
Single-Cell Transcriptomics Reveals Core Regulatory Programs That Determine the Heterogeneity of Circulating and Tissue-Resident Memory CD8+ T Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and LCMV Infection
2.2. Isolation of Lymphocytes from Spleen and Small Intestines
2.3. Cell Sorting
2.4. Single-Cell RNA Sequencing and Analysis
2.5. SCENIC Analysis
2.6. Bulk RNA-Sequencing and Analysis
3. Results
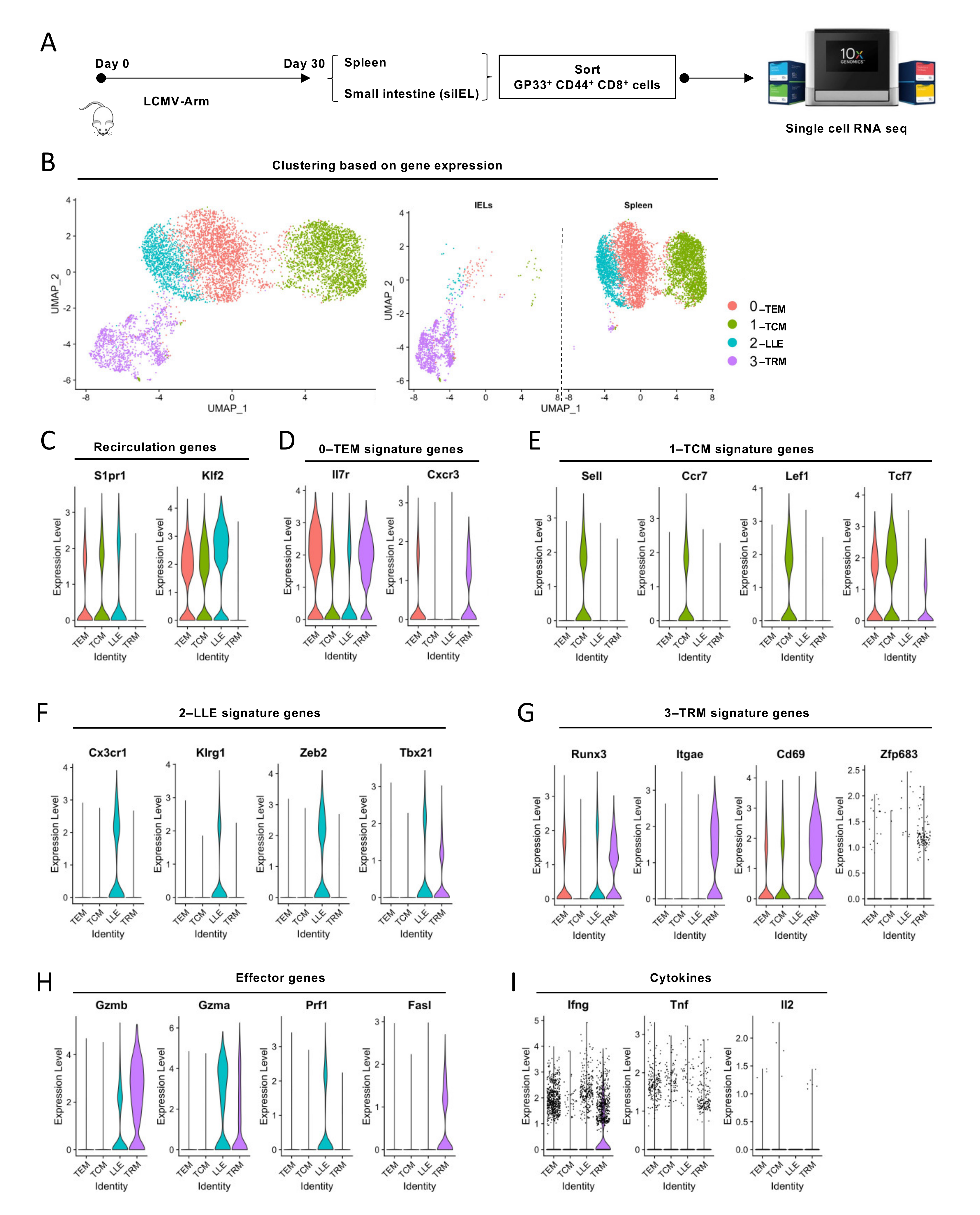
3.1. Single-Cell Transcriptomics Probes Heterogeneity within Memory CD8+ T-Cell Populations
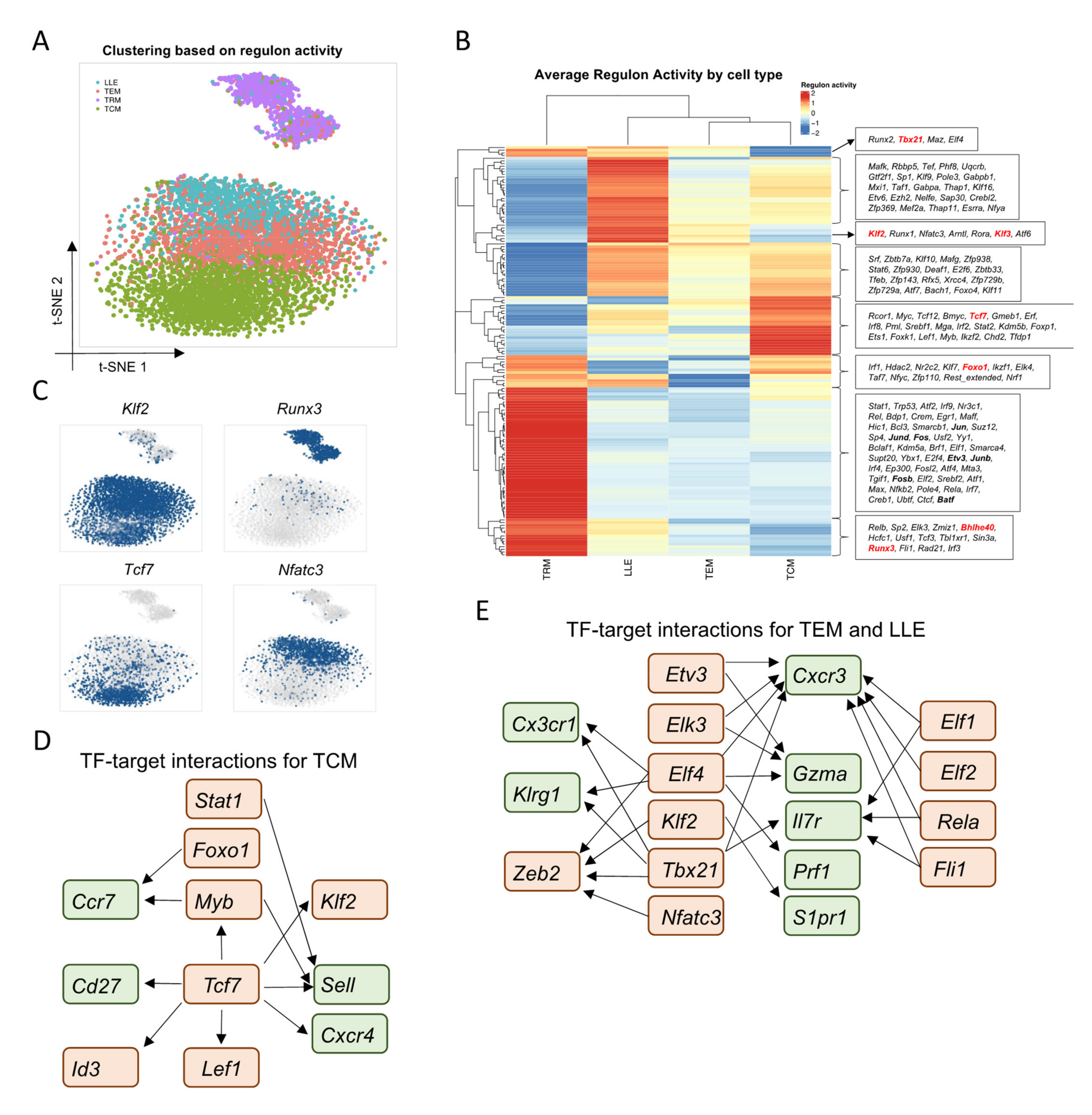
3.2. Single-Cell Network Inference Reveals Candidate Regulators of Memory CD8+ T-Cell Populations
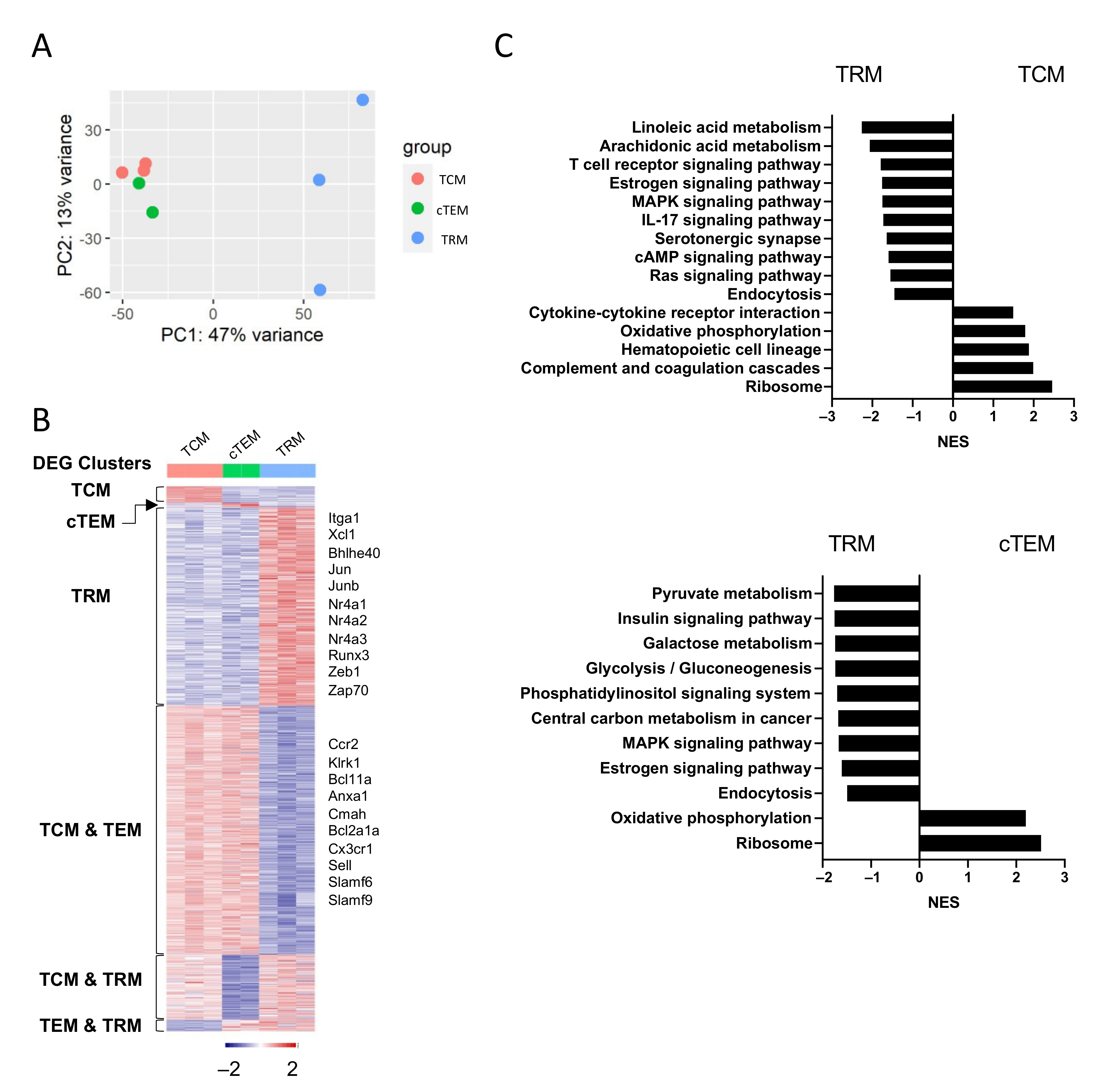
3.3. Bulk RNA-Seq Reveals the Unique Transcriptional Profiles of TRM Cells
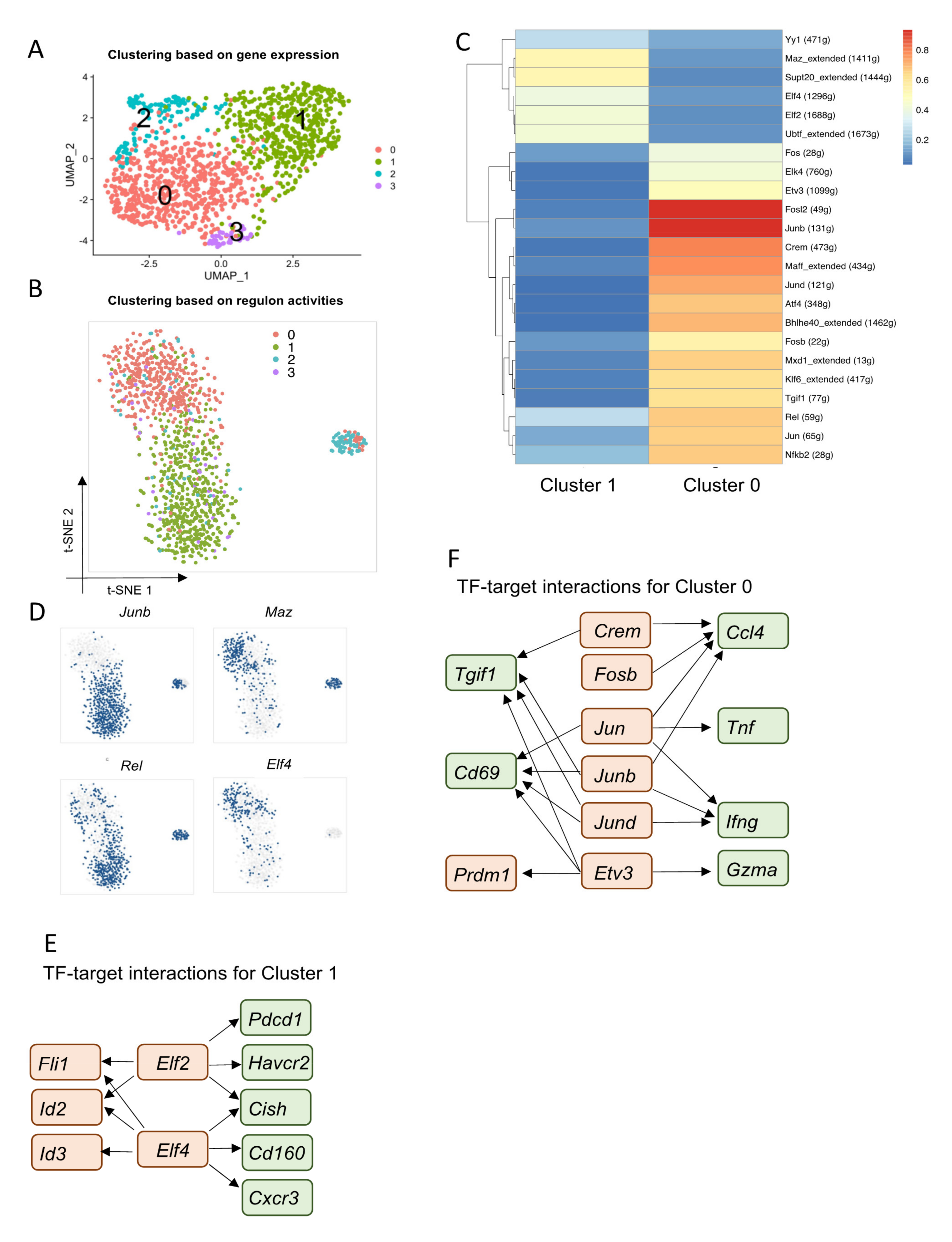
3.4. Core Regulatory Programs That Determine Heterogeneous TRM Populations in siIELs
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nat. Cell Biol. 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Gerlach, C.; Moseman, E.A.; Loughhead, S.M.; Alvarez, D.; Zwijnenburg, A.J.; Waanders, L.; Garg, R.; de la Torre, J.C.; von Andrian, U.H. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 2016, 45, 1270–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renkema, K.R.; Huggins, M.A.; Da Silva, H.B.; Knutson, T.P.; Henzler, C.; Hamilton, S.E. KLRG1+ Memory CD8 T Cells Combine Properties of Short-Lived Effectors and Long-Lived Memory. J. Immunol. 2020, 205, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.; Heath, W.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Masson, F.; Minnich, M.; Olshansky, M.; Bilic, I.; Mount, A.M.; Kallies, A.; Speed, T.P.; Busslinger, M.; Nutt, S.; Belz, G.T. Id2-Mediated Inhibition of E2A Represses Memory CD8+ T Cell Differentiation. J. Immunol. 2013, 190, 4585–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.Y.; Best, J.A.; Knell, J.; Yang, E.; Sheridan, A.; Jesionek, A.K.; Li, H.S.; Rivera, R.R.; Lind, K.C.; D’Cruz, L.M.; et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 2011, 12, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Gordon, S.; Intlekofer, A.; Paley, M.; Mooney, E.C.; Lindsten, T.; Wherry, E.J.; Reiner, S.L. Cutting Edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 2010, 185, 4988–4992. [Google Scholar] [CrossRef] [Green Version]
- Ichii, H.; Sakamoto, A.; Kuroda, Y.; Tokuhisa, T. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 2004, 173, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.R.; Li, Q.; Bupp, M.R.G.; Shrikant, P.A. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity 2012, 36, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Jeannet, G.; Boudousquie, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 2010, 107, 9777–9782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Yu, S.; Zhao, D.-M.; Harty, J.; Badovinac, V.; Xue, H.-H. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nat. Cell Biol. 2017, 552, 253–257. [Google Scholar] [CrossRef]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef]
- Li, C.; Zhu, B.; Son, Y.M.; Wang, Z.; Jiang, L.; Xiang, M.; Ye, Z.; Beckermann, K.E.; Wu, Y.; Jenkins, J.; et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 2019, 51, 491–507.e7. [Google Scholar] [CrossRef]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.J.; Toma, C.; He, Z.; Kurd, N.S.; Nguyen, Q.P.; McDonald, B.; Quezada, L.; Widjaja, C.E.; Witherden, D.A.; Crowl, J.T.; et al. Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity 2020, 52, 808–824.e7. [Google Scholar] [CrossRef]
- Kurd, N.S.; He, Z.; Louis, T.L.; Milner, J.J.; Omilusik, K.D.; Jin, W.; Tsai, M.S.; Widjaja, C.E.; Kanbar, J.N.; Olvera, J.G.; et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. 2020, 5, eaaz6894. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.; Bjorklund, K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, M.I.L.R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Baeyens, A.A.; Schwab, S.R. Finding a Way Out: S1P Signaling and Immune Cell Migration. Annu. Rev. Immunol. 2020, 38, 759–784. [Google Scholar] [CrossRef]
- Ferreira, D.P.; Silva, J.G.; Wyss, T.; Marraco, S.A.F.; Scarpellino, L.; Charmoy, M.; Maas, R.; Siddiqui, I.; Tang, L.; Joyce, J.A.; et al. Central memory CD8+ T cells derive from stem-like Tcf7hi effector cells in the absence of cytotoxic differentiation. Immunity 2020, 53, 985–1000.e11. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.-Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef] [PubMed]
- Omilusik, K.D.; Best, J.A.; Yu, B.; Goossens, S.; Weidemann, A.; Nguyen, J.V.; Seuntjens, E.; Stryjewska, A.; Zweier, C.; Roychoudhuri, R.; et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015, 212, 2027–2039. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, B.M.; Juedes, A.; Szabo, S.J.; von Herrath, M.; Glimcher, L.H. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA 2003, 100, 15818–15823. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.J.; Goldrath, A.W. Transcriptional programming of tissue-resident memory CD8+ T cells. Curr. Opin. Immunol. 2018, 51, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Mackay, L. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2015, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nat. Cell Biol. 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Park, C.S.; Mamonkin, M.; Lacorazza, H.D.; Lacorazza, D. Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Krüppel-like factors KLF4 and KLF2. Nat. Immunol. 2009, 10, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Moon, K.D.; Vacchio, M.S.; Hathcock, K.S.; Hodes, R.J. Downmodulation of Tumor Suppressor p53 by T Cell Receptor Signaling Is Critical for Antigen-Specific CD4+ T Cell Responses. Immunity 2014, 40, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Acharya, N.; Madi, A.; Zhang, H.; Klapholz, M.; Escobar, G.; Dulberg, S.; Christian, E.; Ferreira, M.; Dixon, K.O.; Fell, G.; et al. Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity 2020, 53, 658–671.e6. [Google Scholar] [CrossRef]
- Chen, Z.; Arai, E.; Khan, O.; Zhang, Z.; Ngiow, S.F.; He, Y.; Huang, H.; Manne, S.; Cao, Z.; Baxter, A.E.; et al. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 2021, 184, 1262–1280.e22. [Google Scholar] [CrossRef]
- Rincon, M.; Flavell, R.A.; Davis, R.J. Signal transduction by MAP kinases in T lymphocytes. Oncogene 2001, 20, 2490–2497. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Väänänen, K.; Watford, W.T.; Chen, Z. Estrogen receptor α contributes to T cell–mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018, 11, eaap9415. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nat. Cell Biol. 2009, 460, 103–107. [Google Scholar] [CrossRef]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.C.; Guittard, G.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; van Panhuys, N.; Klebanoff, C.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Nickel, H.; Moncrieffe, H.; Klemm, P.; Scheufen, A.; Föll, D.; Wixler, V.; Schippers, A.; Wagner, N.; Wedderburn, L.R.; et al. The transcription factor CREM drives an inflammatory phenotype of T cells in oligoarticular juvenile idiopathic arthritis. Pediatr. Rheumatol. 2018, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.; Rahimpour, A.; Ma, J.; Collins, N.C.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.; Stefanovic, T.; et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Miron, M.; Farber, D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019, 4, eaas9673. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Weiss, A.; Kumar, G.; Wang, S.; Nel, A. The T-cell antigen receptor utilizes Lck, Raf-1, and MEK-1 for activating mitogen-activated protein kinase. Evidence for the existence of a second protein kinase C-dependent pathway in an Lck-negative Jurkat cell mutant. J. Biol. Chem. 1994, 269, 17349–17357. [Google Scholar] [CrossRef]
- Weiss, A.; Littman, D.R. Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [Google Scholar] [CrossRef]
- D’Souza, W.N.; Chang, C.-F.; Fischer, A.M.; Li, M.; Hedrick, S.M. The Erk2 MAPK Regulates CD8 T Cell Proliferation and Survival. J. Immunol. 2008, 181, 7617–7629. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Farsakoglu, Y.; McDonald, B.; Kaech, S.M. Motility Matters: How CD8+ T-Cell Trafficking Influences Effector and Memory Cell Differentiation. Cold Spring Harb. Perspect. Biol. 2021, a038075. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Shen, J.; Kasmani, M.Y.; Topchyan, P.; Cui, W. Single-Cell Transcriptomics Reveals Core Regulatory Programs That Determine the Heterogeneity of Circulating and Tissue-Resident Memory CD8+ T Cells. Cells 2021, 10, 2143. https://doi.org/10.3390/cells10082143
Chen Y, Shen J, Kasmani MY, Topchyan P, Cui W. Single-Cell Transcriptomics Reveals Core Regulatory Programs That Determine the Heterogeneity of Circulating and Tissue-Resident Memory CD8+ T Cells. Cells. 2021; 10(8):2143. https://doi.org/10.3390/cells10082143
Chicago/Turabian StyleChen, Yao, Jian Shen, Moujtaba Y. Kasmani, Paytsar Topchyan, and Weiguo Cui. 2021. "Single-Cell Transcriptomics Reveals Core Regulatory Programs That Determine the Heterogeneity of Circulating and Tissue-Resident Memory CD8+ T Cells" Cells 10, no. 8: 2143. https://doi.org/10.3390/cells10082143