
Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy

 ,
,
Abstract
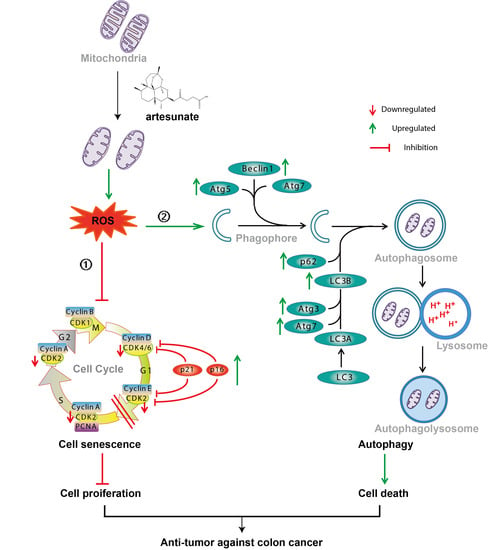
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Drugs and Antibodies
2.2. Cell Viability Assay
2.3. ROS Detection and Quantification
2.4. Cell Apoptosis Detection
2.5. Cell Cycle Analysis
2.6. Senescence-Associated β-Galactosidase (SA-β-gal) Activity Assay
2.7. Cell Proliferation Assay
2.8. Mitochondrial Membrane Potential Assay
2.9. Mitophagy Detection
2.10. Lysosomal Acidification Detection
2.11. Plasmid Transfection and Enhanced Green Fluorescent Protein (EGFP)-LC3B Quantification
2.12. Cellular Calcium Detection
2.13. Western Blotting
2.14. CT26-Derived Implanted Tumor Model
2.15. Immunohistochemistry (IHC)
2.16. Statistical Analysis
3. Results
3.1. Artesunate Inhibited Cell Viabilities Partially due to Excessive Mitochondrial ROS
3.2. Artesunate-Induced Mild Apoptosis Did Not Help to Inhibit the Viability of SW480 and HCT116 Cells
3.3. Artesunate Induced Cell Senescence ROS-Dependently
3.4. Artesunate Promoted Mitochondrial Depolarization and Mitophagy
3.5. Artesunate Induced Autophagy to Inhibit Cell Proliferation Mainly due to the Excessive ROS Generation
3.6. Artesunate Induced Endoplasmic Reticulum Stress (ER Stress) and Activated the Unfolded Protein Response (UPR) via IRE1α Signaling
3.7. Artesunate Increased the Cellular Free Ca2+ Level
3.8. Artesunate Inhibited the Growth of CT26-Derived Tumors in Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLO-BOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABA(A) Receptor Signaling and Impair α Cell Identity. Cell 2017, 168, 86–100. [Google Scholar] [CrossRef]
- Fan, M.; Li, Y.; Yao, C.; Liu, X.; Liu, J.; Yu, B. DC32, a Dihydroartemisinin Derivative, Ameliorates Collagen-Induced Arthritis Through an Nrf2-p62-Keap1 Feedback Loop. Front. Immunol. 2018, 9, 2762. [Google Scholar] [CrossRef]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Krishna, S.; Ganapathi, S.; Ster, I.C.; Saeed, M.E.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P.G.; Efferth, T.; et al. A Randomised, Double Blind, Placebo-Controlled Pilot Study of Oral Artesunate Therapy for Colorectal Cancer. eBioMedicine 2014, 2, 82–90. [Google Scholar] [CrossRef]
- Gopalakrishnan, A.M.; Kumar, N. Antimalarial Action of Artesunate Involves DNA Damage Mediated by Reactive Oxygen Species. Antimicrob. Agents Chemother. 2015, 59, 317–325. [Google Scholar] [CrossRef]
- Wilson, C.; Muñoz-Palma, E.; González-Billault, C. From birth to death: A role for reactive oxygen species in neuronal de-velopment. Semin. Cell Dev. Biol. 2018, 80, 43–49. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Reactive oxygen species, oxidative damage and cell death. In Immunity and Inflammation in Health and Disease; Chatterjee, S., Jungraithmayr, W., Bagchi, D., Eds.; Elsevier: Amsterdam, Netherlands, 2018; pp. 45–55. [Google Scholar]
- Schmidt, H.H.; Ghezzi, P.; Cuadrado, A. Reactive Oxygen Species: Network Pharmacology and Therapeutic Applications; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of Reactive Oxygen Species: An Emerging Approach for Cancer Therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Gao, W.; Xiao, F.; Wang, X.; Chen, T. Artemisinin induces A549 cell apoptosis dominantly via a reactive oxygen spe-cies-mediated amplification activation loop among caspase-9, -8 and -3. Apoptosis 2013, 18, 1201–1213. [Google Scholar] [CrossRef]
- Zhang, Q.; Yi, H.; Yao, H.; Lu, L.; He, G.; Wu, M.; Zheng, C.; Li, Y.; Chen, S.; Li, L.; et al. Artemisinin Derivatives Inhibit Non-small Cell Lung Cancer Cells Through Induction of ROS-dependent Apoptosis/Ferroptosis. J. Cancer 2021, 12, 4075–4085. [Google Scholar] [CrossRef]
- Li, S.; Chaudhary, S.C.; Zhao, X.; Gaur, U.; Fang, J.; Yan, F.; Zheng, W. Artemisinin Protects Human Retinal Pigmented Epi-thelial Cells Against Hydrogen Peroxide-induced Oxidative Damage by Enhancing the Activation of AMP-active Protein Ki-nase. Int. J. Biol. Sci. 2019, 15, 2016–2028. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; Mcnamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; Dimaio, D.; Hwang, E.S. Senes-cence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Banks, H.T.; Sutton, K.L.; Thompson, C.; Bocharov, G.; Roose, D.; Schenkel, T.; Meyerhans, A. Estimation of Cell Proliferation Dynamics Using CFSE Data. Bull. Math. Biol. 2010, 73, 116–150. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Miettinen, T.P.; Peltier, J.; Härtlova, A.; Gierliński, M.; Jansen, V.M.; Trost, M.; Björklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Gallage, S.; Gil, J. Mitochondrial Dysfunction Meets Senescence. Trends Biochem. Sci. 2016, 41, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To Be or Not to Be? How Selective Autophagy and Cell Death Govern Cell Fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2017, 69, 62–72. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Ermolenko, E.; Savidov, N.; Gloriozova, T.A.; Proroikov, V.V. Antiprotozoal and Antitumor Activity of Natural Polycyclic Endoperoxides: Origin, Structures and Biological Activity. Molecules 2021, 26, 686. [Google Scholar] [CrossRef]
- Bu, M.; Yang, B.B.; Hu, L. Natural Endoperoxides as Drug Lead Compounds. Curr. Med. Chem. 2016, 23, 383–405. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.-X.; Jiang, X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2019, 27, 242–254. [Google Scholar] [CrossRef]
- Fulda, S.; Kögel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef]
- Graceffa, V. Therapeutic Potential of Reactive Oxygen Species: State of the Art and Recent Advances. SLAS Technol. Transl. Life Sci. Innov. 2020, 26, 140–158. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Gupta, R.; Sinha, S.; Paul, R.N. The impact of microsatellite stability status in colorectal cancer. Curr. Probl. Cancer 2018, 42, 548–559. [Google Scholar] [CrossRef]
- Gonnella, R.; Montani, M.G.; Guttieri, L.; Romeo, M.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Lett. 2020, 490, 76–88. [Google Scholar] [CrossRef]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1–JNK pathway. Cell Death Differ. 2011, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Koyanagi, S.; Okamoto, A.; Suzuki, S.O.; Matsunaga, N.; Ohdo, S. Stress-Regulated Transcription Factor ATF4 Promotes Neoplastic Transformation by Suppressing Expression of the INK4a/ARF Cell Senescence Factors. Cancer Res. 2012, 72, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hu, T.; Liang, Y.; Li, P.; Chen, X.; Zhang, J.; Ma, Y.; Hao, Q.; Wang, J.; Zhang, P.; et al. Neddylation Inhibition Ac-tivates the Extrinsic Apoptosis Pathway through ATF4-CHOP-DR5 Axis in Human Esophageal Cancer Cells. Clin. Cancer Res. 2016, 22, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Meng, Y.; Sun, Q.; Zhang, Z.; Guo, X.; Sheng, X.; Tai, G.; Cheng, H.; Zhou, Y. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis. 2016, 7, e2334. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2014, 4, 184–192. [Google Scholar] [CrossRef]
- Yan, C.; Liu, J.; Gao, J.; Sun, Y.; Zhang, L.; Song, H.; Xue, L.; Zhan, L.; Gao, G.; Ke, Z.; et al. IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Zhan, Y.; Wang, K.; Li, Q.; Zou, Y.; Chen, B.; Gong, Q.; Ho, H.I.; Yin, T.; Zhang, F.; Lu, Y.; et al. The Novel Autophagy Inhibitor Alpha-Hederin Promoted Paclitaxel Cytotoxicity by Increasing Reactive Oxygen Species Accumulation in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3221. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Gan, S.; Zhuang, X.; Chen, Y.; Lu, L.; Wang, Y.; Qi, X.; Feng, Q.; Huang, Q.; Du, B.; et al. Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy. Cells 2022, 11, 2472. https://doi.org/10.3390/cells11162472
Huang Z, Gan S, Zhuang X, Chen Y, Lu L, Wang Y, Qi X, Feng Q, Huang Q, Du B, et al. Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy. Cells. 2022; 11(16):2472. https://doi.org/10.3390/cells11162472
Chicago/Turabian StyleHuang, Zhiying, Shu Gan, Xuerong Zhuang, Yao Chen, Linlin Lu, Ying Wang, Xiaoxiao Qi, Qian Feng, Qiuju Huang, Biaoyan Du, and et al. 2022. "Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy" Cells 11, no. 16: 2472. https://doi.org/10.3390/cells11162472