
Oxygen Consumption and Basal Metabolic Rate as Markers of Susceptibility to Malignant Hyperthermia and Heat Stroke

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vivo Experiments
- -
- Basal indirect calorimetry. The basal metabolic rate of C57bl6, CASQ1-null, and Y522S mice was registered by indirect calorimetry. Briefly, each mouse was individually placed inside registration chamber of the calorimetry system (Pan Lab/Harvard Instruments, Cornellà, Spain) for 48 h. Measurement obtained during the first 24 h were discarded, because of the adaptation time needed for both mice and system. After adaptation, the volume of carbon dioxide production (VCO2) and the volume of oxygen consumption (VO2) were registered during the following 24 h using the Oxyletpro calorimeter system for mice (Pan Lab/Harvard Instruments, Cornellà, Spain). VCO2 production was then used to estimate O2 consumption and other parameters, as previously described [21]. The analysis of the respiratory quotient (RQ) and the energy expenditure (EE) were performed using Metabolism software (Pan Lab/Harvard Instruments, Spain). In detail, they were calculated as follows: RQ = VCO2/VO2; and EE = (3.815 + (1.32 × RQ)) × VO2 × 1.44. After collection, data were interpolated using Prism 9 software (GraphPad Software), to obtain 24 data (one per hour), and, finally, they were plotted as mean curve.
- -
- Heat-stress protocol and core temperature recording. To detect changes in metabolic rate and core temperature of mice during heat stress, animals were subjected to the heat-stress protocol during indirect calorimetry experiments. Briefly, mice were placed individually in the registration chambers intended for indirect calorimetry (Pan Lab/Harvard Instruments, Cornellà, Spain) and then they were placed inside an environmental chamber (custom made), where temperature and humidity can be controlled. Considering the different susceptibilities of CASQ1-null and Y522S mice to heat, we designed two different protocols of heat stress in order to avoid lethal crisis [18]. In detail, CASQ1-null mice were subjected to 41 °C for 20 min, while Y522S mice were exposed to 37 °C for 20 min. Two different WT mice groups were used as control, being exposed alternatively to 41 °C or 37 °C, for appropriated comparison with CASQ1-null mice and Y522S, respectively. During heat-stress protocol, indirect calorimetry parameters (see previous paragraph) were registered every 4 min and then plotted as VO2 consumption curve. In addition, immediately before and after the protocol, core temperature of each mouse was measured using a rectal thermometer (four channels thermometer TM-946, XS instruments, Milan, Italy). Finally, rise in core temperature during the test was plotted as delta variation.
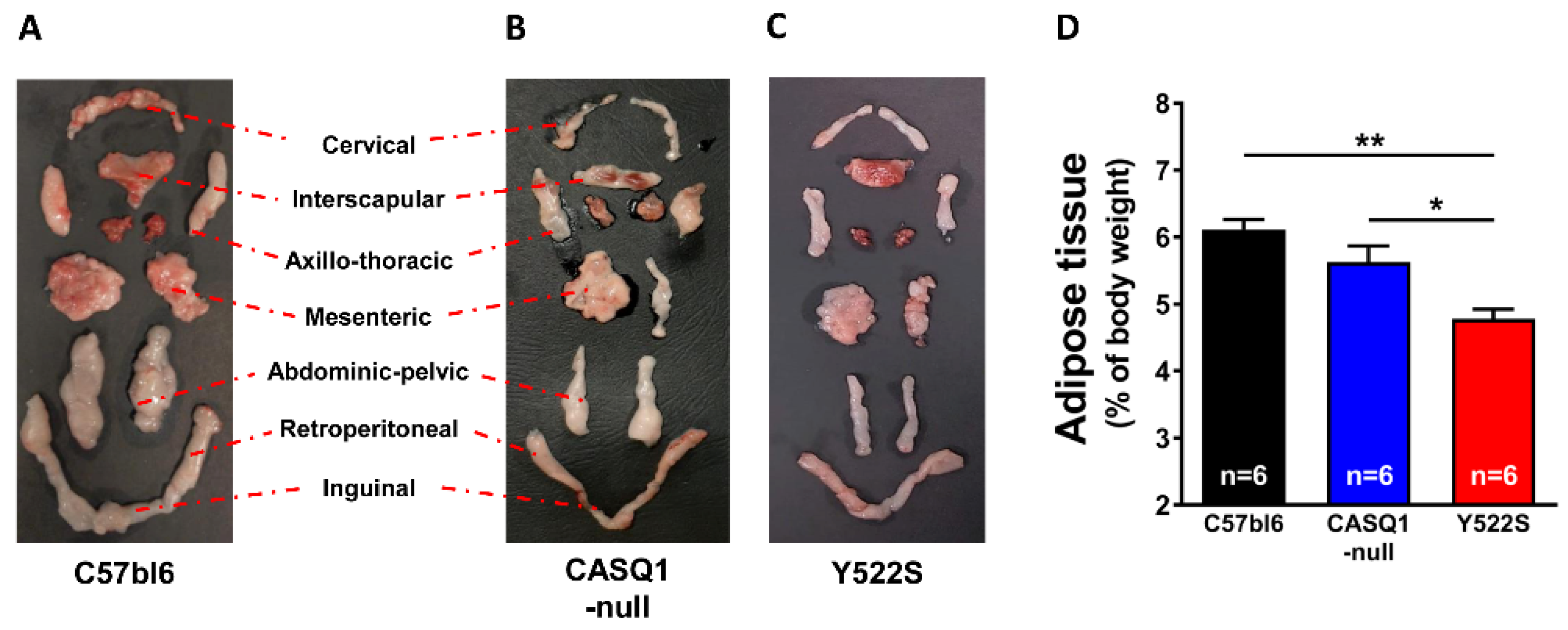
2.3. Body Composition
2.4. Western Blot
2.5. Cytochrome-c Oxidase Activity
2.6. Ultrastructural Analyses
2.7. Statistical Analysis
3. Results
3.1. CASQ1-Null and Y522S Mice Display Increased Metabolic Rate at Rest
3.2. Increased Oxygen Consumption and Body Temperature during Heat Stress in CASQ1-Null and Y522Smice
- (a)
- in CASQ1-null mice, the final temperature reached at the end of the stress protocol at 41 °C (was only slightly higher than in control animals): CASQ1-null: 36.49 ± 0.07 °C to 38.93 ± 0.13 °C (vs. WT: 36.0 ± 0.10 °C to 38.49 ± 0.07 °C), with no difference in ΔT (ΔT = +2.44 °C in CASQ1-null vs. ΔT = +2.47 °C in WT). These results seem in disagreement with previous publications showing the susceptibility of CASQ1-null mice to heat stress. However, we should consider that in previous publications the challenge was applied for 1 h instead of only 20 min.
- (b)
- in Y522S mice, both final temperature and ΔT were significantly greater at the end of the stress protocol at 37 °C: Y522S: 36.50 ± 0.07 °C to 40.61 ± 0.15 °C (vs. WT: 36.00 ± 0.11 °C to 37.98 ± 0.07 °C), with ΔT in Y522S = +4.11 °C (vs. ΔT in WT = +1.98 °C).
3.3. Analysis of Mitochondria and SR Revealed Structural Alterations and Changes in Protein Expression in CASQ1-Null and Y522S Mice
4. Discussion
4.1. Main Findings of the Study
4.2. Hypermetabolism as a Marker of Mice Susceptibility to MH and HS
4.3. Discussing the Role of Mitochondria in the Hypermetabolism of Y522S and CASQ1-Null Mice
4.4. Discussing the Role of SR in the Hypermetabolism of Y522S and CASQ1-Null Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Denborough, M. Malignant hyperthermia. Lancet 1998, 352, 1131–1136. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Phillips, M.S. Malignant hyperthermia. Science 1992, 256, 789–794. [Google Scholar] [CrossRef]
- Wedel, D.J.; Quinlan, J.G.; Iaizzo, P.A. Clinical effects of intravenously administered dantrolene. Mayo Clin. Proc. 1995, 70, 241–246. [Google Scholar] [CrossRef]
- Rosenberg, H.; Davis, M.; James, D.; Pollock, N.; Stowell, K. Malignant hyperthermia. Orphanet J. Rare Dis. 2007, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, H.; Pollock, N.; Schiemann, A.; Bulger, T.; Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Riazi, S.; Kraeva, N.; Hopkins, P.M. Updated guide for the management of malignant hyperthermia. Can. J. Anesth. 2018, 65, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.L.; Monnier, N.; Wolz, W.; Jung, M.; Reis, A.; Nuernberg, G.; Curran, J.L.; Monsieurs, K.; Stieglitz, P.; Heytens, L.; et al. A genome wide search for susceptibility loci in three European malignant hyperthermia pedigrees. Hum. Mol. Genet. 1997, 6, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Bouchama, A.; Knochel, J.P. Heat Stroke. N. Engl. J. Med. 2002, 346, 1978–1988. [Google Scholar] [CrossRef]
- Hopkins, P.M.; Ellis, F.R.; Halsall, P.J. Evidence for related myopathies in exertional heat stroke and malignant hyperthermia. Lancet 1991, 338, 1491–1492. [Google Scholar] [CrossRef]
- Bourdon, L.; Canini, F. On the nature of the link between malignant hyperthermia and exertional heatstroke. Med. Hypotheses 1995, 45, 268–270. [Google Scholar] [CrossRef]
- Ryan, J.F.; Tedeschi, L.G. Sudden unexplained death in a patient with a family history of malignant hyperthermia. J. Clin. Anesth. 1997, 9, 66–68. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Klingler, W.; Jurkat-Rott, K. Nonanesthetic malignant hyperthermia. Anesthesiology 2011, 115, 915–917. [Google Scholar] [CrossRef] [PubMed]
- McGeehin and Mirabelli. The potential impacts of climate variability and change on temperature-related morbidity and mortality in the United States. Environ. Health Perspect. 2001, 109, 185–189. [Google Scholar]
- Centers for Disease Control. Heat-related Deaths-United States, 1999–2003, Mortality and Morbidity Weekly Report published by the Centers for Disease Control. Weekly 2006, 55, 796–798. [Google Scholar]
- Chelu, M.G.; Goonasekera, S.A.; Durham, W.J.; Tang, W.; Lueck, J.D.; Riehl, J.; Pessah, I.N.; Zhang, P.; Bhattacharjee, M.B.; Dirksen, R.T.; et al. Heat- and Anesthesia-Induced Malignant Hyperthermia in an RyR1 Knock-in Mouse. FASEB J. 2006, 20, 329–330. [Google Scholar] [CrossRef]
- Durham, W.J.; Aracena-Parks, P.; Long, C.; Rossi, A.E.; Goonasekera, S.A.; Boncompagni, S.; Galvan, D.L.; Gilman, C.P.; Baker, M.R.; Shirokova, N.; et al. RyR1 S-Nitrosylation Underlies Environmental Heat Stroke and Sudden Death in Y522S RyR1 Knockin Mice. Cell 2008, 133, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Paolini, C.; Quarta, M.; Nori, A.; Boncompagni, S.; Canato, M.; Volpe, P.; Allen, P.D.; Reggiani, C.; Protasi, F. Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J. Physiol. 2007, 583, 767–784. [Google Scholar] [CrossRef]
- Dainese, M.; Quarta, M.; Lyfenko, A.D.; Paolini, C.; Canato, M.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Anesthetic- and Heat-Induced Sudden Death in Calsequestrin-1-Knockout Mice. FASEB J. 2009, 23, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Protasi, F.; Paolini, C.; Dainese, M. Calsequestrin-1: A New Candidate Gene for Malignant Hyperthermia and Exertional/Environmental Heat Stroke. J. Physiol. 2009, 587, 3095–3100. [Google Scholar] [CrossRef]
- Canato, M.; Scorzeto, M.; Giacomello, M.; Protasi, F.; Reggiani, C.; Stienen, G.J. Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc. Natl. Acad. Sci. USA 2010, 107, 22326–22331. [Google Scholar] [CrossRef] [Green Version]
- Even, P.C.; Nadkarni, N.A. Indirect Calorimetry in Laboratory Mice and Rats: Principles, Practical Considerations, Interpretation and Perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R459–R476. [Google Scholar] [CrossRef] [PubMed]
- Babušíková, E.; Kaplán, P.; Lehotský, J.; Jesenák, M.; Dobrota, D. Oxidative modification of rat cardiac mitochondrial membranes and myofibrils by hydroxyl radicals. Gen. Physiol. Biophys. 2004, 23, 327–335. [Google Scholar] [PubMed]
- Gilmour, M.V.; Lemberg, M.R.; Chance, B. Cytochrome oxidase and its derivatives. IX. Spectrophotometric studies on the rapid reaction of ferrous cytochrome c oxidase with molecular oxygen under conditions of complete and partial oxygenation. Biochim. Biophys Acta 1969, 172, 37–51. [Google Scholar] [CrossRef]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-Dependent Uncoupling of Mitochondria from Ca2+ Release Units in Skeletal Muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrangelo, L.; Michelucci, A.; Ambrogini, P.; Sartini, S.; Guarnier, F.A.; Fusella, A.; Zamparo, I.; Mammucari, C.; Protasi, F.; Boncompagni, S. Muscle activity prevents the uncoupling of mitochondria from Ca2+ Release Units induced by ageing and disuse. Arch Biochem. Biophys. 2019, 663, 22–33. [Google Scholar] [CrossRef]
- Loud, A.V. A method for the quantitative estimation of cytoplasmic structures. J. Cell Biol. 1968, 3, 481–487. [Google Scholar] [CrossRef]
- Mobley, B.A.; Eisenberg, B.R. Sizes of components in frog skeletal muscle measured by methods of stereology. J. Gen. Physiol. 1975, 66, 31–45. [Google Scholar] [CrossRef]
- Michelucci, A.; Paolini, C.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Strenuous Exercise Triggers a Life-Threatening Response in Mice Susceptible to Malignant Hyperthermia. FASEB J. 2017, 31, 3649–3662. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Paolini, C.; Canato, M.; Wei-Lapierre, L.; Pietrangelo, L.; de Marco, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Antioxidants Protect Calsequestrin-1 Knockout Mice from Halothane- and Heat-Induced Sudden Death. Anesthesiology 2015, 123, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Boncompagni, S.; Canato, M.; Reggiani, C.; Protasi, F. Estrogens Protect Calsequestrin-1 Knockout Mice from Lethal Hyperthermic Episodes by Reducing Oxidative Stress in Muscle. Oxid. Med. Cell. Longev. 2017, 2017, 6936897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnier, F.A.; Michelucci, A.; Serano, M.; Pietrangelo, L.; Pecorai, C.; Boncompagni, S.; Protasi, F. Aerobic Training Prevents Heatstrokes in Calsequestrin-1 Knockout Mice by Reducing Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 4652480. [Google Scholar] [CrossRef] [Green Version]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Hamilton, S.L.; Dirksen, R.T.; Franzini-Armstrong, C.; Protasi, F. Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. USA 2009, 106, 21996–22001. [Google Scholar] [CrossRef] [Green Version]
- Paolini, C.; Quarta, M.; Wei-LaPierre, L.; Michelucci, A.; Nori, A.; Reggiani, C.; Dirksen, R.T.; Protasi, F. Oxidative Stress, Mitochondrial Damage, and Cores in Muscle from Calsequestrin-1 Knockout Mice. Skelet. Muscle 2015, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signaling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.; Affourtit, C.; Esteves, T.; Green, K.; Lambert, A.; Miwa, S.; Pakay, J.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Rudolf, R.; Mongillo, M.; Magalhães, P.J.; Pozzan, T. In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J. Cell Biol. 2004, 166, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Patron, M.; Granatiero, V.; Rizzuto, R. Molecules and roles of mitochondrial calcium signaling. Biofactors 2011, 37, 219–227. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Canato, M.; Capitanio, P.; Cancellara, L.; Leanza, L.; Raffaello, A.; Reane, D.V.; Marcucci, L.; Michelucci, A.; Protasi, F.; Reggiani, C. Excessive Accumulation of Ca2+ in Mitochondria of Y522S-RYR1 Knock-in Mice: A Link Between Leak From the Sarcoplasmic Reticulum and Altered Redox State. Front. Physiol. 2019, 10, 1142. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Ježek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Uncoupling: New approaches to an old problem of bioenergetics. Biochim. Biophys. Acta 1998, 1363, 100–124. [Google Scholar] [CrossRef] [Green Version]
- Goglia, F.; Skulachev, V.P. A function for novel uncoupling proteins: Antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. 2003, 17, 1585–1591. [Google Scholar] [CrossRef] [Green Version]
- Schrauwen, P.; Hoeks, J.; Hesselink, M.K. Putative function and physiological relevance of the mitochondrial uncoupling protein-3: Involvement in fatty acid metabolism? Prog. Lipid Res. 2006, 45, 17–41. [Google Scholar] [CrossRef]
- Hilse, K.E.; Rupprecht, A.; Egerbacher, M.; Bardakji, S.; Zimmermann, L.; Wulczyn, A.; Pohl, E.E. The Expression of Uncoupling Protein 3 Coincides with the Fatty Acid Oxidation Type of Metabolism in Adult Murine Heart. Front. Physiol. 2018, 9, 747. [Google Scholar] [CrossRef]
- Toyoshima, C.; Inesi, G. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 269–292. [Google Scholar] [CrossRef]
- Block, B. Thermogenesis in muscle. Annu. Rev. Physiol. 1994, 56, 535–577. [Google Scholar] [CrossRef]
- da Costa, D.; Landeira-Fernandez, A. Thermogenic activity of the Ca2+-ATPase from blue marlin heater organ: Regulation by KCl and temperature. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1460–R1468. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serano, M.; Pietrangelo, L.; Paolini, C.; Guarnier, F.A.; Protasi, F. Oxygen Consumption and Basal Metabolic Rate as Markers of Susceptibility to Malignant Hyperthermia and Heat Stroke. Cells 2022, 11, 2468. https://doi.org/10.3390/cells11162468
Serano M, Pietrangelo L, Paolini C, Guarnier FA, Protasi F. Oxygen Consumption and Basal Metabolic Rate as Markers of Susceptibility to Malignant Hyperthermia and Heat Stroke. Cells. 2022; 11(16):2468. https://doi.org/10.3390/cells11162468
Chicago/Turabian StyleSerano, Matteo, Laura Pietrangelo, Cecilia Paolini, Flavia A. Guarnier, and Feliciano Protasi. 2022. "Oxygen Consumption and Basal Metabolic Rate as Markers of Susceptibility to Malignant Hyperthermia and Heat Stroke" Cells 11, no. 16: 2468. https://doi.org/10.3390/cells11162468