
PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Culture
2.3. NET Tumor Spheroids
2.4. Radiotherapy
2.5. Immunohistochemistry
2.6. Immunoblotting
2.7. Sulforhodamine B (SRB) proliferation assay
2.8. Colorimetric cell survival assay
2.9. CCK-8 Viability Assay
2.10. DNA Fragmentation ELISA Photometric Enzyme Immunoassay
2.11. Statistical Analysis
3. Results
3.1. PAkt (Ser473) Expression in GEP-NET Tumors
3.2. Cellular Profiling of PI3K/mTOR Inhibitors, PF-04691502 and PKI-402, in GEP-NET Cancer Cell Lines
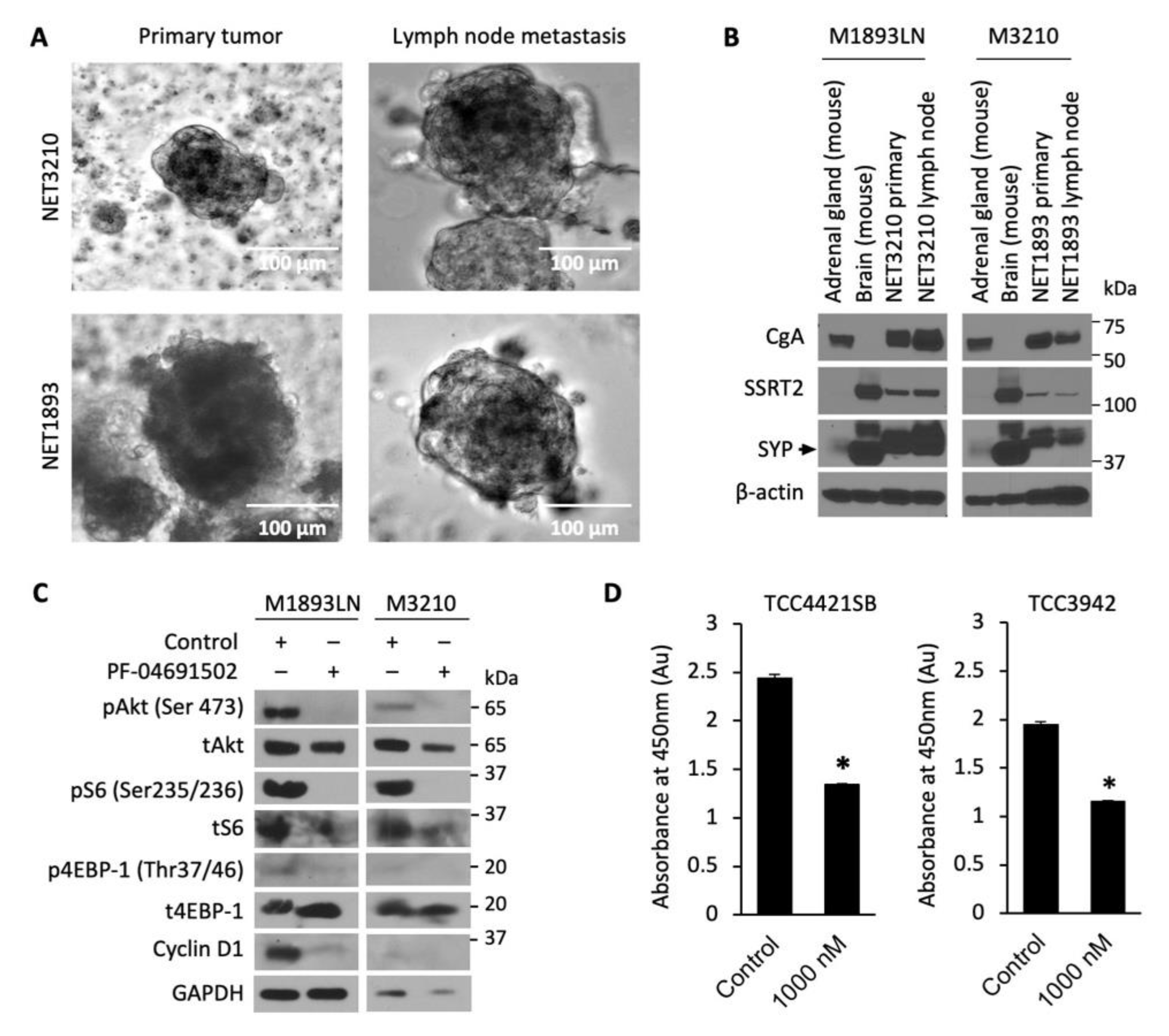
3.3. Cellular Profiling of PF-04691502 in Patient-Derived Tumor Spheroid Model
3.4. Enhanced Radiosensitization of GEP-NET Cells via Schedule Dependent PF-04691502 Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Modlin, I.M.; Moss, S.F.; Chung, D.C.; Jensen, R.T.; Snyderwine, E. Priorities for Improving the Management of Gastroenteropancreatic Neuroendocrine Tumors. JNCI J. Natl. Cancer Inst. 2008, 100, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Campana, D.; Tomassetti, P. Incidence, Epidemiology, Aetiology and Staging, Classification, Clinical Presentation/Signs and Symptoms, Diagnosis, Staging Procedures/Investigation. In PET/CT in Neuroendocrine Tumors; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–5. [Google Scholar]
- Baum, R.P.; Kluge, A.W.; Kulkarni, H.; Schorr-Neufing, U.; Niepsch, K.; Bitterlich, N.; van Echteld, C.J. [177Lu-DOTA] 0-D-Phe1-Tyr3-octreotide (177Lu-DOTATOC) for peptide receptor radiotherapy in patients with advanced neuroendocrine tumours: A phase-II study. Theranostics 2016, 6, 501. [Google Scholar] [CrossRef] [PubMed]
- Ezziddin, S.; Attassi, M.; Guhlke, S.; Ezziddin, K.; Palmedo, H.; Reichmann, K.; Ahmadzadehfar, H.; Biermann, K.; Krenning, E.; Biersack, H.-J. Targeted radiotherapy of neuroendocrine tumors using Lu-177-DOTA octreotate with prolonged intervals. J. Nucl. Med. 2007, 48, 394P. [Google Scholar]
- Limouris, G.; Poulantzas, V.; Trompoukis, N.; Karfis, I.; Chondrogiannis, S.; Triantafyllou, N.; Gennimata, V.; Moulopoulou, L.-E.; Patsouris, E.; Nikou, G. Comparison of 111in-[DTPA0] octreotide versus non carrier added 177lu-[DOTA0, Tyr3]-octreotate efficacy in patients with GEP-NET treated intra-arterially for liver metastases. Clin. Nucl. Med. 2016, 41, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Giovacchini, G.; Müller-Brand, J.; Forrer, F. Targeted radiotherapy with radiolabeled somatostatin analogs. Endocrinol. Metab. Clin. 2011, 40, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Wolin, E.M.; Chasen, B.; Kulke, M.H.; Bushnell, D.L.; Caplin, M.E.; Baum, R.P.; Kunz, P.L.; Hobday, T.J.; Hendifar, A.E. NETTER-1 Phase III: Progression-Free Survival, Radiographic Response, and Preliminary Overall Survival Results in Patients with Midgut Neuroendocrine Tumors Treated with 177-Lu-Dotatate; American Society of Clinical Oncology: Alexandria, VA, USA, 2016. [Google Scholar]
- Van Essen, M.; Krenning, E.P.; Kam, B.L.; De Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide-receptor radionuclide therapy for endocrine tumors. Nat. Rev. Endocrinol. 2009, 5, 382. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptor expression in GEP-NET. Virchows Archiv. 2007, 451, 47–50. [Google Scholar] [CrossRef]
- van Essen, M.; Krenning, E.P.; Kam, B.L.; de Herder, W.W.; van Aken, M.O.; Kwekkeboom, D.J. Report on short-term side effects of treatments with 177 Lu-octreotate in combination with capecitabine in seven patients with gastroenteropancreatic neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 743–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bison, S.M.; Haeck, J.C.; Bol, K.; Koelewijn, S.; Groen, H.; Melis, M.; Veenland, J.; Bernsen, M.; de Jong, M. Optimization of combined temozolomide and peptide receptor radionuclide therapy (PRRT) in mice after multimodality molecular imaging studies. EJNMMI Res. 2015, 5, 62. [Google Scholar] [CrossRef] [Green Version]
- Kong, G.; Johnston, V.; Ramdave, S.; Lau, E.; Rischin, D.; Hicks, R.J. High-administered activity In-111 octreotide therapy with concomitant radiosensitizing 5FU chemotherapy for treatment of neuroendocrine tumors: Preliminary experience. Cancer Biother. Radiopharm. 2009, 24, 527–533. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Lombard-Bohas, C.; Baudin, E.; Kvols, L.K.; Rougier, P.; Ruszniewski, P.; Hoosen, S.; Peter, J.S.; Haas, T.; Lebwohl, D. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J. Clin. Oncol. 2010, 28, 69. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Hörsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Pavel, M.; Baudin, E.; Öberg, K.; Hainsworth, J.; Voi, M.; Rouyrre, N.; Peeters, M.; Gross, D.; Yao, J.C. Efficacy of everolimus plus octreotide LAR in patients with advanced neuroendocrine tumor and carcinoid syndrome: Final overall survival from the randomized, placebo-controlled phase 3 RADIANT-2 study. Ann. Oncol. 2017, 28, 1569–1575. [Google Scholar] [CrossRef]
- Yao, J.C.; Pavel, M.; Lombard-Bohas, C.; Van Cutsem, E.; Voi, M.; Brandt, U.; He, W.; Chen, D.; Capdevila, J.; De Vries, E.G. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: Overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J. Clin. Oncol. 2016, 34, 3906. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; Exner, S.; Erdmann, S.; Brenner, W.; Groetzinger, C. Somatostatin agonist and mTOR inhibitors as potential radioprotectors or radiosensitizers in neuroendocrine tumor cells. J. Nucl. Med. 2016, 57, 1435. [Google Scholar]
- Aljubran, A.H.; Alrowaili, M.; Raef, H.; Bazarbashi, S.; Alzahrani, A.M.; Almuhaideb, A.; Almanea, H.; Badran, A.a.; Al-Dalee, A.; Tuli, M. Combination of Everolimus and lu-177 PRRT in Treatment of G1-2 Neuroendocrine Tumors (NET): Phase 1-2 Study; American Society of Clinical Oncology: Alexandria, VA, USA, 2019. [Google Scholar]
- Claringbold, P.G.; Turner, J.H. NeuroEndocrine tumor therapy with lutetium-177-octreotate and everolimus (NETTLE): A phase I study. Cancer Biother. Radiopharm. 2015, 30, 261–269. [Google Scholar] [CrossRef]
- Yuan, J.; Mehta, P.P.; Yin, M.-J.; Sun, S.; Zou, A.; Chen, J.; Rafidi, K.; Feng, Z.; Nickel, J.; Engebretsen, J. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol. Cancer Ther. 2011, 10, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Britten, C.D.; Adjei, A.A.; Millham, R.; Houk, B.E.; Borzillo, G.; Pierce, K.; Wainberg, Z.A.; LoRusso, P.M. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Investig. New Drugs 2014, 32, 510–517. [Google Scholar] [CrossRef] [PubMed]
- del Campo, J.M.; Birrer, M.; Davis, C.; Fujiwara, K.; Gollerkeri, A.; Gore, M.; Houk, B.; Lau, S.; Poveda, A.; González-Martín, A. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol. Oncol. 2016, 142, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Koehler, M.; Millham, R.; Borzillo, G.; A’Hern, R.; Pierce, K.; Barton, J.; Giorgetti, C. 104P Phase II Randomized Study of Pre-Operative Pf-04691502 Plus Letrozole Compared with Letrozole (L) In Patients with Estrogen Receptor-Positive, Her2-Negative Early Breast Cancer (Bc). Ann. Oncol. 2012, 23, ii44. [Google Scholar] [CrossRef]
- Fazio, N.; Buzzoni, R.; Baudin, E.; Antonuzzo, L.; Hubner, R.A.; Lahner, H.; De Herder, W.W.; Raderer, M.; Teulé, A.; Capdevila, J. A phase II study of BEZ235 in patients with everolimus-resistant, advanced pancreatic neuroendocrine tumours. Anticancer Res. 2016, 36, 713–719. [Google Scholar] [PubMed]
- CytoScanTM SRB Cell Cytotoxicity Assay. Available online: https://www.gbiosciences.com/image/pdfs/protocol/786-213_protocol.pdf (accessed on 14 May 2021).
- Evers, B.M.; Townsend, C.M., Jr.; Upp, J.R.; Allen, E.; Hurlbut, S.C.; Kim, S.W.; Rajaraman, S.; Singh, P.; Reubi, J.C.; Thompson, J.C. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology 1991, 101, 303–311. [Google Scholar] [CrossRef]
- Parekh, D.; Ishizuka, J.; Townsend, C.M., Jr.; Haber, B.; Beauchamp, R.D.; Karp, G.; Kim, S.W.; Rajaraman, S.; Greeley, G., Jr.; Thompson, J.C. Characterization of a human pancreatic carcinoid in vitro: Morphology, amine and peptide storage, and secretion. Pancreas 1994, 9, 83–90. [Google Scholar] [CrossRef]
- Doihara, H.; Nozawa, K.; Kojima, R.; Kawabata-Shoda, E.; Yokoyama, T.; Ito, H. QGP-1 cells release 5-HT via TRPA1 activation; a model of human enterochromaffin cells. Mol. Cell. Biochem. 2009, 331, 239–245. [Google Scholar] [CrossRef]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M. Establishment of the first well-differentiated human pancreatic neuroendocrine tumor model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Molloy, J.; Izumi, T.; Sterpin, E. Impact of backscatter material thickness on the depth dose of orthovoltage irradiators for radiobiology research. Phys. Med. Biol. 2019, 64, 055001. [Google Scholar] [CrossRef]
- Rychahou, P.G.; Jackson, L.N.; Silva, S.R.; Rajaraman, S.; Evers, B.M. Targeted molecular therapy of the PI3K pathway: Therapeutic significance of PI3K subunit targeting in colorectal carcinoma. Ann. Surg. 2006, 243, 833–842; discussion 843–844. [Google Scholar] [CrossRef]
- Allred, D.C.; Clark, G.M.; Elledge, R.; Fuqua, S.A.; Brown, R.W.; Chamness, G.C.; Osborne, C.K.; McGuire, W.L. Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J. Natl. Cancer Inst. 1993, 85, 200–206. [Google Scholar] [CrossRef]
- Allred, D.C.; Harvey, J.M.; Berardo, M.; Clark, G.M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 1998, 11, 155–168. [Google Scholar]
- Johnson, J.; Chow, Z.; Napier, D.; Lee, E.; Weiss, H.L.; Evers, B.M.; Rychahou, P. Targeting PI3K and AMPKα Signaling Alone or in Combination to Enhance Radiosensitivity of Triple Negative Breast Cancer. Cells 2020, 9, 1253. [Google Scholar] [CrossRef] [PubMed]
- Sigma-Aldrich. Cell Death Detection ELISAPLUS; Sigma-Aldrich: St. Louis, MO, USA, 2016. [Google Scholar]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.G.; Scott, A.T.; Li, G.; Sherman, S.K.; Ear, P.H.; Howe, J.R. Metastatic pancreatic neuroendocrine tumors have decreased somatostatin expression and increased Akt signaling. Surgery 2020, 169, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Mallon, R.; Hollander, I.; Feldberg, L.; Lucas, J.; Soloveva, V.; Venkatesan, A.; Dehnhardt, C.; Santos, E.D.; Chen, Z.; Dos Santos, O. Antitumor efficacy profile of PKI-402, a dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor. Mol. Cancer Ther. 2010, 9, 976–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capdevila, J.; Casanovas, O.; Salazar, R.; Castellano, D.; Segura, A.; Fuster, P.; Aller, J.; García-Carbonero, R.; Jimenez-Fonseca, P.; Grande, E.; et al. Translational research in neuroendocrine tumors: Pitfalls and opportunities. Oncogene 2017, 36, 1899–1907. [Google Scholar] [CrossRef]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [Green Version]
- Bundschuh, R.A.; Habacha, B.; Lütje, S.; Essler, M. Therapy of Patients with Neuroendocrine Neoplasia-Evidence-Based Approaches and New Horizons. J. Clin. Med. 2019, 8, 1474. [Google Scholar] [CrossRef] [Green Version]
- Nisa, L.; Savelli, G.; Giubbini, R. Yttrium-90 DOTATOC therapy in GEP-NET and other SST2 expressing tumors: A selected review. Ann. Nucl. Med. 2011, 25, 75–85. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Kam, B.L.; Van Essen, M.; Teunissen, J.J.; van Eijck, C.H.; Valkema, R.; De Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsma, H.; van Vliet, E.I.; Teunissen, J.J.; Kam, B.L.; de Herder, W.W.; Peeters, R.P.; Krenning, E.P.; Kwekkeboom, D.J. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Brighi, N.; Maggio, I.; Manuzzi, L.; Peterle, C.; Ambrosini, V.; Ricci, C.; Casadei, R.; Campana, D. The role of mTOR in neuroendocrine tumors: Future cornerstone of a winning strategy? Int. J. Mol. Sci. 2018, 19, 747. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Li, F.; Cardelli, J.; Martin, K.; Blenis, J.; Huang, S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene 2006, 25, 7029–7040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Jang, Y.H.; Chau, G.C.; Pyo, S.; Um, S.H. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Mod. Pathol. 2013, 26, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Robb, V.A.; Astrinidis, A.; Henske, E.P. Frequent of ribosomal protein S6 hyperphosphorylation in lymphangioleiomyomatosis-associated angiomyolipomas. Mod. Pathol. 2006, 19, 839–846. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuger, S.; Graus, D.; Brendtke, R.; Günther, N.; Katzer, A.; Lutyj, P.; Polat, B.; Chatterjee, M.; Sukhorukov, V.L.; Flentje, M. Radiosensitization of glioblastoma cell lines by the dual PI3K and mTOR inhibitor NVP-BEZ235 depends on drug-irradiation schedule. Transl. Oncol. 2013, 6, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Shimomura, M.; Kobayashi, K.; Kojima, S.; Nakatsura, T. Growth inhibition by NVP-BEZ235, a dual PI3K/mTOR inhibitor, in hepatocellular carcinoma cell lines. Oncol. Rep. 2011, 26, 1273–1279. [Google Scholar]
- Wong, C.H.; Loong, H.H.; Hui, C.W.; Lau, C.P.; Hui, E.P.; Ma, B.B.; Chan, A.T. Preclinical evaluation of the PI3K-mTOR dual inhibitor PF-04691502 as a novel therapeutic drug in nasopharyngeal carcinoma. Investig. New Drugs 2013, 31, 1399–1408. [Google Scholar] [CrossRef]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Xia, M.; Wang, J.; Yu, H.; Chai, J.; Zhang, Z.; Sun, Y.; Su, J.; Sun, L. Dual PI3K/mTOR inhibitor PKI-402 suppresses the growth of ovarian cancer cells by degradation of Mcl-1 through autophagy. Biomed. Pharmacother. 2020, 129, 110397. [Google Scholar] [CrossRef]
- Oishi, T.; Itamochi, H.; Kudoh, A.; Nonaka, M.; Kato, M.; Nishimura, M.; Oumi, N.; Sato, S.; Naniwa, J.; Sato, S. The PI3K/mTOR dual inhibitor NVP-BEZ235 reduces the growth of ovarian clear cell carcinoma. Oncol. Rep. 2014, 32, 553–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlo, M.I.; Molina, A.M.; Lakhman, Y.; Patil, S.; Woo, K.; DeLuca, J.; Lee, C.-H.; Hsieh, J.J.; Feldman, D.R.; Motzer, R.J. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist 2016, 21, 787. [Google Scholar] [CrossRef] [Green Version]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klümpen, H.J.; Capdevila, J. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naïve Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, 766. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef]
- Olive, P.L. The role of DNA single-and double-strand breaks in cell killing by ionizing radiation. Radiat. Res. 1998, 150, S42–S51. [Google Scholar] [CrossRef]
- Zhuang, H.-Q.; Bo, Q.-F.; Yuan, Z.-Y.; Wang, J.; Zhao, L.-J.; Wang, P. The different radiosensitivity when combining erlotinib with radiation at different administration schedules might be related to activity variations in c-MET-PI3K-AKT signal transduction. OncoTargets Ther. 2013, 6, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Hu, Y.; Xi, M.; He, L.; Zhao, L.; Liu, M. Sorafenib modulates the radio sensitivity of hepatocellular carcinoma cells in vitro in a schedule-dependent manner. BMC Cancer 2012, 12, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend, C.M., Jr.; Anthony, L.; Wasilchenko, C.; Melton, M.L.; et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. https://doi.org/10.3390/cells10051261
Chow Z, Johnson J, Chauhan A, Izumi T, Cavnar M, Weiss H, Townsend CM Jr., Anthony L, Wasilchenko C, Melton ML, et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells. 2021; 10(5):1261. https://doi.org/10.3390/cells10051261
Chicago/Turabian StyleChow, Zeta, Jeremy Johnson, Aman Chauhan, Tadahide Izumi, Michael Cavnar, Heidi Weiss, Courtney M. Townsend, Jr., Lowell Anthony, Carrigan Wasilchenko, Matthew L. Melton, and et al. 2021. "PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors" Cells 10, no. 5: 1261. https://doi.org/10.3390/cells10051261