through oxidative stress in a mouse model of asthma. Food Chem. Toxicol. 2014, 71, 272–281. [Google Scholar] [CrossRef]
- Wójtowicz, A.K.; Sitarz-Głownia, A.M.; Szczęsna, M.; Szychowski, K.A. The Action of Di-(2-Ethylhexyl) Phthalate (DEHP) in Mouse Cerebral Cells Involves an Impairment in Aryl Hydrocarbon Receptor (AhR) Signaling. Neurotox. Res. 2019, 35, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornehag, C.G.; Nanberg, E. Phthalate exposure and asthma in children. Int. J. Androl. 2010, 33, 333–345. [Google Scholar] [CrossRef]
- Ito, T.; Inoue, K.; Nishimura, N.; Takano, H. Phthalate esters modulate the differentiation and maturation of mouse peripheral blood mononuclear cell-derived dendritic cells. J. Appl. Toxicol. 2012, 32, 142–148. [Google Scholar] [CrossRef]
- Zou, Q.-Y.; Hong, S.-L.; Kang, H.-Y.; Ke, X.; Wang, X.-Q.; Li, J.; Shen, Y. Effect of di-(2-ethylhexyl) phthalate (DEHP) on allergic rhinitis. Sci. Rep. 2020, 10, 14625. [Google Scholar] [CrossRef]
- Manz, P.; Cadeddu, R.-P.; Wilk, M.; Fischer, J.C.; Fritz, B.; Haas, R.; Wenzel, F. Influence of Di(2-ethylhexyl)phthalate on migration rate and differentiation of human hematopoietic stem and progenitor cells (CD34+). Clin. Hemorheol. Microcirc. 2015, 61, 111–118. [Google Scholar] [CrossRef]
- Kaiser, L.; Weinschrott, H.; Quint, I.; Blaess, M.; Csuk, R.; Jung, M.; Kohl, M.; Deigner, H.-P. Metabolite Patterns in Human Myeloid Hematopoiesis Result from Lineage-Dependent Active Metabolic Pathways. Int. J. Mol. Sci. 2020, 21, 6092. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.N.; Al-Karradi, S.N.H.; Kragstrup, T.W.; Hokland, M. Elimination of erroneous results in flow cytometry caused by antibody binding to Fc receptors on human monocytes and macrophages. Cytom. Part A 2016, 89, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bode, C.; Gräler, M.H. Quantification of sphingosine-1-phosphate and related sphingolipids by liquid chromatography coupled to tandem mass spectrometry. Methods Mol. Biol. 2012, 874, 33–44. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Xia, W.; Hou, M. Macrophage migration inhibitory factor rescues mesenchymal stem cells from doxorubicin-induced senescence though the PI3K-Akt signaling pathway. Int. J. Mol. Med. 2018, 41, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, H.-W. Di(2-ethylhexyl) phthalate (DEHP) metabolites: Mono (2-ethyl-5-hydroxyhexyl) phthalate (5OH-MEHP), mono(2-ethyl-5-oxohexyl) phthalate (5oxo-MEPH), mono(2-ethyl-5-carboxypentyl) phthalate (5cx-MEPP), mono(2-ethylhexyl) phthlate (MEHP). In The MAK-Collection for Occupational Health and Safety; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 211–236. [Google Scholar]
- Tankiewicz, M.; Olkowska, E.; Berg, A.; Wolska, L. Advancement in Determination of Phthalate Metabolites by Gas Chromatography Eliminating Derivatization Step. Front. Chem. 2020, 7, 928. [Google Scholar] [CrossRef] [Green Version]
- Graíño, S.G.; Sendón, R.; Hernández, J.L.; de Quirós, A.R.B. GC-MS screening analysis for the identification of potential migrants in plastic and paper-based candy wrappers. Polymers 2018, 10, 802. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Enviroment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [Green Version]
- Marsee, D.K.; Pinkus, G.S.; Yu, H. CD71 (Transferrin Receptor). Am. J. Clin. Pathol. 2010, 134, 429–435. [Google Scholar] [CrossRef]
- Gutknecht, M.; Geiger, J.; Joas, S.; Dörfel, D.; Salih, H.R.; Müller, M.R.; Grünebach, F.; Rittig, S.M. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun. Signal. 2015, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, M.V.; Vaziri, N.D.; Yuan, J.; Adler, S.G. Upregulation of monocyte/macrophage HGFIN (Gpnmb/Osteoactivin) expression in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, L.; Pourfarzad, F.; Ullrich, S.; Merkel, A.; Were, F.; Carrillo-de-Santa-Pau, E.; Yi, G.; Hiemstra, I.H.; Tool, A.T.J.; Mul, E.; et al. Dynamics of Transcription Regulation in Human Bone Marrow Myeloid Differentiation to Mature Blood Neutrophils. Cell Rep. 2018, 24, 2784–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antal-Szalmas, P.; van Strijp, J.A.G.; Weersink, A.J.L.; Verhoef, J.; van Kessel, K.P.M. Quantitation of surface CD14 on human monocytes and neutrophils. J. Leukoc. Biol. 1997, 61, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Oburoglu, L.; Romano, M.; Taylor, N.; Kinet, S. Metabolic regulation of hematopoietic stem cell commitment and erythroid differentiation. Curr. Opin. Hematol. 2016, 23, 198–205. [Google Scholar] [CrossRef]
- Blaess, M.; Claus, R.A.; Deigner, H.-P.P. HPLC separation and ultrasensitive optical quantification of ceramide species applying 7-(diethylamino)coumarin-3-carbonyl azide derivatisation. J. Chromatogr. B 2015, 986–987, 123–128. [Google Scholar] [CrossRef]
- Blaess, M.; Le, H.; Claus, R.; Kohl, M.; Deigner, H.-P. Stereospecific induction of apoptosis in tumor cells via endogenous C16-ceramide and distinct transcripts. Cell Death Discov. 2015, 1, 15013. [Google Scholar] [CrossRef] [Green Version]
- Siskind, L.J. Mitochondrial Ceramide and the Induction of Apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Obeid, L.M. Ceramide and Apoptosis: Exploring the Enigmatic Connections between Sphingolipid Metabolism and Programmed Cell Death. Anticancer. Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Beverly, L.J.; Siskind, L.J. Sphingolipids and mitochondrial apoptosis. J. Bioenerg. Biomembr. 2016, 48, 153–168. [Google Scholar] [CrossRef]
- Dbaibo, G.S.; Perry, D.K.; Gamard, C.J.; Platt, R.; Poirier, G.G.; Obeid, L.M.; Hannun, Y.A. Cytokine response modifier A (CrmA) inhibits ceramide formation in response to tumor necrosis factor (TNF)-α: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J. Exp. Med. 1997, 185, 481–490. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 275–281. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced Apoptosis Can Occur through a Ceramide-independent Pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e5. [Google Scholar] [CrossRef] [Green Version]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Wu, Y.J.; Chen, W.C.; Lee, T.S.; Tsou, T.C.; Chang, H.C.; Lo, S.W.; Chen, S.L. MEHP interferes with mitochondrial functions and homeostasis in skeletal muscle cells. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, J.; Wakx, A.; Gil, S.; Fournier, T.; Auzeil, N.; Rat, P.; Laprévote, O. Lipidome-wide disturbances of human placental JEG-3 cells by the presence of MEHP. Biochimie 2018, 149, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Ziouzenkova, O.; Brown, J.; Devchand, P.; Francis, S.; Kadakia, M.; Kanda, T.; Orasanu, G.; Sharlach, M.; Zandbergen, F.; et al. PPARs and their metabolic modulation: New mechanisms for transcriptional regulation? J. Int. Med. 2007, 262, 184–198. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef]
- Shan, A.; Leng, L.; Li, J.; Luo, X.; Fan, Y.; Yang, Q.; Xie, Q.; Chen, Y.; Ni, C.; Guo, L.; et al. TCDD-induced antagonism of MEHP-mediated migration and invasion partly involves aryl hydrocarbon receptor in MCF7 breast cancer cells. J. Hazard. Mater. 2020, 398, 122869. [Google Scholar] [CrossRef]
- Dietrich, C. Antioxidant Functions of the Aryl Hydrocarbon Receptor. Stem Cells Int. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, S.; Zhao, T.; Yang, L.; Guo, S.; Shi, Y.; Zhang, X.; Zhou, L.; Ye, L. Mono-2-ethylhexyl phthalate (MEHP) promoted lipid accumulation via JAK2/STAT5 and aggravated oxidative stress in BRL-3A cells. Ecotoxicol. Environ. Saf. 2019, 184, 109611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shen, X.Y.; Zhang, W.W.; Chen, H.; Xu, W.P.; Wei, W. The effects of di 2-ethyl hexyl phthalate (DEHP) on cellular lipid accumulation in HepG2 cells and its potential mechanisms in the molecular level. Toxicol. Mech. Methods 2017, 27, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Craig, Z.R.; Basavarajappa, M.S.; Hafner, K.S.; Flaws, J.A. Mono-(2-Ethylhexyl) Phthalate Induces Oxidative Stress and Inhibits Growth of Mouse Ovarian Antral Follicles1. Biol. Reprod. 2012, 87, 152. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wu, C.; Ye, Y.; Zeng, J.; Zhu, J.; Li, Y.; Wang, W.; Zhang, W.; Chen, Y.; Xie, H.; et al. The Increase of ROS Caused by the Interference of DEHP with JNK/p38/p53 Pathway as the Reason for Hepatotoxicity. Int. J. Environ. Res. Public Health 2019, 16, 356. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2020, 2019, 6175804. [Google Scholar] [CrossRef]
- Dagher, Z.; Ruderman, N.; Tornheim, K.; Ido, Y. Acute regulation of fatty acid oxidation and AMP-activated protein kinase in human umbilical vein endothelial cells. Circ. Res. 2001, 88, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Viragova, S.; Kim, M.J.; Rothman, N.J.; Liu, K.; Reizis, B.; Reiner, S.L. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol. Cell Biol. 2018, 96, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Grünig, D.; Duthaler, U.; Krähenbühl, S. Effect of Toxicants on Fatty Acid Metabolism in HepG2 Cells. Front. Pharmacol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.T.; Hansen, J.S.; Hansen, E.W.; Clausen, P.A.; Nielsen, G.D. Airway inflammation and adjuvant effect after repeated airborne exposures to di-(2-ethylhexyl)phthalate and ovalbumin in BALB/c mice. Toxicology 2007, 235, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, A.M.; Laskin, D.L.; Archer, F.; Syed, K.; Gray, J.P.; Laskin, J.D.; Nwebube, N.; Weinberger, B. Inflammatory effects of phthalates in neonatal neutrophils. Pediatr. Res. 2010, 68, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef]
- Kwak, H.J.; Liu, P.; Bajrami, B.; Xu, Y.; Park, S.Y.; Nombela-Arrieta, C.; Mondal, S.; Sun, Y.; Zhu, H.; Chai, L.; et al. Myeloid Cell-Derived Reactive Oxygen Species Externally Regulate the Proliferation of Myeloid Progenitors in Emergency Granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive Oxygen Species in the Immune System. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.-C.; Pietersz, G.A.; Tang, C.K.; Ramsland, P.A.; Apostolopoulos, V. Reactive Oxygen Species Level Defines Two Functionally Distinctive Stages of Inflammatory Dendritic Cell Development from Mouse Bone Marrow. J. Immunol. 2010, 184, 2863–2872. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M.; Arimitsu, N.; Ito, T.; Sekimizu, K. Antioxidant N-acetyl-l-cysteine inhibits erythropoietin-induced differentiation of erythroid progenitors derived from mouse fetal liver. Cell Biol. Int. 2007, 31, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. 2020, 32, 101511. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, N.; Kukidome, D.; Sada, K.; Motoshima, H.; Furukawa, N.; Matsumura, T.; Nishikawa, T.; Araki, E. Low glucose induces mitochondrial reactive oxygen species via fatty acid oxidation in bovine aortic endothelial cells. J. Diabetes Investig. 2017, 8, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Heier, C.; Kien, B.; Vesely, P.W.; Tang, Z.; Sexl, V.; Schoiswohl, G.; Strießnig-Bina, I.; Hoefler, G.; Zechner, R.; et al. Adipose triglyceride lipase activity regulates cancer cell proliferation via AMP-kinase and mTOR signaling. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2020, 1865, 158737. [Google Scholar] [CrossRef]
- Ferguson, K.K.; Loch-Caruso, R.; Meeker, J.D. Exploration of Oxidative Stress and Inflammatory Markers in Relation to Urinary Phthalate Metabolites: NHANES 1999–2006. Environ. Sci. Technol. 2012, 46, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Lucas, A.; Lam, D.; Cabrales, P. Doxorubicin-loaded red blood cells reduced cardiac toxicity and preserved anticancer activity. Drug Deliv. 2019, 26, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Shou, W.; Payne, R.M.; Caldwell, R.; Field, L.J. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr. Res. 2008, 64, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Al-Gubory, K.H. Environmental pollutants and lifestyle factors induce oxidative stress and poor prenatal development. Reprod. Biomed. Online 2014, 29, 17–31. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, L.; Quint, I.; Csuk, R.; Jung, M.; Deigner, H.-P. Lineage-Selective Disturbance of Early Human Hematopoietic Progenitor Cell Differentiation by the Commonly Used Plasticizer Di-2-ethylhexyl Phthalate via Reactive Oxygen Species: Fatty Acid Oxidation Makes the Difference. Cells 2021, 10, 2703. https://doi.org/10.3390/cells10102703
Kaiser L, Quint I, Csuk R, Jung M, Deigner H-P. Lineage-Selective Disturbance of Early Human Hematopoietic Progenitor Cell Differentiation by the Commonly Used Plasticizer Di-2-ethylhexyl Phthalate via Reactive Oxygen Species: Fatty Acid Oxidation Makes the Difference. Cells. 2021; 10(10):2703. https://doi.org/10.3390/cells10102703
Chicago/Turabian StyleKaiser, Lars, Isabel Quint, René Csuk, Manfred Jung, and Hans-Peter Deigner. 2021. "Lineage-Selective Disturbance of Early Human Hematopoietic Progenitor Cell Differentiation by the Commonly Used Plasticizer Di-2-ethylhexyl Phthalate via Reactive Oxygen Species: Fatty Acid Oxidation Makes the Difference" Cells 10, no. 10: 2703. https://doi.org/10.3390/cells10102703