
Cytosolic HMGB1 Mediates LPS-Induced Autophagy in Microglia by Interacting with NOD2 and Suppresses Its Proinflammatory Function

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatment
2.2. siRNA Transfection
2.3. Generation of HMGB1 Knockdown (KD) Stable Cell Line
2.4. Nuclear Extract Preparation
2.5. Cell Culture Media Preparation
2.6. Immunofluorescence Staining
2.7. Immunoblotting
2.8. Immunoprecipitation-Linked Immunoblot Analysis
2.9. RNA Preparation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.10. Statistical Analyses
3. Results
3.1. Temporal Profile of Autophagy Induction in LPS-Treated Microglia
3.2. NOD2 Plays a Critical Role in LPS-Induced Autophagy in BV2 Cells
3.3. Cytoplasmic HMGB1 Binds to NOD2 and ATG16 in LPS-Treated BV2 Cells
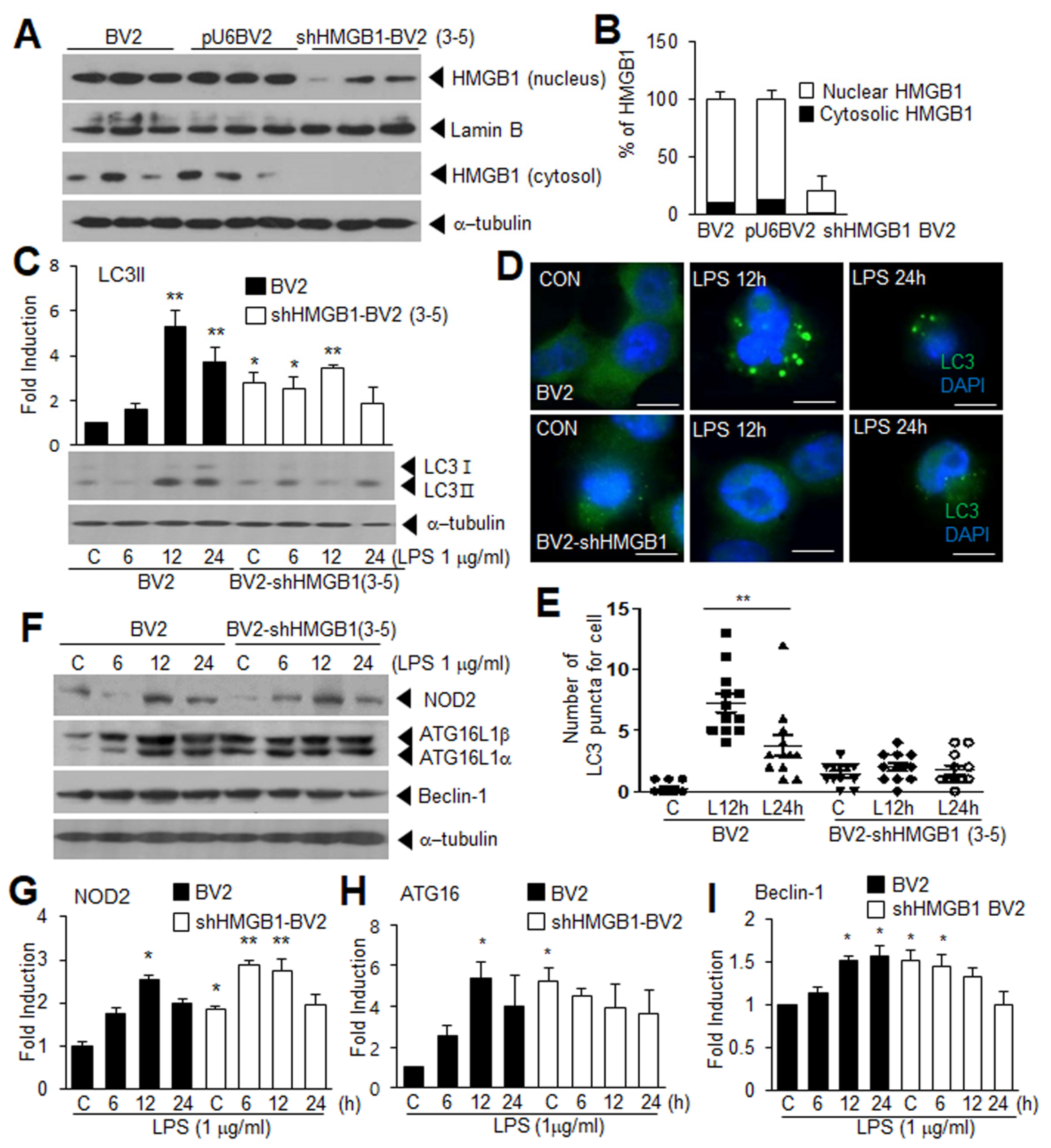
3.4. Suppression of LPS-Induced Autophagy in HMGB1 KD BV2 Cells
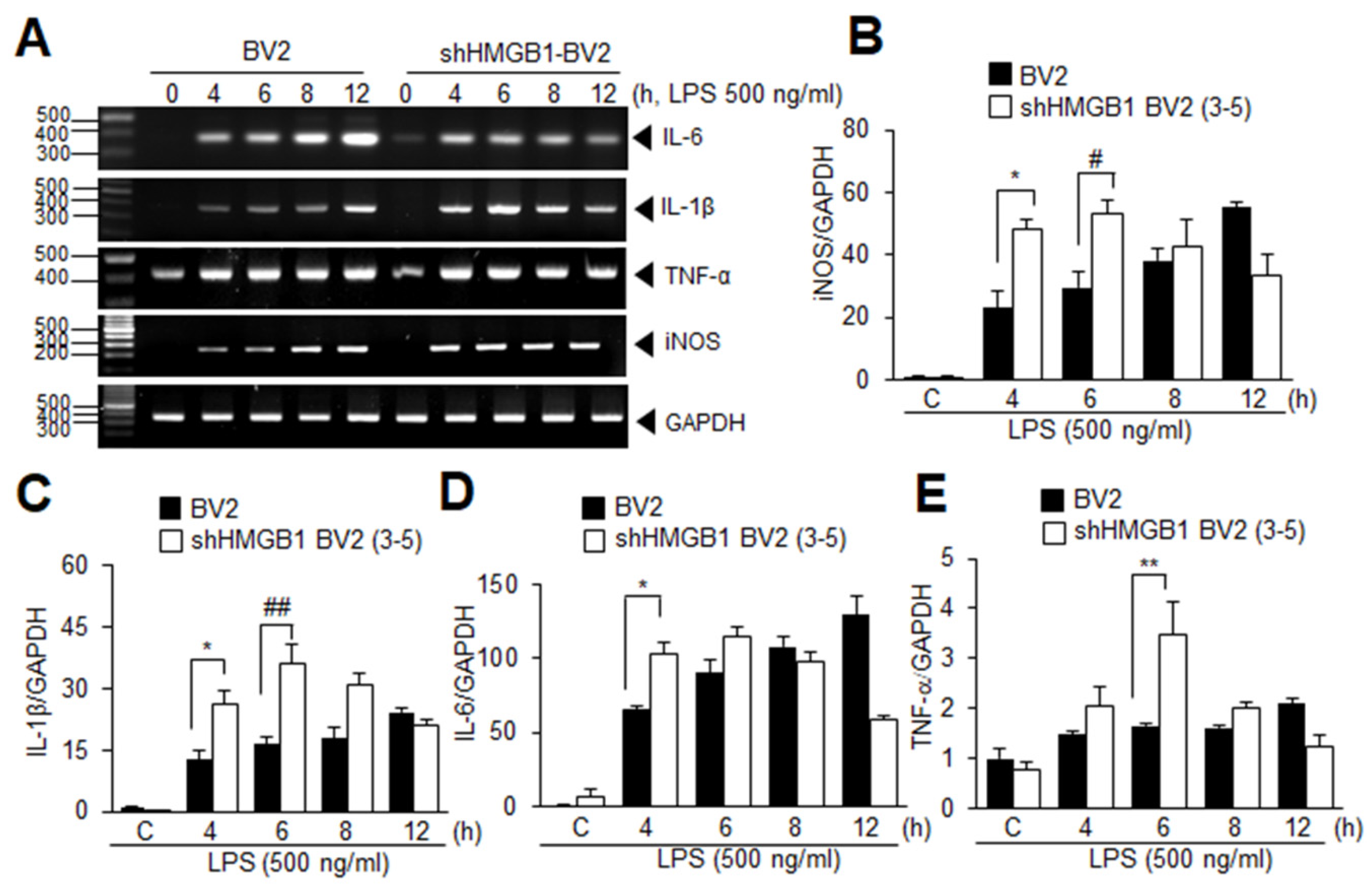
3.5. HMGB1 KD Augments Inflammation in LPS-Treated BV2 Cells at Early Time Points
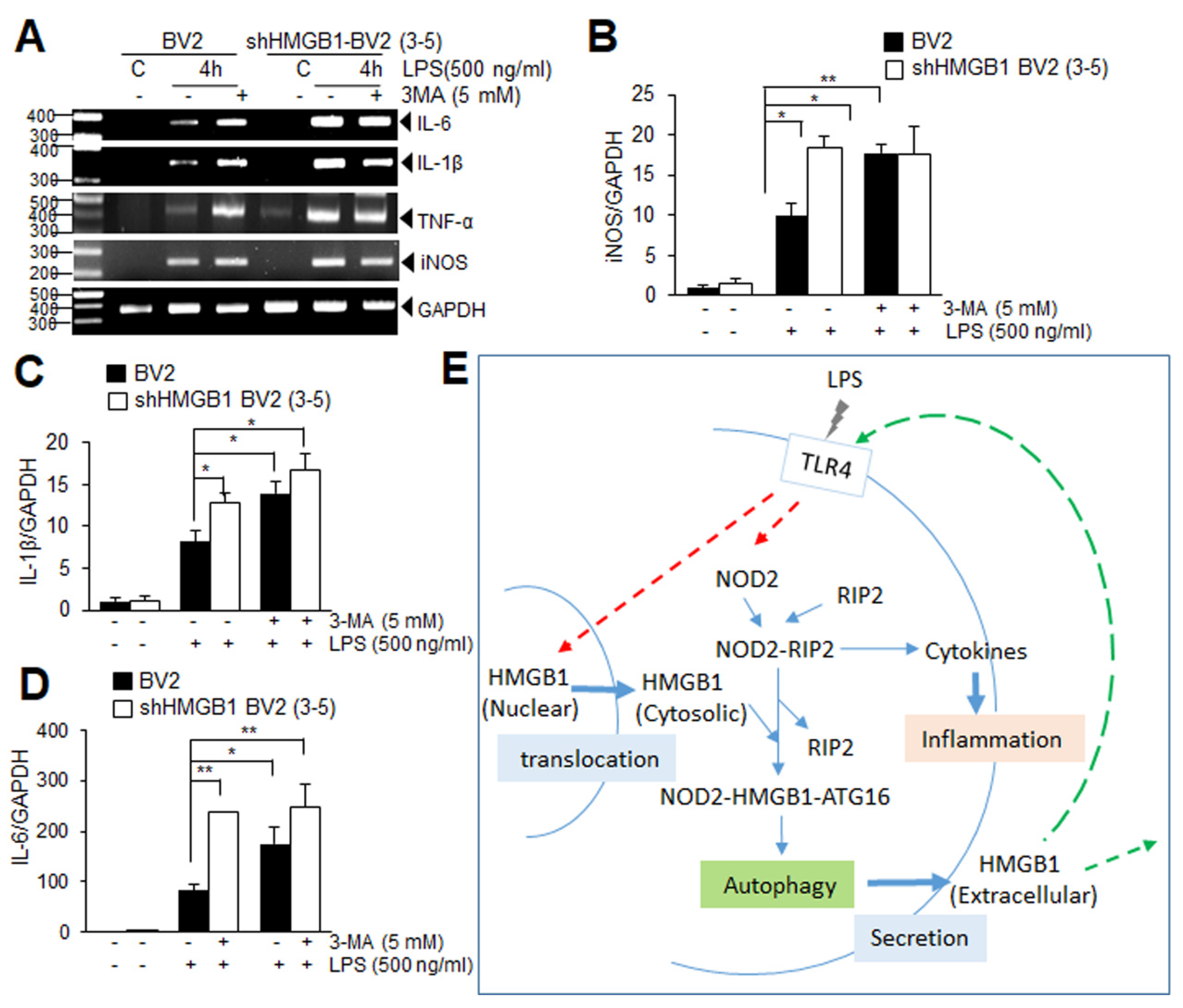
3.6. Suppression of Autophagy Augments Proinflammatory Response in LPS-Treated BV2 Cells at Early Time Points
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Javaherian, K.; Liu, J.F.; Wang, J.C. Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 1978, 199, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.A.; Marekov, L.N.; Beltchev, B.G. Involvement of protein HMG1 in DNA replication. FEBS Lett. 1984, 178, 153–155. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Gu, L.; Guo, S.; Wang, C.; Li, G.M. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J. Biol. Chem. 2004, 279, 20935–20940. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-B.; Choi, J.S.; Min-Hyung, L.; Nam, K.; Piao, C.-S.; Kim, S.-W.; Lee, M.-H.; Han, P.-L.; Park, J.-S.; Lee, J.-K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Investig. 2015, 125, 1098–1110. [Google Scholar] [CrossRef] [Green Version]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., 3rd; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Park, J.H.; Shaw, M.H.; Marina-Garcia, N.; Chen, G.; Kim, Y.G.; Nunez, G. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 2008, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [Green Version]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef] [Green Version]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kwak, M.S.; Lee, B.; Shin, J.M.; Aum, S.; Park, I.H.; Lee, M.G.; Shin, J.S. Secretory autophagy machinery and vesicular trafficking are involved in HMGB1 secretion. Autophagy 2021, 17, 2345–2362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Jin, Y.; Shin, J.H.; Kim, I.D.; Lee, H.K.; Park, S.; Han, P.L.; Lee, J.K. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti-inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol. Dis. 2012, 46, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Lee, H.K.; Lee, H.B.; Jin, Y.; Lee, J.K. Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci. Lett. 2014, 558, 159–163. [Google Scholar] [CrossRef]
- Kim, I.D.; Lee, H.; Kim, S.W.; Lee, H.K.; Choi, J.; Han, P.L.; Lee, J.K. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Dis. 2018, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Iyaswamy, A.; Wang, X.; Krishnamoorthi, S.; Kaliamoorthy, V.; Sreenivasmurthy, S.G.; Durairajan, S.S.K.; Song, J.-X.; Tong, B.C.-K.; Zhu, Z.; Su, C.-F.; et al. Theranostic F-SLOH mitigates Alzheimer’s disease pathology involving TFEB and ameliorates cognitive functions in Alzheimer’s disease models. Redox Biol. 2022, 51, 102280. [Google Scholar] [CrossRef]
- Homer, C.R.; Richmond, A.L.; Rebert, N.A.; Achkar, J.P.; McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010, 139, 1630–1641.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Fu, Y.; Ma, K.; Liu, J.; Liu, X. NOD2 regulates microglial inflammation through the TAK1-NF-kappaB pathway and autophagy activation in murine pneumococcal meningitis. Brain Res. Bull. 2020, 158, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J.; et al. LPS Induces Active HMGB1 Release from Hepatocytes into Exosomes through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Cheng, Y.; Meng, Q.; Wei, L.; Li, W.; Zhang, J.; Huang, S. The synergistic effect of NOD2 and TLR4 on the activation of autophagy in human submandibular gland inflammation. J. Oral Pathol. Med. 2019, 48, 87–95. [Google Scholar] [CrossRef]
- Wen, Z.; Fan, L.; Li, Y.; Zou, Z.; Scott, M.J.; Xiao, G.; Li, S.; Billiar, T.R.; Wilson, M.A.; Shi, X.; et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: Role in posthemorrhagic shock acute lung inflammation. J. Immunol. 2014, 193, 4623–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Wang, R.; Yang, S.; Zhang, X.; Dai, J. Role of Microglia Autophagy in Microglia Activation after Traumatic Brain Injury. World Neurosurg. 2017, 100, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Sheng, R.; Zhang, L.S.; Han, R.; Liu, X.Q.; Gao, B.; Qin, Z.H. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010, 6, 482–494. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Reeta, K.H.; Gupta, Y.K. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav. Brain Res. 2011, 225, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Teubner, B.J.W.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar] [CrossRef]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef]
- Tan, X.; Du, X.; Jiang, Y.; Botchway, B.O.A.; Hu, Z.; Fang, M. Inhibition of Autophagy in Microglia Alters Depressive-Like Behavior via BDNF Pathway in Postpartum Depression. Front. Psychiatry 2018, 9, 434. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Qiu, J.; Wang, P.; Liu, J.; Zhao, Y.; Jiang, F.; Lou, H. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav. Immun. 2021, 91, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.; Guo, S.; Pourteymour, S.; Xu, Q.; Gong, J.; Huang, Z.; Shen, Z.; Diabakte, K.; Cao, Z.; et al. Non-lethal sonodynamic therapy facilitates the M1-to-M2 transition in advanced atherosclerotic plaques via activating the ROS–AMPK–mTORC1–autophagy pathway. Redox Biol. 2020, 32, 101501. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Guo, J.; Wang, G.; Ni, Q.; Liu, X. Toll-Like Receptor 2–Mediated Autophagy Promotes Microglial Cell Death by Modulating the Microglial M1/M2 Phenotype. Inflammation 2020, 43, 101501. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Zhang, Y.; Li, J.; Zhang, J.; Li, Y.; Dang, C.; Li, C.; Fan, Y.; Yu, J.; Pei, Z.; et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012, 8, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, B.; Shen, H.; Lin, L.; Su, T.; Zhong, L.; Yang, Z. Autophagy Promotes Microglia Activation Through Beclin-1-Atg5 Pathway in Intracerebral Hemorrhage. Mol. Neurobiol. 2017, 54, 115–124. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (GenBank Accession No.) | Oligonucleotide Primer Sequences | PCR Product Size (bp) | Tm |
|---|---|---|---|
| IL-1β (M98820) | 5′-AGC ATC CAG CTT CAA ATC TCA-3′ 5′-CGA GGC ATT TTT GTT GTTCAT-3′ | 271 | 54 |
| IL-6 (BC132458) | 5′-GGA AAT GAG AAA AGA GTT GTG CAA T-3′ 5′-CCT TAG CCA CTC CTT CTG TGA-3′ | 370 | 54 |
| TNF-α (NM012675) | 5′-CTC AAA ACT CGA GTG ACA AG-3′ 5′-CTC CGT GAT GTC TAA GTA CT-3′ | 422 | 46 |
| iNOS (AY090567) | 5′-AAGAACGTGTTCACCATGAGG-3′ 5′-CCAGTAGCTGCCACTCTCATC-3′ | 363 | 54 |
| GAPDH (DQ403053) | 5′-TCA TTG ACC TCA ACT ACA TGG T-3′ 5′-CTA AGC AGT TGG TGG TGC AG-3′ | 252 | 54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-W.; Oh, S.-A.; Seol, S.-I.; Davaanyam, D.; Lee, J.-K. Cytosolic HMGB1 Mediates LPS-Induced Autophagy in Microglia by Interacting with NOD2 and Suppresses Its Proinflammatory Function. Cells 2022, 11, 2410. https://doi.org/10.3390/cells11152410
Kim S-W, Oh S-A, Seol S-I, Davaanyam D, Lee J-K. Cytosolic HMGB1 Mediates LPS-Induced Autophagy in Microglia by Interacting with NOD2 and Suppresses Its Proinflammatory Function. Cells. 2022; 11(15):2410. https://doi.org/10.3390/cells11152410
Chicago/Turabian StyleKim, Seung-Woo, Sang-A Oh, Song-I Seol, Dashdulam Davaanyam, and Ja-Kyeong Lee. 2022. "Cytosolic HMGB1 Mediates LPS-Induced Autophagy in Microglia by Interacting with NOD2 and Suppresses Its Proinflammatory Function" Cells 11, no. 15: 2410. https://doi.org/10.3390/cells11152410