
Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases



Department of Molecular Biology, University of Gdansk, Wita Stwosza 59, 80-308 Gdansk, Poland
*
Author to whom correspondence should be addressed.
Cells 2021, 10(2), 365; https://doi.org/10.3390/cells10020365
Submission received: 31 December 2020
/
Revised: 31 January 2021
/
Accepted: 6 February 2021
/
Published: 10 February 2021
(This article belongs to the Special Issue 10th Anniversary of Cells—Advances in Autophagy)
Abstract
:Ferroptosis is one of the recently described types of cell death which is dependent on many factors, including the accumulation of iron and lipid peroxidation. Its induction requires various signaling pathways. Recent discovery of ferroptosis induction pathways stimulated by autophagy, so called autophagy-dependent ferroptosis, put our attention on the role of ferroptosis in lysosomal storage diseases (LSD). Lysosome dysfunction, observed in these diseases, may influence ferroptosis efficiency, with as yet unknown consequences for the function of cells, tissues, and organisms, due to the effects of ferroptosis on physiological and pathological metabolic processes. Modulation of levels of ferrous ions and enhanced oxidative stress, which are primary markers of ferroptosis, are often described as processes associated with the pathology of LSD. Inhibition of autophagy flux and resultant accumulation of autophagosomes in neuronopathic LSD may induce autophagy-dependent ferroptosis, indicating a considerable contribution of this process in neurodegeneration. In this review article, we describe molecular mechanisms of ferroptosis in light of LSD, underlining the modulation of levels of ferroptosis markers in these diseases. Furthermore, we propose a hypothesis about the possible involvement of autophagy-dependent ferroptosis in these disorders.
1. Introduction
Ferroptosis is one of the recently described types of cell death which differs from other well-known types of regulated cell death (apoptosis, necrosis, necroptosis, and others) morphologically, physiologically, and biochemically. Many of its characteristic features, as well as its activation pathways, are still being discovered, but generally, this process is characterized by the accumulation of reactive oxygen species (ROS) as products of increased efficiency of iron metabolism and lipid peroxidation [1,2,3].
The ferroptosis process, as one of regulated cell death, has been known since 2012 [1,4]. It consists of various changes in cellular functions associated with cell morphology, physiology, and biochemistry. In Table 1, crucial ferroptosis features are summarized and compared to the most frequent programmed cell death—apoptosis.
The exact role of ferroptosis has just begun to be the center of interest of researchers. Studies performed to date have indicated its significant role in a few human diseases, including its increased efficiency in neurodegenerative diseases, ischemic reperfusion injury, atherosclerosis, and cancer [2]. However, the efficiency of this process, pathways of its activation, and its role in the development of most diseases remain largely unknown in many disorders. Recently discovered pathways link the autophagy process (lysosomal degradation of macromolecules) and ferroptosis.
In the autophagy process, a degradation-desired macromolecule is engulfed by a membrane called phagophore, forming an autophagosome. This vesicle is fused with lysosome, and lysosomal acid hydrolases digest its content. Autophagy is a physiologically important process which recycles misfolded molecules or dysfunctional organelles, forming precursors of newly synthesized macromolecules. However, too intensive autophagy may also lead to cell death. In fact, increased efficiency of autophagy was observed in response to ferroptosis inductors [3]. These discoveries inspired us to put attention on lysosomal storage diseases (LSD) as disorders in which modulation of ferroptosis might be involved in their molecular pathomechanism.
LSD are a group of 50 or so inherited metabolic diseases caused by mutations in genes encoding lysosomes functions [4]. Dysfunctions in lysosomal ability to convert biologically significant polymers into oligomers and monomers result in the storage of undegraded or partially degraded compounds inside these organelles [4,5]. When considering the molecular mechanisms of LSDs, one can distinguish four major defects leading to these disorders: (i) inactivation of one of the specific lysosomal hydrolases (sphingolipids, glycoproteins, glycosaminoglycans); (ii) defect in a protein involved in the transport of particular compounds through lysosomal membranes; (iii) inactivation of the enzyme that modifies lysosomal proteins, ensuring their proper localization and function; and (iv) lack of specific activators for lysosomal enzymes [6]. Another classification of LSD is based on the nature of stored compound(s). Therefore, the following subgroups of LSDs can be listed: (i) mucopolysaccharidoses, characterized by the accumulation of glycosaminoglycans; (ii) oligosaccharidoses, characterized by the accumulation of oligosaccharides (e.g., mannosidoses, fucosidosis); (iii) glycogen storage diseases (e.g., Pompe disease, Danon disease); (iv) lipidoses in which sphingomyelin is stored (e.g., Niemann–Pick disease); (v) neuronal ceroid lipofuscinoses, characterized by the accumulation of lipopigments; (vi) mucolipidoses, characterized by the accumulation of combinations of lipids and carbohydrates; (vii) sphingolipidoses, characterized by defects of the degradation of lipids containing ceramide (e.g., gangliosidoses GM1 and GM2, Fabry disease, Krabbe disease, metachromatic leukodystrophy, Gaucher disease); and (viii) other diseases in which specific compounds accumulate, such as cystinosis (accumulation of cystine) or Salla disease (accumulation of sialic acid) [7].
Although LSDs are monogenic diseases, specific therapeutic options are available only for relatively few of them. Bone marrow or hematopoietic stem cell transplantations were used in various LSDs; however, their efficacy was very different in particular diseases, and even if some efficacy was observed, it occurred only when the procedure was performed early in life, before the age of 2 years. Substrate reduction therapy, a procedure based on the inhibition of synthesis of a compound which cannot be degraded in lysosomes, is currently available for Gaucher disease and Niemann–Pick disease type C, while studies on the use of small molecules interfering with the syntheses of various compounds are ongoing. Enzyme replacement therapy (intravenous administration of recombinant human enzyme which is otherwise deficient in a patient’s cells) is the most often used specific treatment of LSD, and is currently available for several diseases, including Gaucher disease, Fabry disease, Pompe disease, and mucopolysaccharidoses types I, II, IVA, VI, and VII. However, this therapy is inefficient in treatment of the central nervous system, since intravenously administered enzyme cannot efficiently cross the blood–brain barrier. Finally, gene therapy is a promising option, and there are many studies ongoing which are focused on the use of this procedure in LSD; however, to date, no gene therapy for this group of diseases has been registered [8,9,10].
2. Molecular Mechanisms of Ferroptosis
Determining the exact molecular content of the induction of ferroptosis is one of the challenges posed to cell biologists. Studies of recent years indicated that this process is primarily dependent on (i) iron levels in the cell, (ii) specific metabolic pathways, (iii) the GPX4-dependent pathway, and (iv) ROS levels and lipid peroxidation. Moreover, it can also be regulated by (v) MAP kinases and/or (vi) the intensity of autophagy (in the process called autophagy-dependent ferroptosis) [1,3].
Ferroptosis is primarily dependent on iron concentration [4]. Circulating iron occurs as ferritin-bound Fe3+ ions. Following import to cells by the transferrin receptor TFR1, it is delivered to the endosome where ferrireductase STEAP3 catalyzes the reduction of Fe3+ to Fe2+. The SLC11A2 transporter liberates Fe2+ from the endosome to the cytoplasm where iron excess is stored in the ferritin-bound form, while in the lack of such storage, the Fenton reaction leads to the formation of ROS which are involved in lipid peroxidation, thus inducing ferroptosis. Ferroportin SLC11A3 converts Fe2+ to Fe3+ and participates in iron export from cells [4].
One more pathway that can induce ferroptosis by modulating iron levels is the frataxin-dependent pathway. Lowering the level of this protein results in mitochondrial dysfunction, which in turn leads to iron accumulation in mitochondria and enhanced efficiency of the Fenton reaction. These features may be confirmed by the results of studies on models of Friedreich’s ataxia, a disease caused by mutations in the gene encoding frataxin (the FXN gene), resulting in its decreased expression. An increase in the levels of ferroptosis markers in fibroblasts collected from patients with Friedreich’s ataxia (increased lipid peroxidation, decreased level of antioxidant enzymes, increased protein oxidation) has been reported [11,12]. Such studies were also performed on mouse models. Increased levels of ferroptosis markers were observed in adipocyte precursors in FXN knock-in/knock-out (KIKO) mice (increased lipid peroxidation and decreased glutathione peroxidase 4 activity) [13], and in C2C12 mouse myoblasts after FXN gene silencing (increased expression of pro-ferroptotic and decreased expression of anti-ferroptotic genes) [12,13]. Moreover, fibroblasts collected from Friedreich’s ataxia patients, as well as FXN knockdown human fibrosarcoma HT-1080 cells, were considerably more sensitive to the administration of erastin, one of the best characterized ferroptosis activators [11,14]. The results showing the restoration of resistance to ferroptosis of FXN knockdown cells after blocking the signal of iron starvation indicated the dependence of this phenomenon on intracellular iron concentration [14]. Another disease that could indicate the role of frataxin in ferroptosis is the alcoholic liver disease. Long-term administration of ethanol to mice reduced the expression of the gene encoding frataxin, leading to the accumulation of ROS and the mitochondrial iron pool in primary hepatocytes. Moreover, deficiency of frataxin enhanced ethanol-driven ferroptosis, and restoration of the appropriate level of frataxin reduced the sensitivity of liver to ethanol treatment [15].
The crucial role of iron in ferroptosis induction is also supported by the results of studies on overexpression of the gene coding for TLR1 and repression of the ferritin-coding gene which resulted in the stimulation of this process. Furthermore, impaired expression of the IREB2 transcription factor (that acts to regulate iron levels) caused inhibition of ferroptosis [1,16,17]. The iron-dependent pathway leading to ferroptosis, described above, is depicted in Figure 1.
Apart from iron-dependent ROS generation (described above), enhanced lipid peroxidation can result from changes in glucose and glutamate metabolism. Acyl-CoA synthetase (ACSF2) and citrate synthase (CS), as regulators of the mitochondrial fatty-acid metabolism, participate in the biochemical pathway from glucose to citrate (through glycolysis and Krebs cycle) that can be converted to substrates for lipid synthesis, of which increased levels may lead to their peroxidation [1,16]. Moreover, conversion of glutamate to α-ketoglutarate contributes to the production of citrate [18], causing effects as described above [1,16]. In fact, knock-out mutations in genes coding for ACSF2 or CS halted ferroptosis (induced by erastin), indicating their considerable role in the stimulation of this process [1,16]. In accordance with this model, a small molecule transaminase inhibitor, aminooxyacetic acid, which blocks the conversion of glutamate to α-ketoglutarate, inhibits ferroptosis [16]. Similar effects are caused by inhibition of the pentose-phosphate pathway due to mutations in genes coding for glucose-6-phosphate dehydrogenase and phosphoglycerate dehydrogenase (Figure 2) [1,4].
Glutathione peroxidase 4 (GPX4), one of the antioxidant enzymes, is a key regulator of ferroptosis [1]. A major co-factor of GPX4 is glutathione (GSH), which can be oxidized to glutathione disulfide (GSSG), and then reduced by NADPH/H+-dependent GSH reductase. GSH synthesis is catalyzed by glutamate-cysteine ligase and GSH synthetase, and requires cysteine, glutamate, and glycine as substrates. The direct effect of GSH levels on ferroptosis emerged from the use of erastin (one of the ferroptotic activators), which by lowering GSH levels activates cell death identically to that caused by a lack of GPX4 [19]. So far, a few pathways have been described that could reduce GSH levels in cells, and most of them are dependent on cysteine levels. Intracellular cysteine concentration depends on the activity of cysteine/glutamate antiporter SLC7A11 (Xc− system). Cysteine for GSH synthesis may also come from the degradation of macromolecules in lysosomes, as it can be released from these organelles into the cytoplasm [19]. Thus, glutamate/cysteine exchange by SLC7A11 and the high efficiency of lysosomal degradation of macromolecules are necessary to maintain an adequate level of cysteine. Its decreased level and/or reduced activities of glutathione-synthesizing enzymes can lead to decreased activity of GPX4 which, in turn, causes the accumulation of ROS and lipid peroxidation [19,20]. In fact, inhibition of GPX4 can result in increased ROS concentrations, while GPX4 overexpression cause ROS depletion, modulating ferroptosis efficiency [21].
A protein that has recently been reported to be involved in the regulation of ferroptosis by this pathway is Beclin-1. Under the influence of erastin and other ferroptosis activators, it can bind to the components of the Xc− system, limiting cysteine/glutamate exchange, which leads to a decrease in GSH concentration, inhibition of GPX4, and, as a consequence, the induction of ferroptosis. Binding of Beclin-1 to the Xc− system is determined by its phosphorylation by AMPK kinase at sites S90 and S93. Mutations of the gene coding for Beclin-1, causing modifications in the above mentioned phosphorylation sites, or a reduction in the activity of AMPK kinase, led to the inhibition of erastin-induced ferroptosis, confirming this conclusion [3,22].
NRF2 (nuclear factor erythroid 2-related factor 2) is another protein that has a large impact on the activity of the Xc− system components. Since an increase in its level leads to enhanced expression of genes encoding the SLC7A11 protein and antioxidant proteins, it plays an anti-ferroptotic role. Studies carried out on tumor cell models (hepatocellular carcinoma and head and neck squamous cell carcinoma) have shown that NRF2 overexpression reduces, while knock-out in this gene increases, cell sensitivity to ferroptosis inducers [1,23,24,25]. Reduction in the expression of the NRF2 gene also sensitized the cells to RSL3 (one of the ferroptosis activators that is a GPX4 inhibitor) [24]. These studies also indicated that expression of the gene encoding the p62 protein is necessary to maintain high NRF2 levels after exposure to ferroptosis inducers. This determines the interaction of NRF2 with transcriptional coactivator small v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog proteins, activating the transcription of genes encoding antioxidant enzymes (quinone oxidoreductase-1, heme oxygenase-1, and ferritin heavy chain-1). Silencing of the expression of the p62-coding gene, as well as genes encoding the above mentioned enzymes, led to an increase in the sensitivity of cells to erastin and sorafenib. The same effect was achieved by genetic or pharmacological silencing of the expression of NRF2, both in vitro and in tumor xenograft models [23]. ARF (ARF tumor suppressor, also called p14ARF) is an additional protein that is involved in the regulation of NRF2 activity, and thus has a broad impact on the effectiveness of ferroptosis. This protein, by binding to NRF2, inactivates it, which prevents the positive regulation of SLC7A11 expression, leading to the induction of ferroptosis. A high level of expression of the ARF gene makes cells more sensitive to this type of cell death, while lowering of its expression promotes their survival [25].
The p53 protein has a large impact on the efficiency of ferroptosis, as it regulates the expression of SLC7A11. Recent studies have indicated that its activation by nutlin-3 reduced the expression of SLC7A11 in HT-1080 cells, while mutations in the p53-encoding gene completely canceled this effect. Thus, activation of p53 leads to a blockage of cysteine import, and thus a reduction in GSH levels, inactivation of GPX4, and the induction of ferroptosis [26]. On the other hand, p21 (encoded by the CDKN1A gene) is another p53 target protein. Upregulation of p21 ensures that a high level of GSH is maintained, which is conducive to cell survival, but the exact mechanisms by which this happens are still under investigation. The recycle of oxidized GSH to reduced GSH, decreased export of GSH from the cell, or reduced consumption of GSH were reported as possible molecular mechanisms of the influence of p21 on GSH levels [27,28].
The above described GPX4-dependent pathway, leading to ferroptosis, is presented in Figure 3.
Ferroptosis induction can also result from reactions of ROS with polyunsaturated fatty acids (PUFAs) in lipid membranes. Acyl-CoA synthetase long chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and arachidonate lipoxygenase (ALOX) enzymes are involved in PUFAs metabolism. They acetylate arachidonic acid (AA) and adrenic acid (AdA), catalyze their conversion to membrane phospholipids, and then oxidize them to lipid peroxides [3,29]. Decreased levels of ACSL4 and LPCAT3 in cells prevented the oxidization of sensitive fatty acids in the membrane. On the other hand, depletion of GPX4 led to an abundance of oxidized membranes enriched with arachidonic acid. The role of arachidonic acid appears to be particularly important, as in the absence of GPX4 activity, the liberation of its metabolites (hydroxyeicosatetraenoic acids (HETE): 5-HETE, 11-HETE, and 15-HETE) was observed, in contrast to apoptosis. Thus, their appearance may be specific to ferroptosis [29]. ACSL4 is, therefore, considered as not only a protein which mediates ferroptosis induction, but also as a marker of this process [3]. This pathway, leading to ferroptosis, is presented in Figure 4.
It appears that ferroptosis may also be dependent on MAPK proteins, particularly extracellular signal-regulated kinases (ERK), p38 mitogen-activated protein kinases (p38), and c-Jun N-terminal kinases (JNK), suggesting activation of the Ras/Raf/MEK/ERK pathway. Inhibition of components of this pathway led to a reduced sensitivity to cell death caused by erastin in 12 different sarcoma cell lines [1,30,31,32]. Additional studies on the pathways involved in the induction of ferroptosis indicated the involvement of further genes, the expression of which is necessary for erastin-induced ferroptosis in two cell line models, HT-1080 and Calu-1. These genes included RPL8 (coding for 60S ribosomal protein L8), ATP5G3 (encoding the ATP5G3 protein), CS (coding for citrate synthase), TTC35 (encoding tetratricopeptide repeat domain 35), ACSF2 (encoding acyl-CoA synthetase family member 2), and IREB2 (coding for iron response element binding protein 2). Silencing the expression of these genes abolished the erastin effect on ferroptosis induction in these cell lines [16,33]. HMGB1 (coding for high mobility group box 1 protein) is another gene whose involvement in ferroptosis has been reported. The release of the HMGB1 protein after the use of ferroptosis activators, such as erastin or sorafenib, and a decrease in its secretion after the use of ferroptosis inhibitors (pharmacologically, after treatment with ferrostatin, or genetically, after silencing the expression of the ACSL4 gene) in HT1080 and PANC1 cell lines were demonstrated [3,34]. In addition, it has been proven that in leukemia cells, intracellular HMGB1-triggered ferroptosis also engaged two other proteins, transferrin receptor (TFRC) and advanced glycosylation end-product specific receptor (AGER) [3,31]. However, the link between AGER, TFRC, and HMGB1 in the induction of ferroptosis requires further explanation [3]. The voltage-dependent anion channel (VDAC) also appears to be a positive regulator of ferroptosis as it is a direct target for erastin. Knockdown cells in the VDAC2/3 gene are less sensitive to erastin-induced ferroptosis, while cells with increased expression of this gene appear to be more susceptible to this process [3,32]. Similarly, knockdown of the CARS gene (encoding cysteinyl-tRNA synthetase) inhibited erastin-induced ferroptosis, and CARS overproduction made cells sensitive to ferroptosis. Cystine deprivation was indicated as one of the elements that can link CARS level modulation and the induction of ferroptosis [3,35]. The exact mechanisms of the described phenomenon, however, remain to be elucidated for both these genes. The role of pannexin 1 in the induction of ferroptosis in the renal ischemia/reperfusion injury model has also been studied. Reduced tubular ferroptotic cell death was observed in mice with deletion of the PANX1 gene (encoding pannexin 1) compared to wild-type mice after renal ischemia/reperfusion injury. In addition, silencing of PANX1 expression in cultured human kidney 2 (HK-2) cells was observed to reduce the severity of ferroptosis (as measured by reduced lipid peroxidation and lower iron levels) after incubation in the presence of erastin [30]. It is also worth emphasizing another role of the p53 protein in the regulation of ferroptosis, apart from that already mentioned above. It was indicated that besides interaction with the SLC7A11 and p21 proteins, p53 can interact with another 11 proteins that may affect ferroptosis. Among them, there are products of the following genes: GLS2, SAT1, CDKN1A, and DPP4 [36]. By regulating the expression of GLS2, p53 increases levels of glutaminase 2 (GLS2), the key enzyme involved in the conversion of glutamine to glutamate. Increased GLS2 levels facilitate glutamine metabolism and lower ROS levels, inhibiting ferroptosis. To confirm this hypothesis, experiments were carried out under conditions of silencing the expression of both p53 and GLS2, and an increase in the level of ROS was observed. These studies were performed on six tumor cell lines [36,37]. Another protein upregulated by p53 is diamine acetyltransferase 1 (SAT1) which contributes to lipid peroxidation and ROS-induced ferroptosis. It was demonstrated that this process is dependent on arachidonate 15-lipoxygenase (ALOX15) because inhibition of its activity resulted in a decrease, and overproduction caused an increase, in the sensitivity of oncogenic Ras-expressing cancer cells to erastin- and RSL3-induced ferroptosis [38].
A mechanism for action of p53 in modulating ferroptosis, different from that by regulating transcription, has been observed in human colorectal cancer cells treated with erastin. This mechanism is related to the DPP4 protease (dipeptidyl-peptidase-4), which, under conditions of a lack or low level of p53, forms a complex with the NOX protein (DPP4–NOX), accelerating lipid peroxidation and entrance of cells into the ferroptotic pathway. High levels of p53 protein lead to more efficient DPP4–p53 complex formation, which antagonizes ferroptosis under these conditions. This is a completely different role of p53 in modulating the efficiency of ferroptosis than those described so far and based on the activity of p53 as a transcription factor [39].
3. Implication of Autophagy Process in Ferroptosis (Autophagy-Dependent Ferroptosis)
In recent years, the dependence of ferroptosis on autophagy was discovered, which led to distinguishing autophagy-dependent ferroptosis. The dependence of ferroptosis on autophagy is based on selective kinds of the latter process, i.e., specific lysosomal degradation of particular proteins of organelles which leads to the modulation of ferroptosis efficiency. The selective autophagy processes which influence ferroptosis include (i) ferritinophagy, (ii) lipophagy, (iii) mitophagy, (iv) clockophagy, and (v) chaperon-mediated autophagy. Molecular mechanisms linking both processes are still under investigation [3]. Ferritinophagy, mediated by cargo receptor NCOA4, localized in the forming autophagosome membrane, contributes to ferroptosis induction due to ferritin degradation and iron liberation. This results in enhancement of the Fenton reaction, followed by lipid peroxidation and cell death. On the contrary, decreased levels of NCOA4 inhibited ferritin degradation, preventing ferroptosis [40]. Another example of autophagy–ferroptosis’ relationship is lipophagy induction, i.e., the degradation of lipid droplets (mediated by cargo receptor RAB7A) to form free fatty acids which can be oxidized. Enhanced formation of lipid droplets due to the upregulation of tumor protein D52 (TPD52) prevented ferroptosis induction due to the limitation of lipid peroxidation [41]. On the other hand, an elevated level of RAB7A caused lipophagy activation, and thus, stimulation of lipid peroxidation-mediated ferroptosis [41]. Most cellular ROS derive from mitochondria. Their production is enhanced when mitochondria are damaged or dysfunctional. The role of mitophagy, i.e., selective degradation of mitochondria, is to remove dysfunctional organelles and to decrease ROS levels, preventing lipid peroxidation and reducing ferroptosis efficiency. Until now, over 10 cargo receptors taking part in mitophagy have been identified, including SQSTM1, OPTN, CALCOCO2, TAX1BP1, and others [3]. Recently discovered selective degradation of the ARNTL protein (aryl hydrocarbon receptor nuclear translocator-like), called clockophagy, can also modulate ferroptosis. Clockophagy-mediated decrease in the ARNTL level results in negative regulation of the transcription factor HIF1, responsible for the stimulation of expression of genes whose products are involved in the transport and binding of fatty acids and lipids (mainly FABP3 and FABP7). Lower levels of these proteins (due to HIF1 deficiency) prevent the binding of fatty acids and lipids and their transportation from the cellular membrane to mitochondria, facilitating their peroxidation and thus, stimulating ferroptosis [42]. In chaperone-mediated autophagy, Hsp70 is employed to direct degradation of proteins with the KFERQ amino acid motif. GPX4 is one of such proteins that plays the role of an antioxidant and protects cells against ferroptosis. Thus, chaperone-mediated autophagy of GPX4 results in ferroptosis stimulation [43,44]. All autophagy-dependent ferroptosis’ induction pathways are presented in Figure 5.
The pathways linking autophagy and ferroptosis appear to be very complex and are only at the early stages of studies. Reports that have already been published provided compelling evidence that autophagic pathways may shift to ferroptotic pathways, and this process can be cargo receptors-dependent. Evidence for this is provided by the fact that lowering the level of cargo receptors led to a reduction in the efficiency of autophagy-dependent ferroptosis [3,40,41,42,43,44]. Almost all studies conducted so far indicated autophagy as the initial process that turns into ferroptosis. For this reason, manipulating the intensity of autophagy is already indicated as the key to anti-cancer therapies, not the other way around [45]. However, there are also recently published reports indicating a reverse relationship. The use of ferroptosis activators (artesunate and erastin) led to the induction of autophagy (measured by the number of accumulated autophagosomes and an increase in the level of the LC3-II protein in MEF cells). Moreover, this effect was not observed in cells with a deletion of the Atg5 protein-coding gene, which indicated that this process is mediated by proteins associated with autophagy [46]. It has also been reported that treatment with erastin led to cancer cell death caused by the upregulation of genes encoding autophagous proteins (Beclin-1, Atg5, Atg12, LC3-II, p62). Under conditions of both silencing their expression and lowering iron levels, erastin-induced cell death was not observed. This would suggest an involvement of both autophagy and ferroptosis in cancer cell death [47]. Thus, one can suggest either a two-way action of ferroptosis activators (which would induce autophagy at the same time) or a feedback linkage between ferroptosis and autophagy (initiated by ferroptosis). However, the exact mechanism of the transition from ferroptosis to autophagy has not yet been proposed.
4. Links in the Ferroptosis Induction Network
As indicated in the two preceding sections, there are multiple processes which activate ferroptosis. Particular pathways may lead to ferroptosis independently; however, there are also links between them, forming a specific metabolic network [48]. An example of an interconnection between these pathways is the export of iron (which is stored in a ferritin-bound form) from the endosome to the cytoplasm, mediated by SLC11A2. Increased ferritinophagy, a kind of autophagy leading to the degradation of ferritin, may contribute to the release of large amounts of Fe2+ ions, and thus, to stimulation of the Fenton reaction leading directly to lipid peroxidation [3,4]. Another example is the GPX4 protein which plays a crucial role in maintaining the antioxidant status in the cell, while it is also involved in autophagy-dependent ferroptosis, through activation of HSP90 which results in positive regulation of chaperone-mediated autophagy (CMA) and subsequent lipid peroxidation. It is also worth paying attention to aerobic metabolism [1,19,43,44]. Under conditions of its increased efficiency, release of large amounts of ROS from mitochondria might occur, which contributes to the increased intensity of ferroptosis, also in interaction with components of other pathways [3]. Regardless of the starting point of the signal leading to ferroptosis, all these pathways ultimately lead to the ROS-dependent lipid peroxidation that induces ferroptosis. Therefore, the network of processes demonstrated in Figure 6 indicates the complexity of mechanisms of ferroptosis stimulation, and points to complicated regulations of cellular responses to various factors and agents which may influence the activation or inhibition of ferroptosis.
5. Ferroptosis Disorders as a Mechanism for Pathogenesis of Lysosomal Storage Diseases
Induction of ferroptosis has been investigated, to date, predominantly in light of anti-cancer therapy. Ferroptosis activators cause cell death in various cancers which is promising in light of the development of anti-cancer therapies [1,2]. However, enhanced ferroptosis has been reported in many other diseases, including Alzheimer’s and Parkinson’s diseases, acute kidney failure, liver and heart injury, ischemic reperfusion injury, and atherosclerosis. Treatment with ferroptosis inhibitors caused increased survival of cellular and/or animal models of these diseases [1,2].
A role for ferroptosis, in both physiological and pathological conditions, is still poorly understood and is the subject of intensive studies. Overactivation of this process appears to worsen the course of all diseases tested to date. On the other hand, it was demonstrated that ferroptosis is required for cell proliferation, which might suggest a physiological role for this process [49,50]. The recently discovered dependence of ferroptosis on the autophagy process, in which the lysosomal system is involved, put our attention on LSD. In fact, changes in ferroptosis efficiency arising from modulation of lysosomal activities have been suggested [3]. As mentioned in the introduction, various therapeutic strategies for LSD have been tested; however, apart from the non-neuronopathic type of Gaucher disease, no abolition of symptoms could be achieved in severe and neuronopathic forms of these diseases, and only partial improvement could be achieved in milder forms [51,52,53].
Reports on disturbances of typical ferroptosis markers and other factors influencing them directly appeared quite long ago in the context of LSD, but so far, they have not been associated with possible ferroptosis disorders. It was only due to the discovery of the pathways linking autophagy and ferroptosis that they, once again, became the factors of pathogenesis of these diseases considered in a new aspect.
5.1. Modulation of Iron Levels in Lysosomal Storage Diseases
The first information on iron modulation came from data obtained from patients suffering from neuronal ceroid lipofuscinoses, one of the progressive neurodegenerative LSDs, characterized by excessive accumulation of lipofuscins. These data indicated that the concentration of free iron in cerebrospinal fluids from patients is elevated and increases with disease progression [54,55].
A study on another LSD, Niemann–Pick disease, in which sphingomyelin accumulates in cells, carried out shortly thereafter, showed the surprising result of a complete absence of ferritin, an iron-storing protein, in the visceral organs of four patients [56]. A year later, these results were independently confirmed in more patients, as well as in more biopsy-derived material [57,58]. However, studies on the mouse acid sphingomyelinase-deficient model showed increased levels of the ferritin light chain transcript as well as increased levels of iron in the lungs and brains of animals. These abnormalities were corrected after application of enzyme replacement therapy [59]. If the data on increased mRNA levels for ferritin were confirmed in patients with Niemann–Pick disease, it would mean that the absence of ferritin, as the end product of gene expression, results from translation changes or accelerated protein degradation. Different results were obtained in experiments with the Npc1−/− mouse model. They showed a lower iron content in the liver of mice and decreased expression of ferritin light chain and ferroportin, as well as increased expression of the gene encoding the transferrin receptor in the liver of Npc1−/− mice of different ages [60]. The differences in the analyses performed may result from using different organs as the research material. This hypothesis might be supported by subsequent studies carried out at a later date on the same model, indicating a significant iron load in the brain of mice, which could potentially contribute to neurodegeneration. The authors also hypothesized the effectiveness of deferiprone, a known brain iron chelator, in improving the pathology of Niemann–Pick disease. However, treatment with deferiprone did not bring the expected results, without affecting the course of the disease or the life span of the mice [61].
Increased iron levels have also been found in liver biopsy samples taken from patients with Gaucher disease, one of the most common LSD [62]. These results were confirmed in studies with a large number of patients, showing not only iron but also ferritin overload [63,64,65]. Magnetic resonance imaging allowed precise localization of excess iron in a group of 40 patients in the liver, spine and femoral bone marrow, and spleen, which correlated with increased serum ferritin levels [66]. This problem is more and more often described in the context of the risk of liver fibrosis [66,67,68] or cancer [66,68]. Subsequent studies indicated not only hyperferritinemia and iron accumulation in patients with Gaucher disease, but also the influence of enzyme replacement therapy on these pathological factors. Treatment with the enzyme resulted in a decrease in hyperferritinemia, increased levels of transferrin, and, most importantly, increased levels of hepcidin, a peptide that regulates serum iron levels. The authors confirmed these results by creating a Gaucher disease cell model and by observing the gradual restoration of ferroportin and hepcidin levels from the time of induction of the disease with a glucocerebrosidase inhibitor in macrophages [69]. The effectiveness of ERT in restoring the correct level of ferritin is also evidenced by recently published case reports [70].
Detailed studies on iron metabolism in Gaucher disease were performed, showing not only the deregulation of iron recycling and modulation of ferritin levels, but also the related release of cytokine and the inflammatory response of the organism [71]. The authors indicated that lowering the iron levels by 4-month therapy with its chelators resulted in a decrease in the concentration of iron in the liver and serum ferritin, and also positively influenced the patients’ response to the available treatment, significantly improving their quality of life [71]. An association of inflammatory reaction with iron metabolism was also found by researchers, which indicated hyperferritinemia and elevated levels of TNFα and some interleukins in over 80% of patients with Gaucher disease in the Swedish population [72].
Studies on iron levels in the brain of patients were also carried out in the case of mucopolysaccharidosis type III (Sanfilippo disease), one of the lysosomal storage diseases in which there is storage of heparan sulfate oligosaccharides. Magnetic resonance imaging showed significant iron accumulation in the deep brain nuclei in two siblings with cognitive impairment [73]. Moreover, an attempt was made to discover the mechanism linking inflammation with iron levels in MPS III. In this disease, extensive neuroinflammation is also observed due to the activation of microglia and astrocytes to produce inflammatory cytokines, in which heparan sulfate is involved. In mice with Sanfilippo disease, iron accumulation and elevated levels of hepcidin (a hormone playing a key role in the regulation of iron homeostasis) have been observed, mainly in the cerebral cortex. The authors also indicated a reduced concentration of ferroportin (a protein that exports iron from cells), which contributed to an increase in iron levels. The in vitro studies showed unequivocally that the accumulation of heparan sulfate directly contributes to the above-described phenomena (increased hepcidin levels and decreased ferroportin levels), and that astrocytes and microglia (as opposed to neurons) are the most vulnerable. Cell signaling studies proved the involvement of the TLR4 and STAT3 signaling pathways in the increase in hepcidin concentration, and thus, iron accumulation in cells [74].
Mucolipidosis type IV is one of the LSD caused by mutations in the TRPML1 gene, encoding one of the intracellular late endosomal and lysosomal ion channel proteins. Until recently, the divalent metal transport protein SLC11A2 was thought to be the only known endosomal Fe2+ transporter. Recent findings indicated that the TRPML1 channel can also transport iron. Mutations in the gene coding for this protein led to an increase in iron levels in late endosomes and lysosomes and a decrease in cytosolic iron, which correlated very well with the severity of symptoms in patients with this disease [75,76].
In mouse models of gangliosidosis type I and type II, diseases caused by the accumulation of lipids known as gangliosides, a significant decrease in iron level was observed in the brain [77]. The authors admitted that this result is in stark contrast to the typically elevated levels of iron found in other LSDs. In order to explain the possible mechanism of this phenomenon, they indicated a decrease in the level of transferrin and an increase in the level of hepcidin. Despite disturbances in the levels of proteins influencing iron metabolism in the brain, administering this element to mice prolonged their life by up to 40% and allowed for a smoother disease transition [77].
The reverse discovery was made with fucosidosis. A case report describing a girl with this condition indicated a significant increase in the level of iron in the brain imaged with the use of magnetic resonance imaging, pointing to fucosidosis as another LSD in which changes in iron metabolism, and thus in ferroptosis, may play a significant role in the pathogenesis of the disease [78].
5.2. Lipid Peroxidation in Lysosomal Storage Diseases
The first reports indicating that iron overload may lead to increased lipid peroxidation and, more importantly, that lysosomes may be involved in this phenomenon, appeared already in the 1960s [79]. Those studies already indicated the need to pay attention to the role of disturbances in iron metabolism and lipid peroxidation in the pathogenesis of LSD.
One of the lysosomal diseases in which this problem was highlighted was, as in the case of iron overload, neuronal ceroid lipofuscinosis. Biochemical analyses of patient samples indicated decreases in levels of phosphatidyl ethanolamine polyunsaturated fatty acids (PUFA) and antioxidants [80]. In a canine model of neuronal ceroid lipofuscinoses, an increase in the levels of lipid peroxidation products was also observed [80,81]. An attempt was made to find the cause of ceramide deposition in the tissues of patients, in light of these results. However, no direct evidence of such disease pathogenesis mechanism has been presented.
Studies focusing on lipid peroxidation disorders resulting from changes in the levels of ferritin (which is an antioxidant) also concerned Niemann–Pick disease. The so far described lack of ferritin in patients led to the supposition that an excess of unbound iron could lead to excessive lipid oxidation in ROS-dependent reactions [33]. Studies on this aspect of pathogenesis of Niemann–Pick disease were carried out on fibroblasts collected from patients. Both the concentration of reactive oxygen species and the lipid peroxidation status were increased in these cells compared to cells taken from healthy controls. Moreover, it was noted that the patient-derived fibroblasts were more likely to die from oxidative stress-induced apoptosis. The authors pointed to the participation of NF-κB-dependent signaling pathways in this phenomenon. Moreover, it was shown that silencing the expression of the NPC1 gene in two cell models led to an increase in ROS concentration [82].
Attempts were also made to find pathways linking excessive lipid peroxidation with the storage of various materials in lysosomes in cystinosis, one of the diseases manifested by abnormal accumulation of the amino acid cystin. Studies conducted on a pharmacologically induced rat model have shown lipoperoxidation and carbonylation of proteins, as well as an increase in the activity of oxidative enzymes such as superoxide dismutase, glutathione peroxidase (GPx), and catalase in the kidneys of animals. In addition, administration of cysteamine, used in the treatment of cystinosis, to rats partially alleviated the proven lesions [83]. Interesting research on cystinosis was also carried out on a model of proximal tubular epithelial cells from patients with this disease, in which the authors confirmed disturbances in oxidative status. They also studied the influence of cysteamine on glutathione level and ATP metabolism, thanks to which they indicated increases in glutathione level and restored glutathione redox status in cystinosis cells [84].
Quite extensive research has been carried out on the aspect of lipid peroxidation in models of mucopolysaccharidoses, a group of LSD in which glycosaminoglycans are the stored material. The first such study, indicating a significant role of oxidative stress in the pathology of the disease, was carried out on patients with MPS I. The level of some markers of oxidative stress was assessed in samples from patients at different stages of enzyme replacement therapy (ERT). The results indicated excessive lipid peroxidation, which gradually normalized with the duration of therapy [85]. Similar studies on the same type of MPS were performed with a mouse model [86]. The authors showed an increase in the activity of the enzymes superoxide dismutase and catalase in some organs, such as the cerebellum, lungs, and spleen. Moreover, in these mice, an increased number of carbonyl groups was noted [86]. Studies performed on a small group of patients with MPS type II showed reduced levels of superoxide dismutases and glutamate transporters. However, no changes in the lipid peroxidation process were found [87]. Extended studies on MPS II patients were performed, where oxidative stress parameters were compared in the blood of patients before and after ERT initiation [88,89]. The authors described decreased levels of antioxidant enzymes and increased levels of nitric oxide in plasma prior to enzyme administration. In addition, increased levels of nitrates and nitrites in the urine of patients have been observed. A similar analysis after a few months of therapy showed a significant reduction in the level of malondialdehyde and an increase in the level of sulfhydryl groups. There were no changes in the activity of catalase, superoxide dismutase, glutathione peroxidase, and glutathione reductase. These results indicated a significant exposure of patients with MPS II to the oxidative damage of lipids and proteins and showed a reduction in the amount of antioxidant enzymes [88,89]. Research on oxidative stress was also performed on the MPS IIIB mouse model [90]. Mice at various stages of the disease demonstrated extensive oxidative stress, affecting the CNS from the very first months of life. Additionally, it has been proved that the processes of lipid peroxidation, and protein and DNA oxidation concerned mainly the cerebellum. The authors also showed changes in the expression of genes encoding enzymes involved in oxidation, such as Sod1, Ret, Bmp4, Tgfb, Gzmb, and Prf1, were already at the level of their transcripts. These data indicate, on the one hand, the newly discovered mechanisms leading to the induction of oxidative stress in MPS, and on the other hand, that they appear at a very early stage of the disease, even before symptoms develop [90].
Markers of oxidative stress were also noted in the case of Fabry disease, manifested by an accumulation of glycosphingolipids in cells. Studies carried out on samples taken from patients showed increased lipid peroxidation, and increased levels of nitric oxide and glutathione, as well as decreased levels of glutathione peroxidase and heme oxygenase [91,92]. In patients undergoing enzyme replacement therapy, lipid peroxidation status and nitric oxide levels remained elevated. These results indicated that despite the therapy, oxidative status disturbances were still observed in patients, which may suggest that lipid peroxidation disorders are independent pathogenic factors of this disease [91]. The authors proposed a strategy of inhibiting oxidative stress by pharmacological or nutritional measures as an adjunctive therapy to enzyme replacement therapy in Fabry disease [92].
Other examples of LSD, in which increased lipid peroxidation, ROS accumulation, increased transcription of genes responding to cytotoxic oxidative stress, and decreased levels of antioxidant enzymes were observed, include mucolipidosis type IV (studies on TRPML1-knockdown cells) [93] and Gaucher disease (studies performed on red blood cells collected from patients) [94].
The latest reports also mentioned disturbances in oxidative status in a murine model of Krabbe disease, characterized by the accumulation of psychosin. In twi−/− mice, increased lipid peroxidation and decreased levels of antioxidant enzymes were observed. Supplementation with vitamin D3, as a known antioxidant, resulted in increased expression of genes encoding antioxidant enzymes, decreased lipid peroxidation, decreased inflammation, and delayed psychosin accumulation, which ultimately increased axon integrity in the brain of animals [95].
5.3. Modulation of Activity of the GPX4-GSH-Xc− System in Lysosomal Storage Diseases
Parkinson’s disease is the first neurological disease in which modulation of GPX4 levels has been described. In the case of this disease, both increased oxidative stress and iron accumulation were observed, which may lead to the intensification of the phenomenon of ferroptosis. An increase in expression efficiency of the gene encoding GPX4 has been demonstrated in both in vitro and in vivo models [96]. Changes in glutathione and ROS metabolic pathways have also been studied in myocardial infarction. Studies conducted on a mouse model of this ailment showed a decrease in glutathione metabolism and an increase in ROS levels. One of genes with the most depressed expression was GPX4. The authors confirmed the observed changes by creating a model of H9c2 cardiomyoblasts with silenced expression or depletion of GPX4 in which they observed accumulation of lipid peroxide leading to ferroptotic cell death [97].
In the context of lysosomal storage diseases, there are few reports of modulation of the GPX4-GSH-Xc− system. It is worth paying attention to more and more reports pointing to the connection of the activity and levels of this system to the functions of lysosomes [19,98,99]. However, there are results of experiments indicating the reduction in the level of glutathione peroxidase in the Cln3 knock-in (Cln3 (Deltaex7/8)) mouse model of neuronal ceroid lipofuscinosis [100]. Decreased activity of this enzyme was also noticed in urine and blood samples of patients suffering from Fabry disease, even though those patients were treated with enzyme replacement therapy [101]. Studies on activities or levels of antioxidant enzymes in LSD have typically involved assessing redox potential/oxidative status as an aspect influencing cell dysfunction. More reports indicated a modulation of GSH levels. A decrease in GSH level was observed mainly in cystinosis, as the decrease in the level of cysteine, as one of the substrates for GSH synthesis, is the main cause of these diseases [102,103]. It is worth noting that the decreased level of intracellular cysteine may be the result of dysfunctions of lysosomes which ineffectively degrade macromolecules (especially in LSD), and which in turn may lead to a reduction in GSH levels [19]. Reduced GSH levels have been reported not only in cysteinosis but also in urine and blood samples from patients with Fabry disease and mucopolysaccharidosis type IVA. It is worth noting that in these studies, the investigated patients were treated with enzyme replacement therapy, and yet the levels of GSH remained lower relative to the group of healthy persons [101,104]. These results were confirmed in experiments with the Fabry disease mouse model [105]. This may indicate that the pathogenesis of LSDs is not limited to the primary cause—accumulation of the storage material. Such a hypothesis can be corroborated by recently published results demonstrating dysregulation of expression of hundreds of genes, coding for proteins involved in various cellular processes, in fibroblasts derived from patients suffering from mucopolysaccharidoses [51,106,107,108,109,110,111].
A summary of the modulations of ferroptosis features in LSD is presented in Table 2, with ferroptosis markers defined as features that are necessary and sufficient for the ferroptotic process to take place.
5.4. The Effectiveness of Ferroptosis Modulators in the Course of Lysosomal Storage Diseases: Already Published Reports and Perspectives
An obvious question appeared if there is a potential efficacy of ferroptosis modulators in abolishing or at least alleviating the symptoms of genetic diseases, including LSD. Reports on the potency of ferroptosis inhibitors in human diseases are contradictory. Research on the effectiveness of ferrostatins in Huntington’s disease, periventricular leukomalacia, and kidney proximal tubules cellular models showed an increase in their viability [112]. However, studies on a number of ferroptosis inhibitors in the Parkinson’s disease nerve cell models indicated that only one of them (liproxstatin) was effective in reversing cell death [113].
Similarly, divergent results have been obtained for LSD. Studies on the effectiveness of deferiprone (iron chelator, crossing the blood–brain barrier) showed no changes in the trajectory of the disease and life expectancy in mice with Niemann–Pick disease [61]. On the other hand, iron chelators, deferoxamine or deferasirox, have also been used in patients with Gaucher disease. Short-term drug intake led to a significant reduction in serum ferritin and hepcidin, and iron deposition in the liver. Patients’ clinical and analytical data also improved after long-term follow-up [71].
However, one should be aware that mechanisms of ferroptosis induction are very complicated, as underlined also in this article. These mechanisms may be different in different tissues and organs, and in the case of changes in cell physiology (such as in human diseases), various pathways leading to ferroptosis might be disturbed. This may result in the ineffectiveness of individual ferroptosis inhibitors. Certainly, this aspect requires further research.
6. Autophagy Disorders in Lysosomal Storage Diseases
Disorders of the autophagy process have already been described quite extensively in the context of the pathogenesis of LSD [114,115,116]. The discovery of autophagy-dependent ferroptosis, however, sheds new light on this process. Thus, autophagy should be recognized not only as a cellular process that is altered in performance with LSD, but also as a process that can lead to cell death.
In most of the LSDs described so far, various phenotypes of autophagy have been observed. They can take place at various stages, from autophagosome formation and maturation, through the accumulation of abnormal autophagosomes, to autophagic flux blockage. Such disorders have been observed so far in neuronal ceroid lipofuscinoses, glycogenosis, Niemann–Pick disease, Gaucher disease, mucolipidosis type IV [114], Fabry disease [115], and mucopolysaccharidoses [51].
7. Possible Role of Autophagy in Ferroptosis Modulation in Lysosomal Storage Diseases
Autophagic flux disorders due to lysosomal dysfunction in LSD are inevitable. One of their consequences may be the accumulation of autophagosomes, the appearance of which may induce cellular stress and initiate cellular processes to deal with them. Unfortunately, the accumulation of autophagosomes can trigger further pathological cellular phenomena. The recent discovery of autophagy-dependent ferroptosis points to the autophagosome as the source of cargo receptors, initiating the response leading to ferroptotic cell death. Due to the relatively recent discovery of this way of ferroptosis induction, reports on the modulation of levels of cargo receptors in LSD are so far infrequent and concern a few diseases, but their number is growing rapidly. Some of the popular proteins found to be cargo receptors in autophagy-dependent ferroptosis have already been studied in LSD because of their other important cellular functions, such as involvement in lysosomal and proteasomal degradation (SQSTM1) or endocytosis (Rab7A). The rest remains an open question.
The greatest number of LSDs in which the levels of the proteins mentioned above were measured were indicated for SQSTM1. Studies on mucolipidosis type IV in a model of Mcoln−/− mouse have indicated inclusions formed by SQSTM1 in animal brains [116]. For the same disease, similar results were shown in the Mcoln1−/− neuronal culture model [117]. Moreover, in mucolipidosis type II and III, an increase in the levels of the described protein [118] was indicated in studies on the model of fibroblasts taken from patients. The accumulation of SQSTM1-positive aggregates has also been demonstrated in glycogen storage disease type II [119], nephropathic cystinosis [120], and Gaucher disease [121]. For another cargo receptor, Rab7A, one more LSD, Niemann–Pick disease, was tested. The studies showed an increase in the level of this protein in the liver of the Npc1−/− mice [122]. In all these studies, the authors set the primary goal of research on the effectiveness of autophagy or proteasomal degradation or endocytosis. Of course, the accumulation of SQSTM1 in cells could be the result of the response to the appearance of misfolded proteins, and the accumulation of Rab7A could mean a serious disturbance in endocytic transport. However, at that time, no one imagined that these proteins could also play a key role in cell death.
8. Concluding Remarks
Disturbances in iron homeostasis, lipid peroxidation, as well as the efficiency of the autophagy process and cell death, have been observed for years in LSDs. However, the recent discovery of autophagy-dependent ferroptosis not only drew attention to the problem of ferroptosis as one of the causes of neurodegeneration in LSD (due to the known dysfunction of lysosomes in these diseases) but also led to the assumption that there are pathways that connect the described phenomena, observed in these diseases, and may partially explain the pathophysiology of neurodegeneration. In this paper, we hypothesize that the accumulation of autophagosomal vesicles resulting from a decrease in autophagy efficiency in LSDs may initiate the process of ferroptosis in an autophagy-dependent manner. The so far proven elevated levels of some cargo receptor proteins in the initiation of ferroptosis (SQSTM1, Rab7, and certainly also others) may initiate a number of reactions which result in an increase in iron concentration or oxidative stress, which are the basic markers of ferroptosis, occurring in most of these diseases. Therefore, this work points to autophagy-dependent ferroptosis as one of the additional LSD pathomechanisms that can lead to cell death.
It is worth noting, however, that the presented modulations of the level or activity of features typical for ferroptosis were studied in LSD in a completely different context than the study of its role in the pathogenesis of this group of diseases. The idea that ferroptosis disorders are possible additional aspects of LSD pathology, presented in this paper and based on the recently discovered autophagy–ferroptosis relationships, and the modulation of its markers determined so far, is therefore a novel hypothesis. In fact, there are some reports suggesting that possible ferroptotic disorders may contribute to the pathogenesis of this group of diseases, though they were not devoted to understanding LSD–ferroptosis connections. Taken together, those reports did not provide conclusive evidence for a strong relationship between LSD and ferroptosis; however, they led us to propose the above presented hypothesis of which verification will be necessary in the near future.
Author Contributions
Conceptualization, K.P.; data curation, K.P., E.R. and L.G.; writing—original draft preparation, K.P.; writing—review and editing, G.W.; visualization, E.R.; supervision, K.P., and G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar]
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Classification of lysosomal storage diseases. In Lysosomal Storage Disorders: A Practical Guide; Mehta, A., Winchester, B., Eds.; John Wiley & Sons, Ltd.: Hobokek, NJ, USA, 2012; pp. 37–46. [Google Scholar]
- Peters, H.; Ellaway, C.; Nicholls, K.; Reardon, K.; Szer, J. Treatable lysosomal storage diseases in the advent of disease-specific therapy. Intern. Med. J. 2020, 50, 5–27. [Google Scholar] [CrossRef]
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [PubMed]
- Sheth, J.; Nair, A. Treatment for lysosomal storage disorders. Curr. Pharm. Des. 2020, 26, 5110–5118. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Liu, J.; He, H.; Wang, J.; Guo, X.; Lin, H.; Chen, H.; Jiang, C.; Chen, L.; Yao, P.; Tang, Y. Oxidative stress-dependent frataxin inhibition mediated alcoholic hepatocytotoxicity through ferroptosis. Toxicology 2020, 445, 152584. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cel death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating irondependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X c- Activity. Curr. Biol. 2018, 28, 2388–2399.e5. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell. 2017, 68, 224–232.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Dixon, S. The p53-p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget 2018, 9, 24572–24573. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem. Biol. 2019, 26, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 Mediates Ferroptosis That Contributes to Renal Ischemia/Reperfusion Injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, Y.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Gai, C.; Ding, D.; Wang, F.; Li, W. Targeted p53 on Small-Molecules-Induced Ferroptosis in Cancers. Front. Oncol. 2018, 8, 507. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Shen, S.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar]
- Lee, Y.-S.; Kalimuthu, K.; Park, Y.S.; Makala, H.; Watkins, S.C.; Choudry, M.H.A.; Bartlett, D.L.; Kwon, Y.T.; Lee, Y.J. Ferroptotic agent-induced endoplasmic reticulum stress response plays a pivotal role in the autophagic process outcome. J. Cell Physiol. 2020, 235, 6767–6778. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, X.; Lu, S.; He, C.; Wang, C.; Wang, L.; Wang, X.; Ge, P.; Song, D. Erastin triggers autophagic death of breast cancer cells by increasing intracellular iron levels. Oncol. Lett. 2020, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Brokowska, J.; Węgrzyn, G. Changes in cellular processes occurring in mucopolysaccharidoses as underestimated pathomechanisms of these diseases. Cell Biol. Int. 2019. [Google Scholar] [CrossRef]
- Kant, S.; Atta, M.G. Therapeutic advances in Fabry disease: The future awaits. Biomed. Pharmacother. 2020, 131, 110779. [Google Scholar] [CrossRef]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B.; Westermarck, T. Increased non-protein-bound iron and decreased protection against superoxide-radical damage in cerebrospinal fluid from patients with neuronal ceroid lipofuscinoses. Lancet 1982, 2, 459–460. [Google Scholar] [CrossRef]
- Heiskala, H.; Gutteridge, J.M.; Westermarck, T.; Alanen, T.; Santavuori, P. Bleomycin-detectable iron and phenanthroline-detectable copper in the cerebrospinal fluid of patients with neuronal ceroid-lipofuscinoses. Am. J. Med. Genet. Suppl. 1988, 5, 193–202. [Google Scholar] [CrossRef]
- Christomanou, H.; Kellermann, J.; Linke, R.P.; Harzer, K. Deficient ferritin immunoreactivity in visceral organs from four patients with Niemann-Pick disease type C. Biochem. Mol. Med. 1995, 55, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Christomanou, H.; Harzer, K. Ouchterlony double immunodiffusion method demonstrates absence of ferritin immunoreactivity in visceral organs from nine patients with Niemann-Pick disease type C. Biochem. Mol. Med. 1996, 58, 176–183. [Google Scholar] [CrossRef]
- Christomanou, H.; Vanier, M.T.; Santambrogio, P.; Arosio, P.; Kleijer, W.J.; Harzer, K. Deficient ferritin immunoreactivity in tissues from niemann-pick type C patients: Extension of findings to fetal tissues, H and L ferritin isoforms, but also one case of the rare Niemann-Pick C2 complementation group. Mol. Genet. Metab. 2000, 70, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Dhami, R.; Passini, M.A.; Schuchman, E.H. Identification of novel biomarkers for Niemann-Pick disease using gene expression analysis of acid sphingomyelinase knockout mice. Mol. Ther. 2006, 13, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Argüello, G.; Martinez, P.; Peña, J.; Chen, O.; Platt, F.; Zanlungo, S.; González, M. Hepatic metabolic response to restricted copper intake in a Niemann-Pick C murine model. Metallomics 2014, 6, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Lotan, A.; Yeshurun, S.; Schroeder, A.; Bush, A.I. Iron chelation by deferiprone does not rescue the Niemann-Pick Disease Type C1 mouse model. Biometals 2020, 33, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Iancu, T.C.; Perl, D.P.; Sternlieb, I.; Lerner, A.; Leshinsky, E.; Kolodny, E.H.; Hsu, A.; Good, P.F. The application of laser microprobe mass analysis to the study of biological material. Biometals 1996, 9, 57–65. [Google Scholar] [CrossRef]
- Stein, P.; Yu, H.; Jain, D.; Mistry, P.K. Hyperferritinemia and iron overload in type 1 Gaucher disease. Am. J. Hematol. 2010, 85, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treatment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Faucher, B.; Seguier, J.; Swiader, L.; Cuquemelle, C.; Cerutti, D.; Ebbo, M. Gaucher Disease type 1 mimicking immune thrombocytopenia: Role of hyperferritinemia and hypergammaglobulinemia in the initial evaluation of an isolated thrombopenia. Rev. Med. Interne 2019, 40, 680–683. [Google Scholar] [CrossRef]
- Regenboog, M.; Bohte, A.E.; Akkerman, E.M.; Stoker, J.; Hollak, C.E.M. Iron storage in liver, bone marrow and splenic Gaucheroma reflects residual disease in type 1 Gaucher disease patients on treatment. Br. J. Haematol. 2017, 179, 635–647. [Google Scholar] [CrossRef]
- Bohte, A.E.; van Dussen, L.; Akkerman, E.M.; Nederveen, A.J.; Sinkus, R.; Jansen, P.L.M.; Stoker, J.; Hollak, C.E.M. Liver fibrosis in type I Gaucher disease: Magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS ONE 2013, 8, e57507. [Google Scholar] [CrossRef] [Green Version]
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; Dahl, S.V.; Häussinger, D.; Müller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J. Inherit. Metab. Dis. 2018, 41, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, T.; Reihani, N.; Daher, R.; de Villemeur, T.B.; Belmatoug, N.; Rose, C.; Colin-Aronovicz, Y.; Puy, H.; Le Van Kim, C.; Franco, M.; et al. Involvement of hepcidin in iron metabolism dysregulation in Gaucher disease. Haematologica 2018, 103, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Marchi, G.; Nascimbeni, F.; Motta, I.; Busti, F.; Carubbi, F.; Cappellini, M.D.; Pietrangelo, A.; Corradini, E.; Piperno, A.; Girelli, D. Hyperferritinemia and diagnosis of type 1 Gaucher disease. Am. J. Hematol. 2020, 95, 570–576. [Google Scholar] [CrossRef]
- Medrano-Engay, B.; Irun, P.; Gervas-Arruga, J.; Andrade-Campos, M.; Andreu, V.; Alfonso, P.; Pocovi, M.; Giraldo, P. Iron homeostasis and infIammatory biomarker analysis in patients with type 1 Gaucher disease. Blood Cells Mol. Dis. 2014, 53, 171–175. [Google Scholar] [CrossRef]
- Lorenz, F.; Pawłowicz, E.; Klimkowska, M.; Beshara, S.; Bulanda Brustad, A.; Skotnicki, A.B.; Wahlin, A.; Machaczka, M. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol. Dis. 2018, 68, 35–42. [Google Scholar] [CrossRef]
- Brady, J.; Trehan, A.; Landis, D.; Toro, C. Mucopolysaccharidosis type IIIB (MPS IIIB) masquerading as a behavioural disorder. BMJ Case Rep. 2013, 2013, bcr2013009592. [Google Scholar] [CrossRef] [Green Version]
- Puy, V.; Darwiche, W.; Trudel, S.; Gomila, C.; Lony, C.; Puy, L.; Lefebvre, T.; Vitry, S.; Boullier, A.; Karim, Z.; et al. Predominant role of microglia in brain iron retention in Sanfilippo syndrome, a pediatric neurodegenerative disease. Glia 2018, 66, 1709–1723. [Google Scholar] [CrossRef]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselyov, K.; Colletti, G.A.; Terwilliger, A.; Ketchum, K.; Lyons, C.W.P.; Quinn, J.; Muallem, S. TRPML: Transporters of metals in lysosomes essential for cell survival? Cell Calcium 2011, 50, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M.; Williams, I.; Smith, D.; Cox, T.M.; Platt, F.M. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol. Dis. 2009, 34, 406–416. [Google Scholar] [CrossRef]
- Gautschi, M.; Merlini, L.; Calza, A.-M.; Hayflick, S.; Nuoffer, J.-M.; Fluss, J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur. J. Paediatr. Neurol. 2014, 18, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Britton, R.S.; Bacon, B.R.; Recknagel, R.O. Lipid peroxidation and associated hepatic organelle dysfunction in iron overload. Chem. Phys. Lipids 1987, 45, 207–239. [Google Scholar] [CrossRef]
- Rider, J.A.; Dawson, G.; Siakotos, A.N. Perspective of biochemical research in the neuronal ceroid-lipofuscinosis. Am. J. Med. Genet. 1992, 42, 519–524. [Google Scholar] [CrossRef]
- Evans, J.; Katz, M.L.; Levesque, D.; Shelton, G.D.; de Lahunta, A.; O’Brien, D. A variant form of neuronal ceroid lipofuscinosis in American bulldogs. J. Vet. Intern. Med. 2005, 19, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mellon, S.H.; Butters, T.D.; Nevyjel, M.; Covey, D.F.; Bembi, B.; Dardis, A. Oxidative stress in NPC1 deficient cells: Protective effect of allopregnanolone. J. Cell Mol. Med. 2009, 13, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Cielo Rech, V.; Feksa, L.R.; Arevalo do Amaral, M.F.; Waltereith Koch, G.; Wajner, M.; Dutra-Filho, C.S.; de Souza Wyse, A.T.; Duval Wannmacher, C.M. Promotion of oxidative stress in kidney of rats loaded with cystine dimethyl ester. Pediatr. Nephrol. 2007, 22, 1121–1128. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Kluijtmans, L.A.J.; van der Velden, T.J.; Willems, P.H.; Scheffer, P.G.; Masereeuw, R.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim. Biophys. Acta 2011, 1812, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves Pereira, V.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef]
- Reolon, G.K.; Reinke, A.; de Oliveira, M.R.; Braga, L.M.; Camassola, M.; Andrades, M.E.; Fonseca Moreira, J.C.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in oxidative markers in the cerebellum and peripheral organs in MPS I mice. Cell Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Filippon, L.; Vanzin, C.C.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; do Carmo R Peralba, M.; Schwartz, I.V.D.; Giugliani, R.; Vargas, C.R. Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef]
- Diaz Jacques, C.E.; Donida, B.; Mescka, C.P.; Rodrigues, D.G.B.; Marchetti, D.P.; Bitencourt, F.H.; Burin, M.G.; de Souza, C.F.M.; Giugliani, R.G.; Vargas, C.R. Oxidative and nitrative stress and pro-inflammatory cytokines in Mucopolysaccharidosis type II patients: Effect of long-term enzyme replacement therapy and relation with glycosaminoglycan accumulation. Biochim. Biophys. Acta 2016, 1862, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.R.D.; Di Domenico, C.; Musella, A.; Cecere, F.; Di Napoli, D.; Di Natale, P. Mucopolysaccharidosis IIIB: Oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res. 2009, 1279, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Biancini, G.B.; Jacques, C.E.; Hammerschmidt, T.; de Souza, H.M.; Donida, B.; Deon, M.; Vairo, F.P.; Lourenço, C.M.; Giugliani, R.; Vargas, C.R. Biomolecules damage and redox status abnormalities in Fabry patients before and during enzyme replacement therapy. Clin. Chim. Acta 2016, 461, 41–46. [Google Scholar] [CrossRef]
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calò, L.A. Oxidative stress and the altered reaction to it in Fabry disease: A possible target for cardiovascular-renal remodeling? PLoS ONE 2018, 13, e0204618. [Google Scholar] [CrossRef] [PubMed]
- Coblentz, J.; St Croix, C.; Kiselyov, K. Loss of TRPML1 promotes production of reactive oxygen species: Is oxidative damage a factor in mucolipidosis type IV? Biochem. J. 2014, 457, 361–368. [Google Scholar] [CrossRef]
- Moraitou, M.; Dimitriou, E.; Dekker, N.; Monopolis, I.; Aerts, J.; Michelakakis, H. Gaucher disease: Plasmalogen levels in relation to primary lipid abnormalities and oxidative stress. Blood Cells Mol. Dis. 2014, 53, 30–33. [Google Scholar] [CrossRef]
- Paintlia, M.K.; Singh, I.; Singh, A.K. Effect of vitamin D3 intake on the onset of disease in a murine model of human Krabbe disease. J. Neurosci. Res. 2015, 93, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Park, T.-J.; Park, J.H.; Lee, G.S.; Lee, J.-Y.; Shin, J.-H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.-J.; Han, B.-S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, P.; Druckrey-Fiskaaen, C.; Kouznetsova, E.; Heinitz, K.; Bigl, M.; Cotman, S.L.; Schliebs, R. Developmental impairments of select neurotransmitter systems in brains of Cln3(Deltaex7/8) knock-in mice, an animal model of juvenile neuronal ceroid lipofuscinosis. J. Neurosci. Res. 2008, 86, 1857–1870. [Google Scholar] [CrossRef]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.O.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chol, M.; Nevo, N.; Cherqui, S.; Antignac, C.; Rustin, P. Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem. Biophys. Res. Commun. 2004, 324, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, L.; Pastore, A.; Rizzo, C.; Piemonte, F.; Rizzoni, G.; Emma, F. Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr. Res. 2006, 59, 332–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.-S.; Arning, E.; West, M.L.; Day, T.S.; Chen, S.; Meng, X.-L.; Forni, S.; McNeill, N.; Goker-Alpan, O.; Wang, X.; et al. Tetrahydrobiopterin deficiency in the pathogenesis of Fabry disease. Hum. Mol. Genet. 2017, 26, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Hoinkis, D.; Rintz, E.; Brokowska, J.; Cyske, Z.; Wegrzyn, G. Underestimated Aspect of Mucopolysaccharidosis Pathogenesis: Global Changes in Cellular Processes Revealed by Transcriptomic Studies. Int. J. Mol. Sci. 2020, 21, 1204. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Krzelowska, K.; Piotrowska, E.; Węgrzyn, G. Changes in expressions of genes involved in the regulation of cellular processes in mucopolysaccharidoses as assessed by fibroblast culture-based transcriptomic analyses. Metab Brain Dis. 2020, 35, 1353–1360. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Jankowska, E.; Rintz, E.; Witkowska, J.; Wieczerzak, E.; Podlacha, M.; Węgrzyn, G. Proteasome Composition and Activity Changes in Cultured Fibroblasts Derived From Mucopolysaccharidoses Patients and Their Modulation by Genistein. Front. Cell Dev. Biol. 2020, 8, 540726. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Podlacha, M.; Węgrzyn, G. Genetic Base of Behavioral Disorders in Mucopolysaccharidoses: Transcriptomic Studies. Int. J. Mol. Sci. 2020, 21, 1156. [Google Scholar] [CrossRef] [Green Version]
- Brokowska, J.; Pierzynowska, K.; Gaffke, L.; Rintz, E.; Węgrzyn, G. Expression of genes involved in apoptosis is dysregulated in mucopolysaccharidoses as revealed by pilot transcriptomic analyses. Cell Biol. Int. 2020. [Google Scholar] [CrossRef]
- Rintz, E.; Gaffke, L.; Podlacha, M.; Brokowska, J.; Cyske, Z.; Węgrzyn, G.; Pierzynowska, K. Transcriptomic Changes Related to Cellular Processes with Particular Emphasis on Cell Activation in Lysosomal Storage Diseases from the Group of Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 3194. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar α-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Laquerrière, L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Micsenyi, M.C.; Dobrenis, K.; Stephney, G.; Pickel, J.; Vanier, M.T.; Slaugenhaupt, S.A.; Walkley, S.U. Neuropathology of the Mcoln1(-/-) knockout mouse model of mucolipidosis type IV. J Neuropathol. Exp. Neurol. 2009, 68, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.A.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Inhibition of autophagosome formation restores mitochondrial function in mucolipidosis II and III skin fibroblasts. Mol. Genet. Metab. 2009, 98, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Fanin, M.; Masiero, E.; Angelini, C.; Sandri, M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ. 2012, 19, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Sansanwal, P.; Sarwal, M.M. p62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatr. Nephrol. 2012, 27, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergande, M.R.; Zarate, E.; Haney-Ball, C.; Davidson, C.D.; Scesa, G.; Givogri, M.I.; Bongarzone, E.R.; Cologna, S.M. Standard-flow LC and thermal focusing ESI elucidates altered liver proteins in late stage Niemann-Pick, type C1 disease. Bioanalysis 2019, 11, 1067–1083. [Google Scholar] [CrossRef] [PubMed]



Figure 4.
Activation of ferroptosis by ROS levels and lipid peroxidation-dependent pathway [1,3,29].




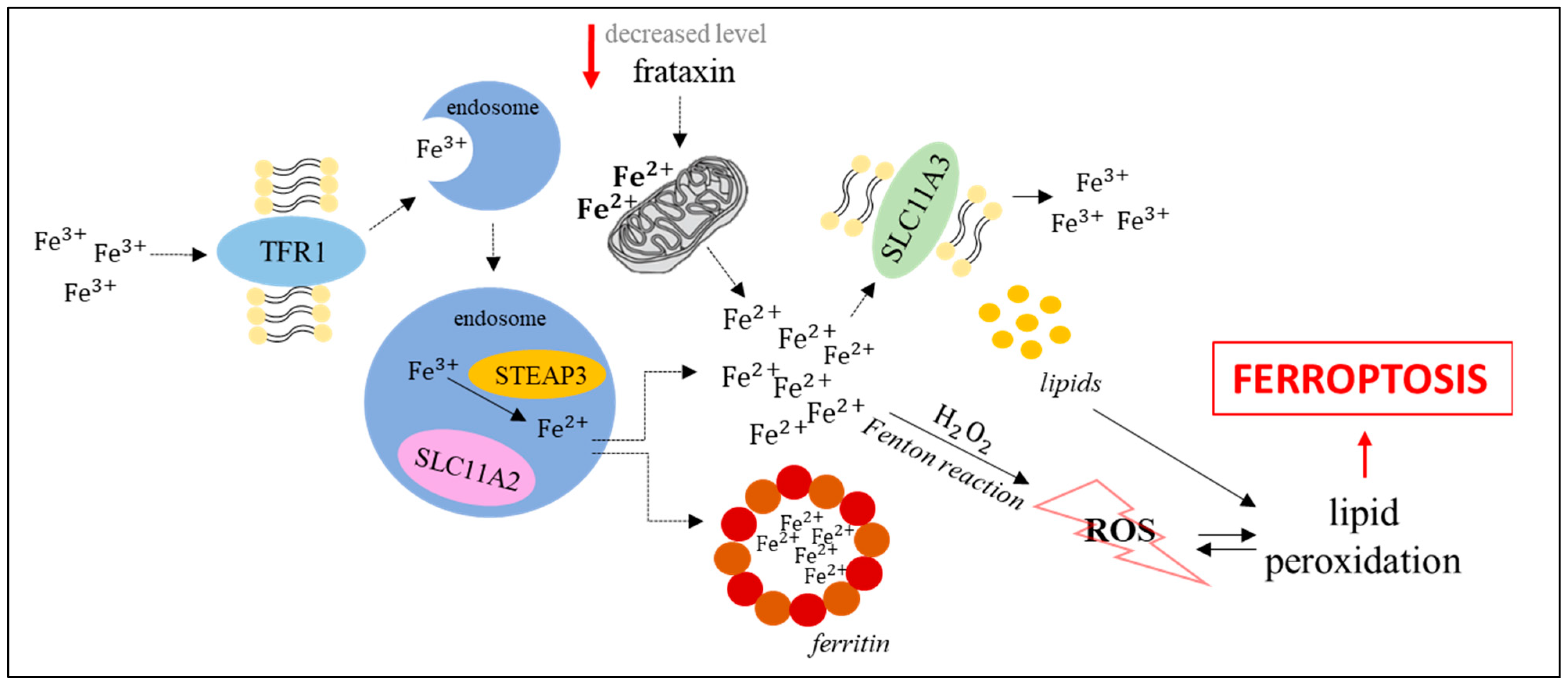{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of major features of apoptosis, autophagy, and ferroptosis (based on [1]).
Table 1.
Comparison of major features of apoptosis, autophagy, and ferroptosis (based on [1]).
| Features | Apoptosis | Autophagy | Ferroptosis | |
|---|---|---|---|---|
| morphology | cell membrane | plasma membrane blebs forming | no changes | cell rounding up (lack of plasma membrane blebs) |
| cytoplasm | pseudopods retraction and cellular volume reduction | accumulation of autophagosomes and autophagolysosomes | reduction in size of mitochondria, condensed densities of mitochondrial membrane, rupture of the mitochondrial membrane, reduction in mitochondrial cristae | |
| nucleus | nuclear volume reduction, fragmentation of nucleus and chromatin condensation | lack of chromatin condensation | lack of nuclear volume reduction and chromatin condensation | |
| biochemistry | caspases activation, DNA fragmentation, exposure of phosphatidylserine, mitochondrial membrane potential (ΔΨm) dissipation | conversion of LC3-I to LC3-II form, increased levels of the LAMP2 protein, increased levels of Atg proteins and the Beclin-1 protein | reactive oxygen species accumulation, lipid peroxidation, iron accumulation, activation of MAP kinases, inhibition of cystine-glutamate antiporter, increased NADPH oxidation, glutathione depletion, arachidonic acid mediators, mitochondrial membrane potential (ΔΨm) dissipation | |
Table 2.
Modulations of ferroptosis features in lysosomal storage diseases.
| Ferroptosis Marker | Disease | Model | Material/Organ | Reference(s) | |
|---|---|---|---|---|---|
| increased iron concentration | NCL | Patients | cerebrospinal fluid | [54,55] | |
| Niemann–Pick Disease | ASMKO mice | lung and brain | [59] | ||
| BALB/cJ Npc1nih (Npc1−/−) mice | brain | [61] | |||
| Gaucher disease | Cells | patient-derived skin fibroblasts | [62] | ||
| Patients | liver or spleen biopsy serum MRI imaging of liver, bone, marrow, spleen | [62,63,67] [64,65,69] [66,71] | |||
| MPS type III | Patients | MRI imaging of brain | [73] | ||
| C57Bl/6 Naglu−/− mice | brain | [74] | |||
| ML type IV | Cells | patient-derived skin fibroblasts | [75] | ||
| Fucosidosis | Patients | MRI imaging of brain | [78] | ||
| increased lipid peroxidation | NCL | Patients | serum, brain | [80] | |
| English setters American bulldogs | brain brain, eyes | [80] [81] | |||
| Niemann–Pick disease | Cells | patient-derived skin fibroblasts | [82] | ||
| Cystinosis | rats loaded with cystine dimethyl ester | kidney | [83] | ||
| Cells | proximal tubule epithelial cells taken from patients | [84] | |||
| MPS type I | Patients | serum | [85] | ||
| C57BL/6 Idua−/− mice | brain, heart, lung, diaphragm, liver, kidney, spleen | [86] | |||
| MPS type II | Patients | plasma urine | [88,89] [89] | ||
| MPS type IIIB | C57Bl/6 Naglu−/− mice | brain | [90] | ||
| Fabry disease | Patients | blood, urine plasma | [91] [92] | ||
| ML type IV | cells | TRPML1−/− cells | [93] | ||
| Gaucher disease | patients | erythrocytes and plasma | [94] | ||
| Krabbe disease | GALC twi−/− mice | brain | [95] | ||
| inhibition of activity or decreased level of GPX4-GSH-Xc− system components | GPX4 | NCL | Cln3Dex7/8 knock-in mice | brain | [100] |
| Fabry disease | patients | urine and blood | [101] | ||
| glutathione (GSH) | infantile cystinosis | cells | patient-derived skin fibroblasts | [102] | |
| nephropathic cystinosis | cells | patient-derived skin fibroblasts | [103] | ||
| Fabry disease | patients | urine and blood | [101] | ||
| B6/129-B57BL/6 Fabry mice | heart, kidney, liver, plasma | [105] | |||
| MPS type IVA | patients | urine and blood | [104] | ||
| Xc− | no data | - | - | - | |
Abbreviations: NCL—neuronal ceroid lipofuscinoses; ML—mucolipidosis; MPS—mucopolysaccharidosis.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pierzynowska, K.; Rintz, E.; Gaffke, L.; Węgrzyn, G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells 2021, 10, 365. https://doi.org/10.3390/cells10020365
AMA Style
Pierzynowska K, Rintz E, Gaffke L, Węgrzyn G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells. 2021; 10(2):365. https://doi.org/10.3390/cells10020365
Chicago/Turabian StylePierzynowska, Karolina, Estera Rintz, Lidia Gaffke, and Grzegorz Węgrzyn. 2021. "Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases" Cells 10, no. 2: 365. https://doi.org/10.3390/cells10020365
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.