

Bioinformatic Analyses of Broad H3K79me2 Domains in Different Leukemia Cell Line Data Sets
by
 , and
, and
Prerna Sharma
1,
Hedieh Sattarifard
1,
Narges Fatemiyan
1,
Ted M. Lakowski
2,3 and
James R. Davie
1,3,* 1
Department of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB R3E 0J9, Canada
2
College of Pharmacy Pharmaceutical Analysis Laboratory, University of Manitoba, Winnipeg, MB R3E 0V9, Canada
3
CancerCare Manitoba Research Institute, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada
*
Author to whom correspondence should be addressed.
Cells 2022, 11(18), 2830; https://doi.org/10.3390/cells11182830
Submission received: 27 June 2022
/
Revised: 6 September 2022
/
Accepted: 7 September 2022
/
Published: 10 September 2022
(This article belongs to the Special Issue Transcriptional Regulatory Mechanisms in Health and Disease)
Abstract
:A subset of expressed genes is associated with a broad H3K4me3 (histone H3 trimethylated at lysine 4) domain that extends throughout the gene body. Genes marked in this way in normal cells are involved in cell-identity and tumor-suppressor activities, whereas in cancer cells, genes driving the cancer phenotype (oncogenes) have this feature. Other histone modifications associated with expressed genes that display a broad domain have been less studied. Here, we identified genes with the broadest H3K79me2 (histone H3 dimethylated at lysine 79) domain in human leukemic cell lines representing different forms of leukemia. Taking a bioinformatic approach, we provide evidence that genes with the broadest H3K79me2 domain have known roles in leukemia (e.g., JMJD1C). In the mixed-lineage leukemia cell line MOLM-13, the HOXA9 gene is in a 100 kb broad H3K79me2 domain with other HOXA protein-coding and oncogenic long non-coding RNA genes. The genes in this domain contribute to leukemia. This broad H3K79me2 domain has an unstable chromatin structure, as was evident by enhanced chromatin accessibility throughout. Together, we provide evidence that identification of genes with the broadest H3K79me2 domain will aid in generating a panel of genes in the diagnosis and therapeutic treatment of leukemia in the future.
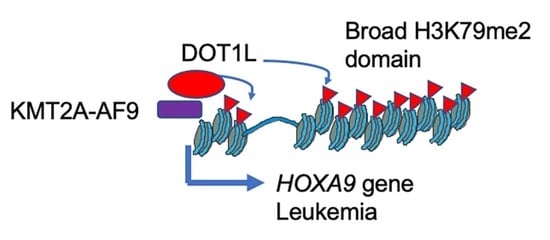
1. Introduction
Leukemia, a blood-borne cancer, typically results from genetic and epigenetic events that deregulate gene expression. The four main types of leukemia include acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphocytic leukemia (ALL), and chronic lymphocytic leukemia. Epigenetics studies molecules and mechanisms that modify gene-activity states in the context of the same DNA sequence [1]. Epigenetic modifiers of chromatin include players categorized as writers, readers, and erasers. Writers introduce chemical modifications on DNA and histones; readers are effector proteins containing, for example, methyl-lysine-binding motifs, which identify and interpret those chemical changes; erasers are enzymes that remove the chemical tags.
Mixed-lineage leukemia (MLL) is a form of leukemia occurring predominantly in children that is a consequence of a chromosomal rearrangement of 11q23 [2,3,4,5]. These rearrangements can fuse otherwise independent genes into a hybrid gene, resulting in fusion proteins containing domains initially encoded by separate genes. In MLL, the KMT2A gene, which codes for an H3K4 lysine methyltransferase [6], fuses with another protein-coding gene, generating an oncoprotein [7]. In this fusion, KMT2A retains its N-terminus (KMT2AN), which is placed in-frame with the C-terminus of one of several fusion partners, creating an oncogenic chimeric protein [7,8]. Hence, the KMT2A fusion protein is composed of KMT2AN and a fusion partner. KMT2AN does not contain the H3K4 methylation SET domain but it does contain a CXXC domain that is involved in binding to nonmethylated CpG-rich promoter regions [2,4,6,8,9,10]. Although many different KMT2A fusion partners have been characterized, some of the most common ones are AFF1/AF4, MLLT10/AF10, MLLT3/AF9, and MLLT1/ENL [3]. For example, the cell lines MOLM-13 and MOLM-14 have the fusion protein KMT2A-AF9 [11]. AF9 then recruits the methyltransferase, Disruptor of Telomeric Silencing 1-Like (DOT1L) [5].
DOT1L is a lysine methyltransferase that catalyzes mono-, di-, and tri-methylations of histone H3 at lysine 79 (H3K79me1, me2, or me3) using S-adenosylmethionine (SAM) as a cofactor in its active site [12,13,14]. Prior ubiquitination of H2B is observed to increase H3K79 methylation efficiency. Genome-wide studies using chromatin immunoprecipitation (ChIP) and ChIP sequencing (ChIP-Seq) indicate that H3K79me2 is associated with active gene transcription in humans [13,15,16]. Knockdown studies of DOT1L show a complete loss of H3K79 methylation, indicating that DOT1L is the only known H3K79 methyltransferase [13].
DOT1L has a C-terminal non-specific DNA-binding domain that aids in its normal interactions with nucleosomes [12]. In AML, DOT1L is aberrantly recruited to inappropriate genomic locations through its interaction with AF9, ENL, AF10, and AF17 [15]. AF9 and ENL, readers that bind acetylated H3K9, recruit DOT1L to open chromatin regions [16,17,18]. The fusion protein KMT2A-AF9 aberrantly recruits DOT1L to genes that are typically the target of KMT2A, such as the HOX genes. This chimeric protein loses its H3K4 methylation function while gaining H3K79 methylation, resulting in high expression of HOX genes like HOXA9. The aberrantly high HOXA9 expression results in MLL, and knockdown of HOXA9 expression results in reduced proliferation and apoptosis [19]. DOT1L was hypothesized to directly interact with the carboxy-terminal domain of the largest RNAP II subunit during transcriptional elongation [13] because of its colocalization with RNAP II [20]. However, recent ChIP-seq, PRO-seq, and 4sUDRB-Seq analyses showed that DOT1L is unlikely to have a role in elongation; instead, it may be involved in transcriptional initiation [20].
Histone modifications associated with the transcriptionally active state of genes include H3K4me1, H3K4me3, and H3K27ac, among many others. For a subset of genes, active marks such as H3K4me3, H3K27ac, and H3K79me2 are associated with a broad stretch of the gene body starting at the 5′ end of the gene [21,22]. This was initially observed with H3K4me3 and was named the broad H3K4me3 domain [22,23]. We and others have shown that genes with the broadest H3K4me3 domains identify cancer-related genes that are specific to the cancer-cell type [24]. For example, Belhocine et al. identified genes with the broad H3K4me3 domain in human thymic T-cell populations versus in T-Acute Lymphoblastic Leukemia (T-ALL) samples [25]. Expressed genes marked with the broad H3K4me3 domain in normal cells were cell-identity genes and tumor-suppressor genes, whereas in cancer, genes marked in this way were oncogenes involved in critical cancer processes.
To date, H3K4me3 is the most-studied broad H3K4me3 domain. Recently, we reported that H3K4ac is also arranged in a broad domain that is associated with critical genes and regulatory regions involved in breast cancer [24]. In this bioinformatic study, we identified genes with the broadest H3K79me2 domain that were unique to different types of leukemic cells. We show that the genes with the broadest H3K79me2 domain have known roles in leukemia and identify genes that should be further characterized as potential drivers of leukemia.
2. Results
2.1. Identification of Genes with a Broad H3K79me2 Domain in Leukemic Cells
To determine whether human leukemic cells have a broad H3K79me2 domain, we first analyzed publicly available H3K79me2 ChIP-Seq data using a representative acute myeloid leukemia cell line (MLL-type) MOLM-13. The MOLM-13 cell line was derived from a 20-year-old male with acute monocytic leukemia (AML-M5a) resulting from the chromosomal insertion (11;9)(q23; p22p23) [11]. This cell line expresses the KMT2A-AF9 fusion protein, which aberrantly recruits the DOT1L H3K79 methyltransferase to HOX genes, resulting in their overexpression. Appropriate GEO files (GSE113191, GSE149183) were identified, and the FASTQ files were entered in the Partek Flow program. The MACS2 peak caller was set in the broad mode. Figure 1 shows that most of the H3K79me2 presented as a narrow peak at the 5′ end of the gene and a lesser number of H3K79me2 peaks were quite broad, up to 471,096 bp long. The gene with the broadest H3K79me2 domain was JMJD1C (chr10:63,163,632-63,634,728) (Figure S1), a known target of the KMT2A-AF9 fusion protein and an oncogene essential to the self-renewal of AML cells [26]. The breadth of the top 5% of broad H3K79me2 domains in MOLM-13 cells ranged from 25,846 to 471,096 bp.
Next, we repeated these analyses with cell lines representing chronic myeloid leukemia (K562), adult acute lymphoblastic leukemia (Loucy), and childhood acute lymphoblastic leukemia (DND-41). K562 cells were derived from a 53-year-old female patient in blast crisis. It was the first immortalized CML cell line, with a hypotriploid karyotype [27,28,29]. Loucy cells were derived from the peripheral blood of a 38-year-old European female adult with T-cell ALL with a t(16:20) and 5q- chromosomal aberrations [30]. DND-41 was derived from the peripheral blood of a 13-year-old male with T-cell ALL and carrying a p53 mutation [31]. None of the cell lines that we used have known mutations to DOT1L. We used the following GEO files—K562, GSE29611; Loucy, GSE96166; DND-41, GSE29611—and loaded the FASTQ files into Partek Flow for analysis as described for MOLM-13 cells. The breadth of the top 5% broadest H3K79me2 peaks varied among the cell lines (DND-41, 27,560–340,185 bp; K562, 18,304–191,997 bp; Loucy, 3241–8467 bp), with the breath of the H3K79me2 region being the shortest in the Loucy cell line (Table S1). Other available H3K79me2 ChIP-Seq data for the Loucy cell line (GEO file GSE76745) were analyzed, and the maximal length of the broad H3K79me2 domains was found to be 3400 to 6077 bp. Together, these results are consistent with the Loucy cell line having the shortest of the broad H3K79me2 domains.
The gene with the broadest H3K79me2 domain in each cell line was SPG7 (chr16:89,301,571-89,493,568) in K562; PTPRC (chr1:198,683,545-198,692,012) in Loucy and Neuroligin 4 X-linked (NLGN4X) (chrX:5,892,371-6,232,556) in DND-41. SPG7 matrix AAA peptidase subunit paraplegin (SPG7) codes for a mitochondrial metalloprotease protein and is upregulated in colorectal cancer [32]. Protein tyrosine phosphatase receptor-type C (PTPRC) gene, a member of the protein tyrosine phosphatase (PTP) family, is abnormally expressed in leukemic cells [33]. Neuroligin 4 X-linked (NLGN4X) codes for a type-B carboxylesterase/lipase protein family member and is associated with poor relapse-free survival in triple-negative breast cancer [34]. Although these genes have known roles in cancer, only the PTPRC gene has a documented role in leukemic cells.
GO-term analysis (biological process) for genes with the top 5% broad H3K79me2 domains in the four cell lines was carried out using Enrichr (Table 1) [35,36]. Phosphorylation as a biological process was prominent for genes in MOLM-13, DND-41, and K562 cells, whereas the Loucy cell line had genes with biological processes involved in RNA splicing.
In comparison, we determined the GO terms for genes that had the shortest H3K79me2 regions (bottom 5%). Protein phosphorylation was among the most significant biological processes for the four cell lines. The most significant GO terms for the bottom 5% of the genes in the four cell lines were as follows: MOLM-13, protein phosphorylation; DND-41, peptidyl-serine phosphorylation; K562, epidermal growth factor receptor-signaling pathway; Loucy, regulation of cell–matrix adhesion.
Publicly available RNA sequencing (RNA-Seq) data for the four cell lines were analyzed with Partek Flow. Appropriate RNA-Seq files were identified (K562-GSE90239, GSE78556, GSE140322; Loucy-GSE155337; DND41-GSE173867, GSE116873; MOLM-13-GSE113191, GSE181003). The RNA-Seq was done in duplicate or triplicate in each study. The FASTQ files were uploaded into the Partek Flow program. For DOT1L expression, the enzyme was expressed at a greater level in MOLM-13 cells compared to the other three cell lines (based on TPM: MOLM-13, 27.0; DND-41, 3.8; K562, 2.5; Loucy, 5.2). Following normalization using TPM (Table S1), the transcript levels in each RNA-Seq study were binned into five groups. The genes in the top 5% broad H3K79me2 domains for each cell line were then cross-referenced with the transcript level of that gene. Approximately 60% of the genes with the broad H3K79me2 domain had transcript levels in the top 40% (K562, 61%; Loucy, 61%; DND-41, 56%; MOLM-13, 61%), and about 80% of the genes with the broad H3K79me2 domain were in the top 60% (K562, 88%; Loucy, 81%; DND-41, 84%; MOLM-13, 89%). These observations show that the genes with the broadest H3K79me2 domains tend to exhibit higher transcript levels.
The top 5% broad H3K79me2 domains in the four leukemic cell lines that were unique to that cell line were identified (Table S1) and are displayed in Figure 2. The GO terms for these genes (biological process) included “negative regulation of myeloid-cell differentiation (MOLM-13)”, “mitotic-chromosome condensation (DND-41)”, “regulation of organelle organization (K562)”, and “RNA splicing (Loucy)” (Table 2).
Figure 3 shows H3K79me2 tracks for genes that were in the top 5% of broadest H3K79me2 domains. A broad H3K79me2 region was observed over the DACH1 gene in MOLM-13 cells but not in the other cell lines (Figure 3A). The dachshund-family transcription factor 1 (DACH1) gene is upregulated by the KMT2A-AF9 fusion protein [37]. DACH1 is a coactivator of HOXA9 and is involved in myeloid leukemogenesis. An intense H3K79me2 region was present over the zinc-finger protein 521 (ZNF521) gene in MOLM-13 and to a lesser extent in Loucy (Figure 3B). DND-41 and K562 cells did not have H3K79me2 over the ZNF521 gene. All four lines showed a sharp H3K79me2 peak at the 5′ end of the SS18 gene, which is next to the ZNF521 gene. In contrast, the PSMA8 gene did not have an associated H3K79me2 peak. KMT2A-AF9 directly interacts with the ZNF521 promoter, activating its transcription [38]. ZNF521 is a critical effector of KMT2A fusion proteins in blocking myeloid differentiation and can be used as a potential therapeutic target for this leukemia subtype [38]. DND-41 and, to a much lesser extent in Loucy, has a broad H3K79me2 domain over the NLGN4X gene (Figure 3C). Neuroligin 4 X-linked (NLGN4X) is a protein-coding gene that encodes a member of the type-B carboxylesterase/lipase protein family [39]. NLGN3/4X activates actin regulators p21-activated kinase 1 and cofilin, which are involved in mediating the effects of adhesion proteins on actin filaments, growth cones, and neuritogenesis [40]. The HIVEP3 gene has a broad H3K79me2 region in DND-41 cells (Figure 3D). HIVEP3 encodes a member of the human immunodeficiency virus type 1 enhancer-binding protein family [39]. It contains multiple zinc-finger and acid-rich (ZAS) domains, as well as serine–threonine-rich regions [39]. The ZAS protein family has been implicated in regulating gene expression of HIV-1 long-terminal repeat and other genes [41]. HIVEP3 is a key regulator that controls (both transcriptionally and post-transcriptionally) innate-like T-lymphocyte-cell development [42]. Next to the HIVEP3 gene is the FOXJ3 gene, which has an H3K79me2 peak at the 5′ end of the gene in the four cell lines. Loucy has a broad H3K79me2 region over the Sprouty RTK-signaling antagonist 1 (SPRY1) gene which was not observed in the other cell lines (Figure 3E). SPRY1 is involved in the negative regulation of the fibroblast growth-factor receptor-signaling pathway [39]. It is one of the many genes overexpressed in p185BCR-ABL-positive ALL [43]. SPRY1 has been observed to be markedly overexpressed in cells of AML patients [44]. AML patients with high SPRY1 expression had poor prognoses [44]. The SGMS1 gene in K562 cells had two broad H3K79me2 regions that were not observed in the other cell lines (Figure 3F). Sphingomyelin synthase 1 is encoded by the SGMS1 gene and metabolizes ceramides into sphingomyelin, the most abundant sphingolipid in mammalian cells [45]. Together these observations identified cell type-specific genes that have broad H3K79me2 domains. Several of these genes have known roles in leukemia.
2.2. MOLM-13 and HOXA9 Cluster
In MOLM-13 cells, the HOXA9 gene is immersed in a broad H3K79me2 domain (Figure 4). The H3K79me2 tracks and RNA tracks for the HOXA9, HOXA10, and HOXA11 genes in MOLM-13 cells are shown in Figures S2 and S3. This gene region was identified as HOXA11-AS1_3 by Partek Flow and as being unique to MOLM-13 with a broad H3K79me2 domain in the top 5%. This domain was also rich in DOT1L. DOT1L appears to be distributed over two sections in this domain. Of interest, the HOXA9 broad H3K79me2 domain has several genes with known roles in leukemia (Table S2). HOXA-cluster members, consisting of 11 genes and non-coding RNA elements, are widely expressed in myeloid cells [46,47]. Among the HOXA gene family are genes encoding DNA-binding proteins that widely control embryo segmentation and later developmental events. Several studies reported that overexpression of HOXA genes is directly involved in carcinoma progression, especially in acute myeloid leukemia and lymphoid leukemia. In this cluster, HOXA9, 10, and 11 had the highest transcript levels in MOLM-13 cells (Figure S3). In addition to the transcription-factor-coding genes, several genes express long non-coding RNAs, many of which are oncogenic lncRNAs. Both HOTTIP and HOTAIRM1, for example, have known roles in leukemia [48,49].
Another interesting feature of the HOXA9 broad H3K79me2 domain is that the domain is hypersensitive to DNase I digestion, which identifies accessible chromatin regions (Figure 4). The DNase I hypersensitivity covers the entire HOXA9 broad H3K79me2 domain and regions on either side of the domain. This observation suggests that this region has an unstable chromatin structure.
The HOXA-AS3 gene region was also identified as having a unique broad H3K79me2 domain in Loucy cells (Figure 5). This region has a very similar broad H3K79me2 domain over the HOXA9 gene cluster to what was observed for MOLM-13 cells; however, MACS2 called these peaks differently (HOXA11-AS1_3 for MOLM-13 and HOXA-AS for Loucy). The SET-NUP214 fusion protein is expressed in the Loucy cell line [50]. This fusion protein binds to the promoters of the HOXA9 and HOXA10 genes and recruits DOT1L to these sites [50,51]. Although the mechanism by which DOT1L is recruited to the HOXA9 cluster is different in MOLM-13 and Loucy cells, the result is similar, with the domain acquiring a broad H3K79me2 domain.
2.3. Broad H3K79me2 Domain in AML Cell Lines
We next explored whether different AML cells (MLL-type) that had the KMT2A fusion protein would have different sets of genes with the broadest H3K79me2 domain. FASTQ files were obtained from available public datasets (MV4-11, GSE79899; THP-1, GSE79899, MOLM-13, GSE149183). The breadth of the top 5% of broadest H3K79me2 peaks varied among the cell lines (MV4-11, 3410–11,073 bp; THP-1, 2601–6194 bp; MOLM-13, 25,846–471,096 bp) (Table S3). The JMJD1C gene and HOXA (including HOXA9) gene clusters were associated with a broad H3K79me2 domain in these three AML (MLL-type) cell lines. The HOXA gene cluster was considerably shorter in THP-1 (45 kb) and MV4-11 (30 kb) cells than in MOLM-13 (116 kb) and Loucy (111 kb) cells. The DACH1 gene had a broad H3K79me2 domain in MOLM-13 and THP-1, but not in MV4-11 cells. As with MOLM-13 cells, NLGN4X, HIVEP3, SPRY1, and SGMS1 did not have an H3K79me2 signature in THP-1 or MV4-11.
GO-term analysis (biological process) of genes with the top 5% of broad H3K79me2 domains in the three AML cell lines was conducted using Enrichr (Table 1 and Table 3). Although the GO term “phosphorylation” was prominent for MOLM-13 genes, MV4-11 and THP-1 had genes involved in RNA splicing.
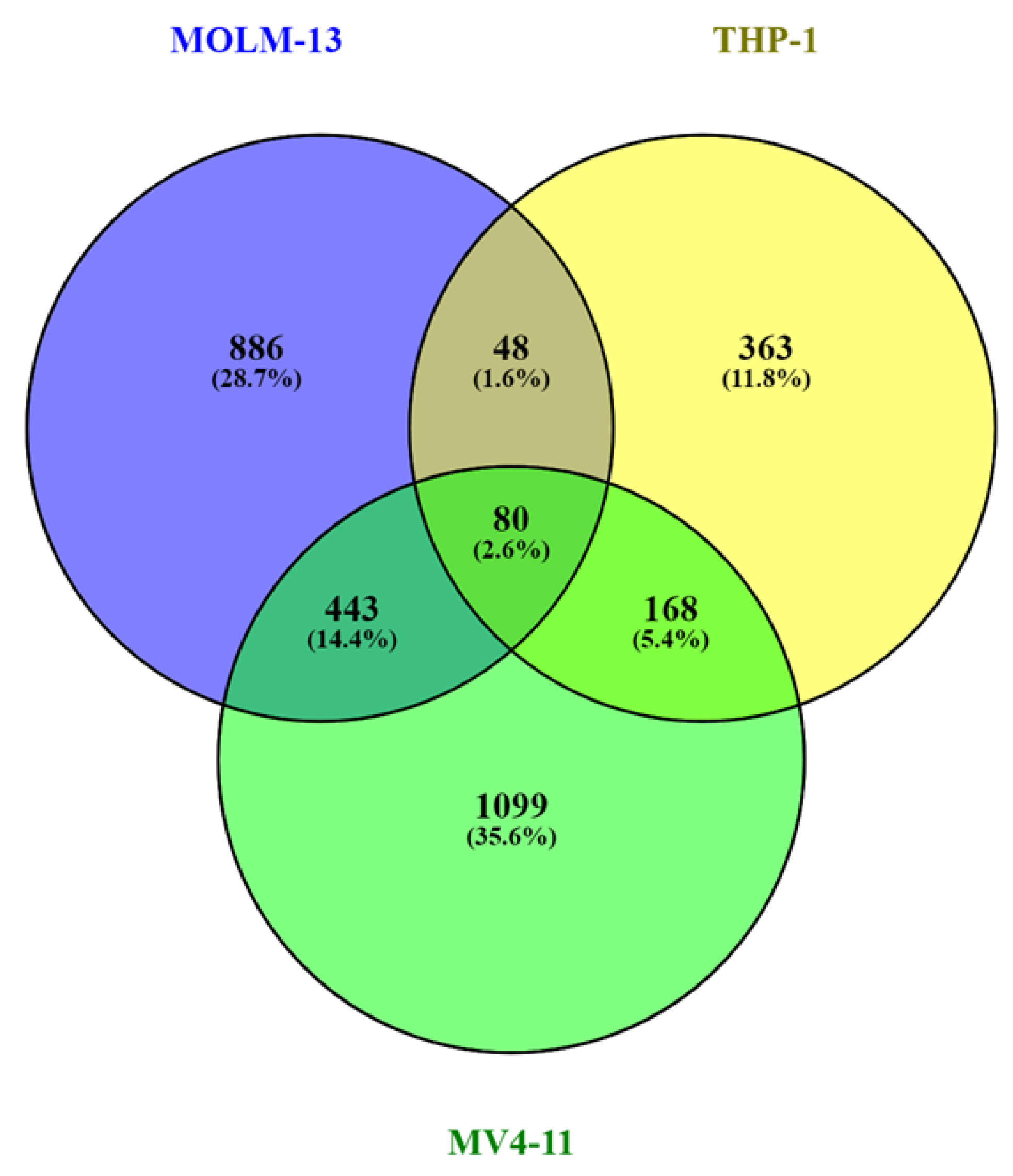
The top 5% of broad H3K79me2 domains in the three AML cell lines that were unique to that cell line were identified (Figure 6, Table S3). These analyses found the following genes in the four cell lines: MV4-11, 1099; THP-1, 363, MOLM-13, 886. The GO terms for these genes (biological process) included “protein phosphorylation (MOLM-13)”, “nuclear import (MV4-11),” and “mitotic spindle organization (THP-1)” (Table 4).
Genes with the broad H3K79me2 domain and listed as present in the top 5% of an AML cell line include CUB And Sushi Multiple Domains 3 (CSMD3, MV4-11), Twist Family BHLH Transcription Factor 1 (TWIST1, THP-1), and Ephrin A5 (EFNA5, MOLM-13). The CSMD3 gene has been investigated for its possible role in cancer progression [52]. The TWIST1 gene is involved in epithelial–mesenchymal transition and is involved in several cancers, including AML [53,54]. The EFNA5 gene plays a role in several cancers, including ovarian cancer and prostate cancer [55,56]. It will be of interest to further explore the role of these genes in AML. In summary, the three AML (MLL-type) cell lines have genes in common and genes that are distinct with the broad H3K79me2 domain.
3. Discussion
DOT1L-mediated methylation of H3K79 is an active mark that is involved in transcription initiation [20]. Mass spectrometry analyses of cellular H3K79 unmodified and methyl forms in a variety of mammalian cells (THP-1, U937, HL60, mouse thymus) showed that K79 is about 88–95% unmodified, 4.4–10.1% is K79me1, and 0.5–1.6% is in K79me2, whereas H3K79me3 is undetectable [57,58]. The extent of H3K79 methylation increases with acetylation of H3, with K79me1 predominating in the tetra-acetylated state [59]. DOT1L methylation of H3K79 is enhanced when the nucleosome has ubiquitinated H2B, which is dependent upon ongoing transcription [60,61]. Acetylation of H3 is also linked to ongoing transcription [62]. Thus, H3K79 methylation is augmented with H3 acetylation and H2B ubiquitination (K120), modifications that are both dependent upon ongoing transcription.
Several studies concluded that H3K79me2 is an indicator of genes involved in leukemogenesis [63,64]. Inhibition of DOT1L activity reduces HOXA9 expression and leukemic cell survival [14]. Together, these observations show the importance of H3K79me2 catalyzed by DOT1L. However, DOT1L is also involved in the destabilization of the nucleosome, which is independent of its catalytic activity [65]. The enhanced chromatin accessibility over the 100 kb HOXA9 gene domain, which is evinced by the DNase sensitivity data in Figure 4, is likely the result of multiple factors that destabilize nucleosome structure, including DOT1L itself, histone modifications such as ubiquitinated H2B and acetylation, and chromatin remodelers. We envisage a dynamic process in which the dissolution of nucleosomes with heavily modified histone octamers would transiently present the accessible DNA to a host of proteins (such as transcription factors), followed by the reassembly of the nucleosome with unmodified histones and specific histone variants (such as the replacement histone variant H3.3) [21,66].
With regards to RNA splicing, it has been reported that DOT1L-mediated H3K79me2 is involved in alternate splicing (skipping exon and alternate 3′ splice site) [67]. Exon skipping was prevalent in AML (MLL) cell types. For genes with a broad H3K79me2 domain that participate in alternative splicing, this histone modification could have a significant impact on the transcript produced and contribute to leukemic transformation [67].
Several histone modifications (H3K4me3, H3K4ac, H3K27ac, H3K79me2) associated with transcribed genes exhibit a broad distribution [23,24,26]. In normal cells, the broadest of the H3K4me3 domains are associated with genes involved in cell identity, whereas in cancer cells, the genes marked with the broadest H3K4me3 domain are cancer-related [21,25]. We recently showed that the genes marked with the broadest H3K4ac domain identify genes important in different breast-cancer types [24]. Here, we applied a similar strategy to identify genes important to different leukemic cell types by finding which genes had the broadest H3K79me2 domain that was present solely in one of the four cell types.
In our initial analyses of genes with H3K79me2 ChIP-Seq data for MOLM-13 cells, the JMJD1C gene was identified as having the broadest H3K79me2 domain. JMJD1C is an H3K9 demethylase with a known oncogenic role in AML and, in particular, those caused by insertions and translocations of KMT2A. JMJD1C is overexpressed in cells with the KMT2A-AF9 and other fusions and is required for self-renewal of leukemic cells [68]. However, JMJD1C has a role in other AML cells, where it is required by RUNX1–RUNX1T1 to increase cell proliferation [69].
The breadths of the H3K79me2 domains in Loucy and THP-1 cells were considerably shorter than those of the other leukemia cells studied. The reason(s) for the shorter H3K79me2 domain, whether biological or technical, requires further investigation.
Identifying genes that have the broadest H3K79me2 domain in only one of the four leukemic cell lines (MOLM-13, K562, Loucy, DND-41) produced an informative list of genes. In MOLM-13 cells, for example, DACH1 and ZNF21 genes are shown to have broad H3K79me2 domains. Both genes play critical roles in AML and are potential therapeutic targets. Several genes in MOLM-13 cells identified in this analysis have been documented as playing critical roles in AML with the KMT2A-AF9 translocation—for example, RUNX2, MEIS1, HOXA9, and PBX3 [70]. Except for PBX3, these genes are unique in having the broadest H3K79me2 domain in MOLM-13 cells. PBX3 is also in the top 5% broadest H3K79me2 domains in Loucy cells, but not in DND-41 or K562 cells.
There are several limitations to this study. Firstly, a limitation to the analyses of broad H3K79me2 domains and broad modified-histone domains in general is the quality of the antibody used [71]. In reviewing the GEO files used in these studies, the source of the anti-H3K79me2 antibody was not always listed. For other GEO entries, there is the concern that the anti-H3K79me2 antibody cross-reacts with H3K79me1, which is more abundant than H3K79me2 [71]. In future studies of the broad H3K79me2 domain, it is critical that the antibody against H3K79me2 undergo a rigorous validation [71]. Secondly, there are several limitations to our observation of the top 5% of broad H3K79me2 domains tending to have a higher level of gene expression. The levels of transcripts are steady-state, and the cellular level of a particular transcript would depend on the gene’s rate of transcription, pre-mRNA splicing, transcript turnover, and many other factors. To accurately and directly determine the impact of the chromatin structure of the broad H3K79me2 on a particular gene, transcription needs to be measured by a nuclear run-on assay. It was noted that one of the features of genes with the broad H3K4me3 domain is transcriptional consistency [22]. Whether genes with the broad H3K79me2 domain exhibit transcriptional consistency remains to be determined. Thirdly, future studies need to be done with leukemia-patient samples.
One of the most interesting broadest H3K79me2 domains includes the HOXA9 gene, which is found in MOLM-13 and Loucy cell lines, which can be explained by the recruitment of DOT1L to HOXA9 genes but through different mechanisms. The HOXA9 genes in K562 and DND-41 cells do not have an associated broad H3K79me2 domain. In MOLM-13 cells, DOT1L is recruited to HOXA9 via the oncogenic fusion protein KMT2A-AF9, whereas in Loucy cells, SET-NUP214 recruits DOT1L. The HOXA9 domain, which is rich in CpG islands, can bind to the CXXC domain of KMT2A-AF9, resulting in the recruitment of DOT1L to these regions, and such binding prevents methylation of these CpG sequences [72]. The broad H3K79me2 domain contains several transcription-factor-coding genes (e.g., HOXA10) and oncogenic lncRNAs. In future studies, targeting multiple sites in this 100 kb H3K79me2 domain through epigenetic-editing methods may result in the silencing of several oncogenic players involved in leukemia.
4. Conclusions
Genes associated with the broadest active histone mark (e.g., H3K4me3, H3K4ac, H3K79me2) have essential functions in cells, and this epigenetic signature can identify genes important in normal and disease states. Through approaches that we describe here, gene panels can be generated to aid in the diagnosis and treatment of disease.
5. Materials and Methods
5.1. Data Collection
Analyses were conducted using publicly available ChIP-Seq data from the Gene Expression Omnibus (GEO) database. The GEO files for the H3K79me2 ChIP-Seq files for the following cell lines were as follows: MOLM-13 (GSE149183, GSE43063, GSE113191), MOLM-14 (GSE76745), K562 (GSE29611), Loucy (GSE96166), DND-41 (GSE29611), MV4-11 (GSE79899), and THP-1 (GSE79899). The GEO files for the DNase I-Seq files for MOLM-13 were from GSE149183. DOT1L ChIP-Seq files for MOLM-13 were from GSE149183. RNA-Seq files for the leukemic cell lines were from the following sources: K562, GSE90239, GSE78556, GSE140322; Loucy, GSE155337; DND-41, GSE173867, GSE116873; MOLM-13, GSE113191, GSE181003.
5.2. ChIP Sequence Analyses (MACS2)
FASTQ files were obtained from the European Nucleotide Archive and downloaded to the Partek Flow analytical program (Partek Incorporated, 100 Chesterfield Business Parkway, Chesterfield, Missouri 63005). The Bowtie 2 aligner was used, with sequences being aligned against the human hg38 assembly. Identification of broad H3K79me2 peaks was carried out using the MACS2 broad-peak function. The peaks were annotated using Ensembl transcript release 91.
5.3. Broad H3K79me2 Domain
The top 5% of the broadest domains were extracted from the H3K79me2 peaks identified by the MACS2 peak caller. We used online tool VENNY 2.1 to classify genes per cell type and to identify the genes that were unique to each cell line. The VENNY 2.1 tool was then applied to identify the unique genes with the broadest H3K79me2 domains. We extracted these exclusive genes and performed gene ontology (GO) (biological process) with Enrichr [35,36].
5.4. RNA Sequence Analyses
FASTQ files for the cell lines were obtained from the European Nucleotide Archive and downloaded to the Partek Flow analytical program (Partek Incorporated, 100 Chesterfield Business Parkway, Chesterfield, Missouri 63005). The HISAT 2.1.0 aligner was used, with sequences being aligned against the human hg38 assembly. Quantification was completed using the Partek Flow “quantify to annotation model (Partek E/M)” and the Ensembl transcript release 104. Normalization was carried out using the TPM method.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/cells11182830/s1, Figure S1: H3K79me2 ChIP-Seq track from JMJD1C gene in MOLM-13 cells; Figure S2: H3K79me2 ChIP-Seq tracks for HOXA9, HOXA10, and HOXA11 genes in MOLM-13 cells; Figure S3: RNA-Seq tracks for HOXA9, HOXA10, and HOXA11 genes in MOLM-13 cells; Table S1: H3K79me2 and RNA seq results from MOLM-13, DND-41, Loucy, and K562; Table S2: Genes in the HOXA9 gene cluster; Table S3: H3K79me2 seq results from MV4-11 and THP-1.
Author Contributions
Conceptualization: J.R.D.; the first draft was prepared by P.S. and J.R.D.; analyses of ChIP Seq and RNA Seq data using MACS2 were completed by J.R.D.; interpretation of the data was carried out by P.S., H.S., N.F., T.M.L. and J.R.D.; Writing of the manuscript was completed by P.S., H.S., N.F., T.M.L. and J.R.D. All authors have read and agreed to the published version of the manuscript.
Funding
Research support by grant from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-05927) to J.R.D. and the CancerCare Manitoba Foundation (761020451) to J.R.D. and T.M.L., and the Canadian Institutes of Health Research (PJT-175226) to T.M.L. and J.R.D. Hedieh Sattarifard was supported in part by Graduate Enhancement of Tri-Council Stipends (GETS) through the University of Manitoba.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The study was done with publicly available datasets which can be obtained through Gene Expression Omnibus.
Acknowledgments
We acknowledge that the CancerCare Manitoba Research Institute and the University of Manitoba campuses are located on original lands of Anishinaabeg, Cree, Oji-Cree, Dakota, and Dene peoples, and on the homeland of the Métis Nation.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Hess, J.L. The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, R. Systematic Classification of Mixed-Lineage Leukemia Fusion Partners Predicts Additional Cancer Pathways. Ann. Lab. Med. 2016, 36, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, R. MLL Leukemia and Future Treatment Strategies. Arch. Pharm. 2015, 348, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Slany, R.K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef]
- Britten, O.; Ragusa, D.; Tosi, S.; Kamel, Y.M. MLL-Rearranged Acute Leukemia with t(4;11)(Q21;Q23)-Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef]
- Yokoyama, A. Leukemogenesis via aberrant self-renewal by the MLL/AEP-mediated transcriptional activation system. Cancer Sci. 2021, 112, 3935–3944. [Google Scholar] [CrossRef]
- Collins, C.T.; Hess, J.L. Role of HOXA9 in leukemia: Dysregulation, cofactors and essential targets. Oncogene 2015, 35, 1090–1098. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Matsuo, Y.; MacLeod, R.; Uphoff, C.; Drexler, H.; Nishizaki, C.; Katayama, Y.; Kimura, G.; Fujii, N.; Omoto, E.; Harada, M.; et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 1997, 11, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Feng, Q.; Li, Z.; Zhang, Y.; Xu, R.-M.; Keck, W.M. Structure of the Catalytic Domain of Human DOT1L, a Non-SET Domain Nucleosomal Histone Methyltransferase Mosomes during the Cell Division (Grewal and Elgin, 2002). A Recent Addition to the Histone Modification Reper-Toire Is Histone H3 Lysine-79 (Lys79) M. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef]
- Sarno, F.; Nebbioso, A.; Altucci, L. DOT1L: A key target in normal chromatin remodelling and in mixed-lineage leukaemia treatment. Epigenetics 2019, 15, 439–453. [Google Scholar] [CrossRef]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.; Li, W.; et al. AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.H.E.; Erb, M.A.; et al. ENL Links Histone Acetylation to Oncogenic Gene Expression in Acute Myeloid Leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef]
- Wang, X.; Chen, C.-W.; Armstrong, S.A. The role of DOT1L in the maintenance of leukemia gene expression. Curr. Opin. Genet. Dev. 2016, 36, 68–72. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; Van Den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 Is Required for Survival in Human MLL-Rearranged Acute Leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef]
- Wu, A.; Zhi, J.; Tian, T.; Cihan, A.; Cevher, M.A.; Liu, Z.; David, Y.; Muir, T.W.; Roeder, R.G.; Yu, M. DOT1L complex regulates transcriptional initiation in human erythroleukemic cells. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Beacon, T.H.; Delcuve, G.P.; López, C.; Nardocci, G.; Kovalchuk, I.; van Wijnen, A.J.; Davie, J.R. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin. Epigenetics 2021, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet. 2015, 47, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.A.; Pollina, E.A.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.D.; Devarajan, K.; Daugherty, A.C.C.; Kundaje, A.B.B.; Mancini, E.; et al. H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell 2014, 158, 673–688. [Google Scholar] [CrossRef]
- López, C.; Barnon, M.T.; Beacon, T.H.; Nardocci, G.; Davie, J.R. The key role of differential broad H3K4me3 and H3K4ac domains in breast cancer. Gene 2022, 826. [Google Scholar] [CrossRef] [PubMed]
- Belhocine, M.; Simonin, M.; Flores, J.D.A.; Cieslak, A.; Manosalva, I.; Pradel, L.; Smith, C.; Mathieu, E.-L.; Charbonnier, G.; Martens, J.H.; et al. Dynamics of broad H3K4me3 domains uncover an epigenetic switch between cell identity and cancer-related genes. Genome Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Chen, M.; Eng, R.; DeJong, J.; Sinha, A.U.; Rahnamay, N.F.; Koche, R.; Al-Shahrour, F.; Minehart, J.; Chen, C.-W.; et al. MLL-AF9– and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Investig. 2016, 126, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Lozzio, B.B.; Lozzio, C.B. Properties and usefulness of the original K-562 human myelogenous leukemia cell line. Leuk. Res. 1979, 3, 363–370. [Google Scholar] [CrossRef]
- Gribble, S.M.; Roberts, I.; Grace, C.; Andrews, K.M.; Green, A.R.; Nacheva, E.P. Cytogenetics of the Chronic Myeloid Leukemia-Derived Cell Line K562. Cancer Genet. Cytogenet. 2000, 118, 1–8. [Google Scholar] [CrossRef]
- Naumann, S.; Reutzel, D.; Speicher, M.; Decker, H.-J. Complete karyotype characterization of the K562 cell line by combined application of G-banding, multiplex-fluorescence in situ hybridization, fluorescence in situ hybridization, and comparative genomic hybridization. Leuk. Res. 2001, 25, 313–322. [Google Scholar] [CrossRef]
- Prokocimer, M.; Peller, S.; Ben-Bassat, H.; Goldfinger, N.; Rotter, V. P53 Gene Mutation in a T-Acute Lymphoblastic Leukemia Cell Line (Loucy) with t(16:20) and 5q-Chromosomal Aberrations. Leuk. Lymphoma 1998, 29, 607–611. [Google Scholar] [CrossRef]
- Minowada, J.; Tsubota, T.; Nakazawa, S.; Srivastava, B.I.S.; Huang, C.C.; Oshimura, M.; Sonta, S.; Han, T.; Sinks, L.F.; Sandberg, A.A. Establishment and Characterization of Leukemic T-Cell Lines, B-Cell Lines, and Null-Cell Line: A Progress Report on Surface Antigen Study of Fresh Lymphatic Leukemias in Man. In Immunological Diagnosis of Leukemias and Lymphomas; Springer: Berlin/Heidelberg, Germany, 1977; Volume 20, pp. 241–251. [Google Scholar] [CrossRef]
- Aziz, N.A.A.; Mokhtar, N.M.; Harun, R.; Mollah, M.M.H.; Rose, I.M.; Sagap, I.; Tamil, A.M.; Ngah, W.Z.W.; Jamal, R. A 19-Gene expression signature as a predictor of survival in colorectal cancer. BMC Med. Genom. 2016, 9, 58. [Google Scholar] [CrossRef]
- Al Barashdi, M.A.; Ali, A.; McMullin, M.F.; Mills, K. Protein tyrosine phosphatase receptor type C (PTPRC or CD45). J. Clin. Pathol. 2021, 74, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Henderson, H.J.; Karanam, B.; Samant, R.; Vig, K.; Singh, S.R.; Yates, C.; Bedi, D. Neuroligin 4X overexpression in human breast cancer is associated with poor relapse-free survival. PLoS ONE 2017, 12, e0189662. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, H.S.; Hwang, J.; Kim, Y.H.; Lim, G.Y.; Sohn, W.J.; Yoon, S.R.; Kim, J.Y.; Park, T.S.; Oh, S.H.; et al. Regulation of HOXA9 Activity by Predominant Expression of DACH1 against C/EBPα and GATA-1 in Myeloid Leukemia with MLL-AF9. Biochem. Biophys. Res. Commun. 2012, 426, 299–305. [Google Scholar] [CrossRef]
- Germano, G.; Morello, G.; Aveic, S.; Pinazza, M.; Minuzzo, S.; Frasson, C.; Persano, L.; Bonvini, P.; Viola, G.; Bresolin, S.; et al. ZNF521 sustains the differentiation block in MLL-rearranged acute myeloid leukemia. Oncotarget 2017, 8, 26129–26141. [Google Scholar] [CrossRef]
- NCBI. Genes & Expression. Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 23 April 2021).
- Gatford, N.J.F.; Deans, P.J.M.; Duarte, R.R.R.; Chennell, G.; Sellers, K.J.; Raval, P.; Srivastava, D.P. Neuroligin-3 and neuroligin-4X form nanoscopic clusters and regulate growth cone organization and size. Hum. Mol. Genet. 2021, 31, 674–691. [Google Scholar] [CrossRef]
- Allen, C.E.; Mak, C.-H.; Wu, L.-C. The κB transcriptional enhancer motif and signal sequences of V(D)J recombination are targets for the zinc finger protein HIVEP3/KRC: A site selection amplification binding study. BMC Immunol. 2002, 3, 10. [Google Scholar] [CrossRef]
- Krovi, S.H.; Zhang, J.; Michaels-Foster, M.J.; Brunetti, T.; Loh, L.; Scott-Browne, J.; Gapin, L. Thymic iNKT single cell analyses unmask the common developmental program of mouse innate T cells. Nat. Commun. 2020, 11, 6238. [Google Scholar] [CrossRef]
- Juric, D.; Lacayo, N.J.; Ramsey, M.C.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Goldstone, A.H.; O’Dwyer, P.J.; Paietta, E.; Sikic, B.I. Differential Gene Expression Patterns and Interaction Networks in BCR-ABL–Positive and –Negative Adult Acute Lymphoblastic Leukemias. J. Clin. Oncol. 2007, 25, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Wang, Y.; Ji, C.; Shi, C.; Li, Y. SPRY1 promotes cell proliferation and inhibits apoptosis by activating Hedgehog pathway in acute myeloid leukemia. Hematology 2021, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bilal, F.; Montfort, A.; Gilhodes, J.; Garcia, V.; Riond, J.; Carpentier, S.; Filleron, T.; Colacios, C.; Levade, T.; Daher, A.; et al. Sphingomyelin Synthase 1 (SMS1) Downregulation Is Associated With Sphingolipid Reprogramming and a Worse Prognosis in Melanoma. Front. Pharmacol. 2019, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Argiropoulos, B.; Humphries, R.K. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–6776. [Google Scholar] [CrossRef] [PubMed]
- Rezsohazy, R.; Saurin, A.; Maurel-Zaffran, C.; Graba, Y. Cellular and molecular insights into Hox protein action. Development 2015, 142, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Gabra, M.M.; Salmena, L. microRNAs and Acute Myeloid Leukemia Chemoresistance: A Mechanistic Overview. Front. Oncol. 2017, 7, 255. [Google Scholar] [CrossRef]
- Chen, L.; Hu, N.; Wang, C.; Zhao, H. HOTAIRM1 knockdown enhances cytarabine-induced cytotoxicity by suppression of glycolysis through the Wnt/β-catenin/PFKP pathway in acute myeloid leukemia cells. Arch. Biochem. Biophys. 2020, 680, 108244. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Van Grotel, M.; Tchinda, J.; Lee, C.; Beverloo, H.B.; Van Der Spek, P.J.; Stubbs, A.; Cools, J.; Nagata, K.; Fornerod, M.; et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 2008, 111, 4668–4680. [Google Scholar] [CrossRef]
- Mendes, A.; Fahrenkrog, B. NUP214 in Leukemia: It’s More than Transport. Cells 2019, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, X.; Jiang, H.; Chen, B. A Promising Glycolysis- and Immune-Related Prognostic Signature for Glioblastoma. World Neurosurg. 2022, 161, e363–e375. [Google Scholar] [CrossRef]
- Dabiri, A.; Sharifi, M.; Sarmadi, A. Knockdown of SOX12 Expression Induced Apoptotic Factors Is Associated with TWIST1 and CTNNB1 Expression in Human Acute Myeloid Leukemia Cells. Int. J. Mol. Cell. Med. 2021, 10, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, W.; Yan, X.J.; Lin, X.Q.; Li, W.; Mi, J.Q.; Li, J.M.; Zhu, J.; Chen, Z.; Chen, S.J. DNMT3A Mutation Leads to Leukemic Extramedullary Infiltration Mediated by TWIST1. J. Hematol. Oncol. 2016, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Jukonen, J.; Moyano-Galceran, L.; Höpfner, K.; Pietilä, E.A.; Lehtinen, L.; Huhtinen, K.; Gucciardo, E.; Hynninen, J.; Hietanen, S.; Grénman, S.; et al. Aggressive and recurrent ovarian cancers upregulate ephrinA5, a non-canonical effector of EphA2 signaling duality. Sci. Rep. 2021, 11, 8856. [Google Scholar] [CrossRef]
- Xie, J.; Xing, S.; Shen, B.-Y.; Chen, H.-T.; Sun, B.; Wang, Z.-T.; Wang, J.-W.; Lu, X.-X. PIWIL1 interacting RNA piR-017061 inhibits pancreatic cancer growth via regulating EFNA5. Hum. Cell 2021, 34, 550–563. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Kakadia, P.M.; Chen, Y.; Li, Y.-Q.; Deshpande, A.J.; Buske, C.; Zhang, K.-L.; Zhang, Y.; Xu, G.-L.; Bohlander, S.K. Global reduction of the epigenetic H3K79 methylation mark and increased chromosomal instability in CALM-AF10–positive leukemias. Blood 2009, 114, 651–658. [Google Scholar] [CrossRef]
- Vlaming, H.; McLean, C.M.; Korthout, T.; Alemdehy, M.F.; Hendriks, S.; Lancini, C.; Palit, S.; Klarenbeek, S.; Kwesi-Maliepaard, E.M.; Molenaar, T.M.; et al. Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. EMBO J. 2019, 38, e101564. [Google Scholar] [CrossRef]
- Zhang, K.; Siino, J.S.; Jones, P.R.; Yau, P.M.; Bradbury, E.M. A mass spectrometric ?Western blot? to evaluate the correlations between histone methylation and histone acetylation. PROTEOMICS 2004, 4, 3765–3775. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Hollander, D.; Voichek, Y.; Ast, G.; Oren, M. Cotranscriptional histone H2B monoubiquitylation is tightly coupled with RNA polymerase II elongation rate. Genome Res. 2014, 24, 1572–1583. [Google Scholar] [CrossRef]
- Davie, J.R.; Murphy, L.C. Level of ubiquitinated histone H2B in chromatin is coupled to ongoing transcription. Biochemistry 1990, 29, 4752–4757. [Google Scholar] [CrossRef]
- Martin, B.J.E.; Brind’Amour, J.; Kuzmin, A.; Jensen, K.N.; Liu, Z.C.; Lorincz, M.; Howe, L.J. Transcription shapes genome-wide histone acetylation patterns. Nat. Commun. 2021, 12, 210. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Fan, G.L.; Liu, J.J.; Liu, L.; Li, Q.Z.; Lin, H. Identification of Key Histone Modifications and Their Regulatory Regions on Gene Expression Level Changes in Chronic Myelogenous Leukemia. Front. Cell Dev. Biol. 2021, 8, 621578. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Shah, R.N.; Ruthenburg, A.J. Non-canonical H3K79me2-dependent pathways promote the survival of MLL-rearranged leukemia. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kang, C.; Yang, H.-S.; Jung, T.; Hebert, H.; Chung, K.Y.; Kim, S.J.; Hohng, S.; Song, J.-J. Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev. 2019, 33, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Black, B.L.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, Q.; Garza, N.; Kornblau, S.; Jin, V.X. Integrative analysis reveals functional and regulatory roles of H3K79me2 in mediating alternative splicing. Genome Med. 2018, 10, 30. [Google Scholar] [CrossRef]
- Izaguirre-Carbonell, J.; Christiansen, L.; Burns, R.; Schmitz, J.; Li, C.; Mokry, R.L.; Bluemn, T.; Zheng, Y.; Shen, J.; Carlson, K.-S.; et al. Critical role of Jumonji domain of JMJD1C in MLL-rearranged leukemia. Blood Adv. 2019, 3, 1499–1511. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, N.; Liu, X.; Laurent, B.; Tang, Z.; Eng, R.; Shi, Y.; Armstrong, S.A.; Roeder, R.G. JMJD1C is required for the survival of acute myeloid leukemia by functioning as a coactivator for key transcription factors. Genes Dev. 2015, 29, 2123–2139. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Wang, G.; Tian, X.; Zhou, J.; Yu, W.; Fan, Z.; Dong, L.; Lu, J.; Xu, J.; et al. Integrated transcriptomic and epigenetic data analysis identifiesaberrant expression of genes in acute myeloid leukemia with MLL-AF9 translocation. Mol. Med. Rep. 2019, 21, 883–893. [Google Scholar] [CrossRef]
- Shah, R.; Grzybowski, A.; Cornett, E.; Johnstone, A.L.; Dickson, B.M.; Boone, B.A.; Cheek, M.A.; Cowles, M.W.; Maryanski, D.; Meiners, M.J.; et al. Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Mol. Cell 2018, 72, 162–177.e7. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the MLL CXXC domain–DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2009, 17, 62–68. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Density distribution of H3K79me2 peaks in MOLM-13 cells. The panel shows the density distribution of H3K79me2 peaks identified by the MACS2 peak caller.
Figure 1.
Density distribution of H3K79me2 peaks in MOLM-13 cells. The panel shows the density distribution of H3K79me2 peaks identified by the MACS2 peak caller.
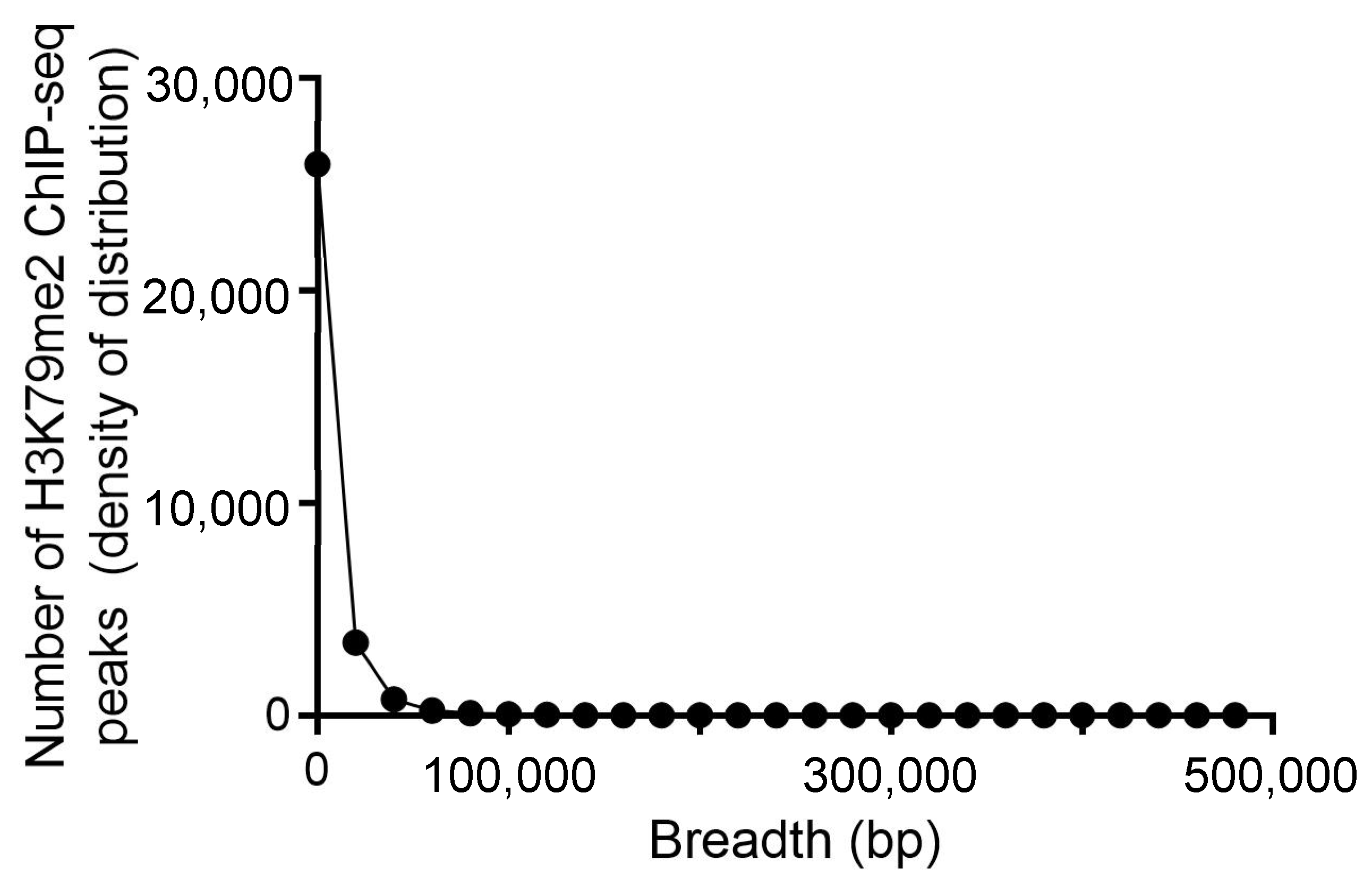
Figure 2.
Distribution of genes with the broad H3K79me2 domain in MOLM-13, K562, Loucy, and DND-41 cells. The number of genes that are unique and in common in the top 5% of genes with the broad H3K79me2 among the four cell lines is shown.
Figure 2.
Distribution of genes with the broad H3K79me2 domain in MOLM-13, K562, Loucy, and DND-41 cells. The number of genes that are unique and in common in the top 5% of genes with the broad H3K79me2 among the four cell lines is shown.
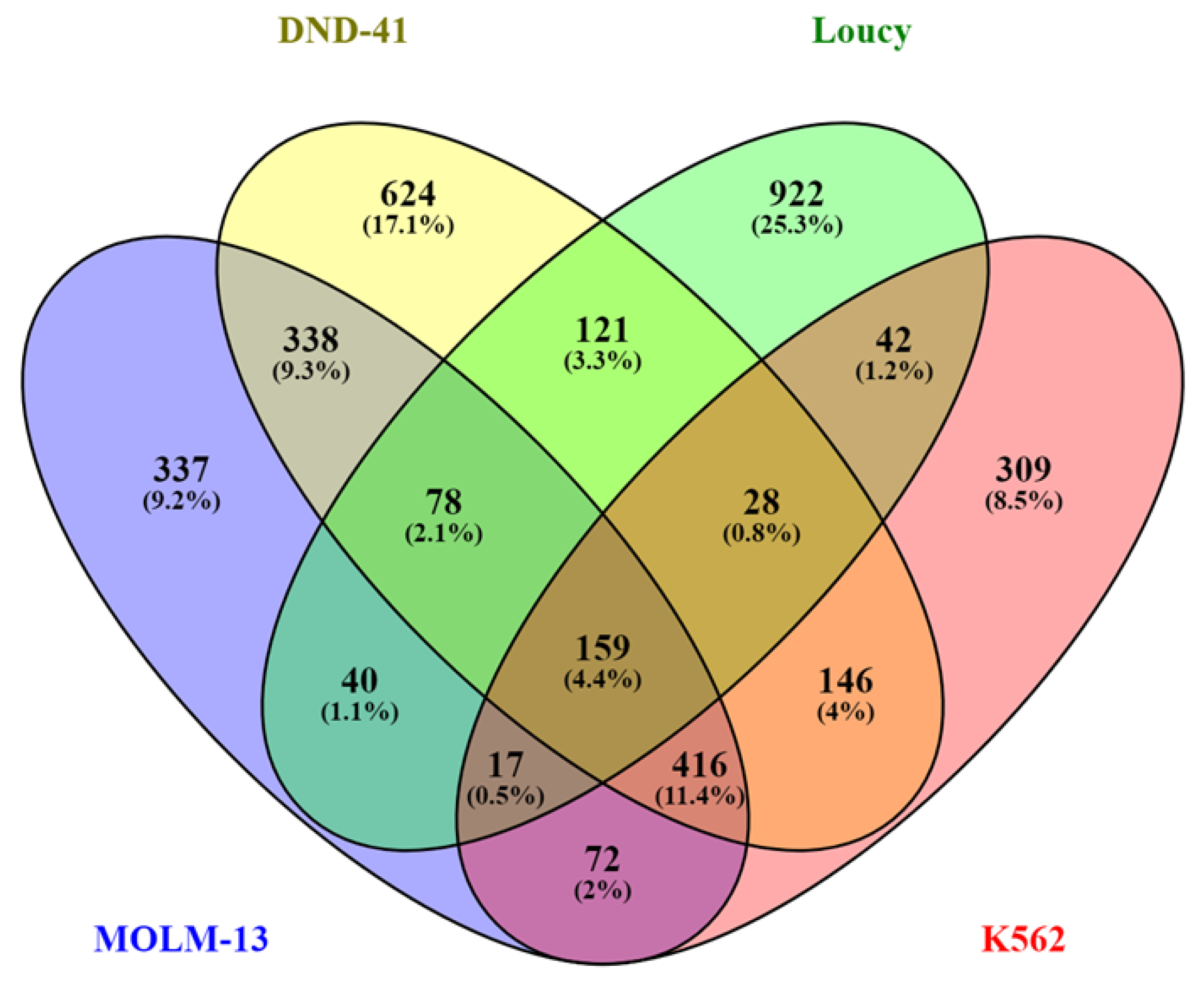
Figure 3.
H3K79me2 ChIP-Seq tracks of genes with the broad H3K79me2 domain. The panel shows the H3K79me2 ChIP-Seq tracks for the four cell lines. (A) DACH1, (B) ZNF21, (C) NLGN4X, (D) HIVEP3, (E) SSPRY1, (F) SGMS1.
Figure 3.
H3K79me2 ChIP-Seq tracks of genes with the broad H3K79me2 domain. The panel shows the H3K79me2 ChIP-Seq tracks for the four cell lines. (A) DACH1, (B) ZNF21, (C) NLGN4X, (D) HIVEP3, (E) SSPRY1, (F) SGMS1.

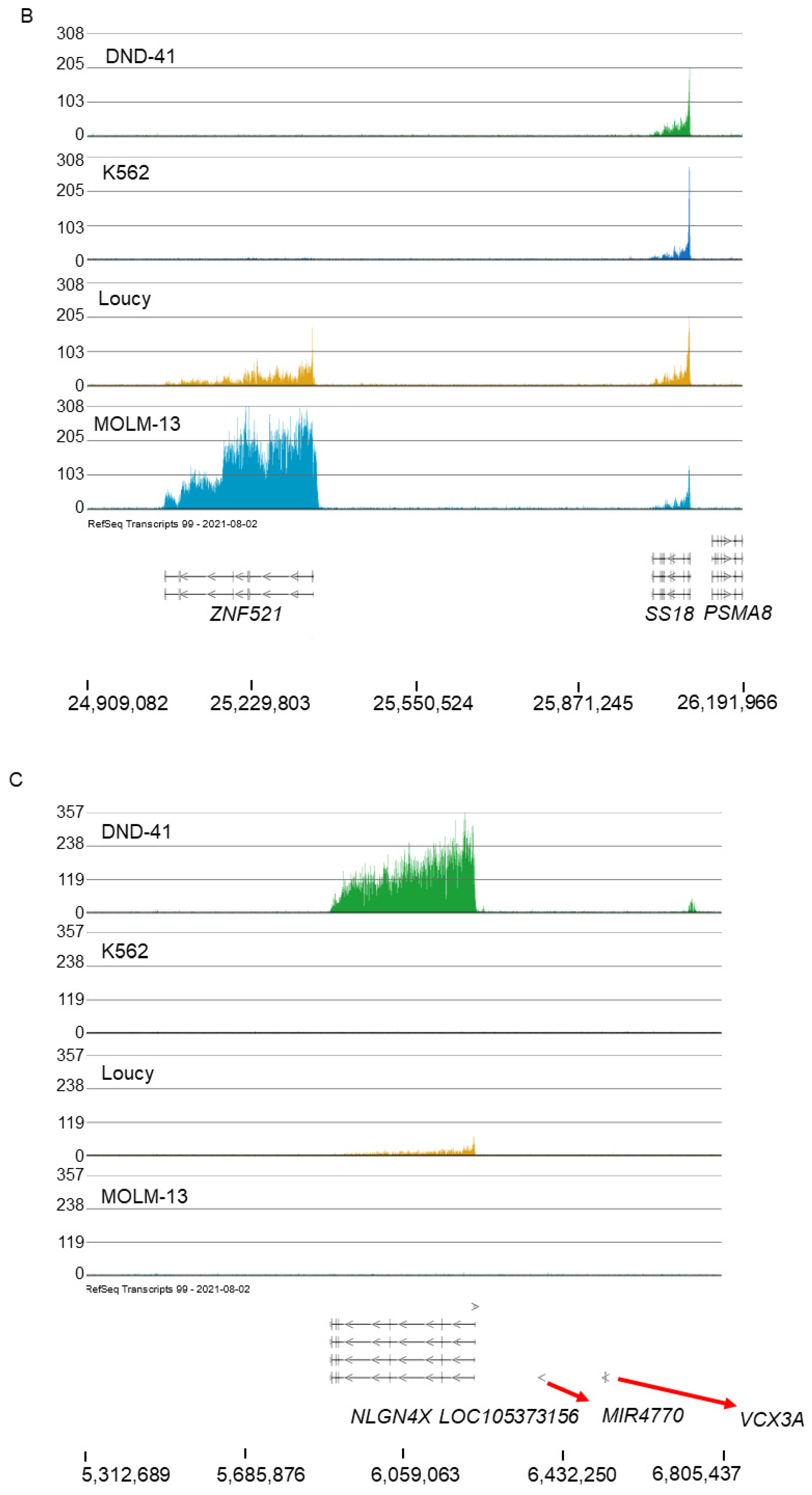

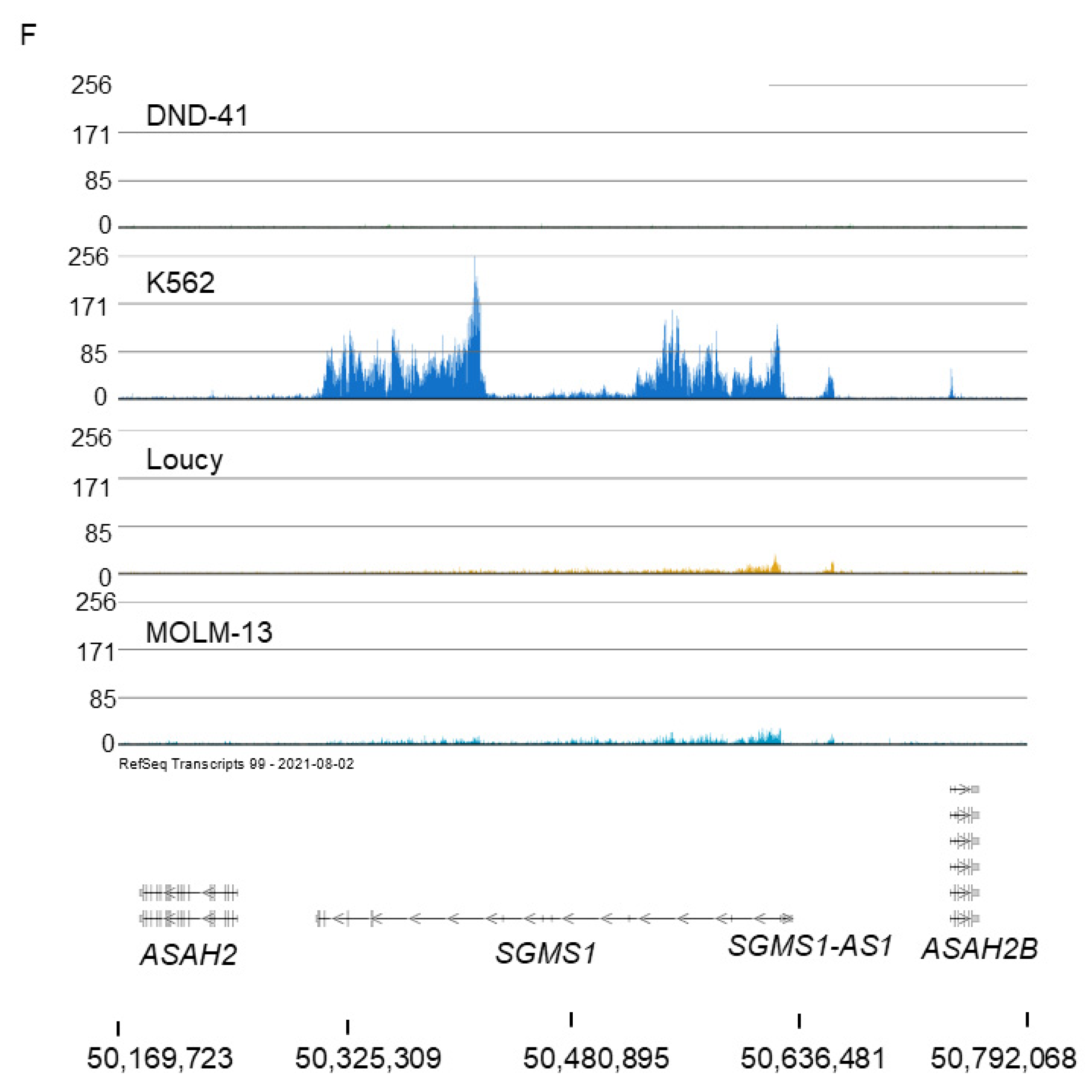
Figure 4.
Broad H3K79me2 domain with the HOXA9 gene in MOLM-13 cells. ChIP-Seq tracks for DOT1L and H3K79me2 are shown. The DNase 1-Seq track shows the chromatin accessibility of the broad H3K79me2 domain with the HOXA9 gene in MOLM-13 cells. Through the CXXC domain, KMT2A-AF9 could be recruited to the numerous CpG islands present in this genomic region. KMT2A-AF9 recruits DOT1L, which methylates H3K79.
Figure 4.
Broad H3K79me2 domain with the HOXA9 gene in MOLM-13 cells. ChIP-Seq tracks for DOT1L and H3K79me2 are shown. The DNase 1-Seq track shows the chromatin accessibility of the broad H3K79me2 domain with the HOXA9 gene in MOLM-13 cells. Through the CXXC domain, KMT2A-AF9 could be recruited to the numerous CpG islands present in this genomic region. KMT2A-AF9 recruits DOT1L, which methylates H3K79.
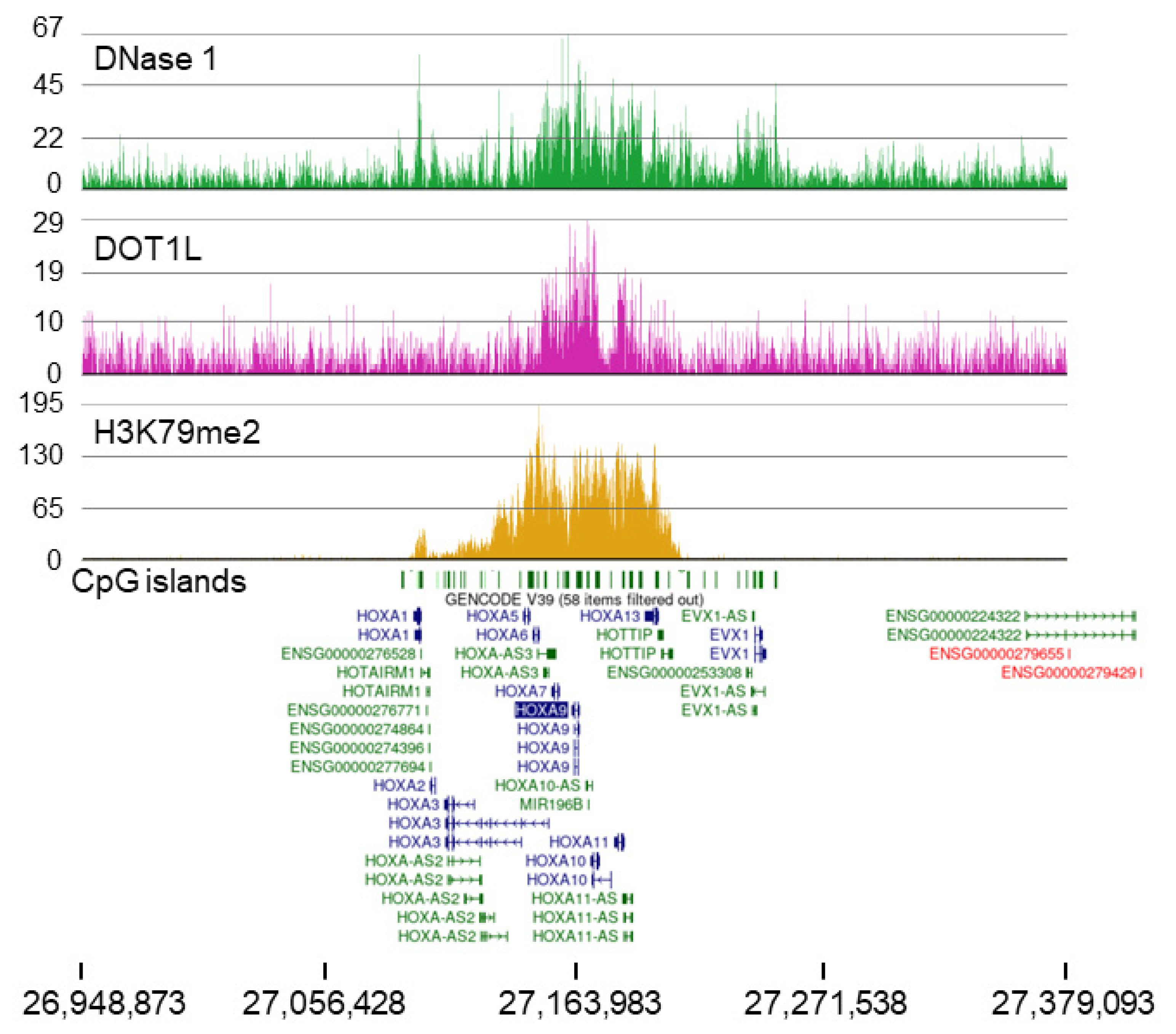
Figure 5.
Broad H3K79me2 domain with the HOXA9 gene in Loucy cells. The panel shows the H3K79me2 ChIP-Seq tracks for HOXA9 in MOLM-13 and Loucy cells.
Figure 5.
Broad H3K79me2 domain with the HOXA9 gene in Loucy cells. The panel shows the H3K79me2 ChIP-Seq tracks for HOXA9 in MOLM-13 and Loucy cells.
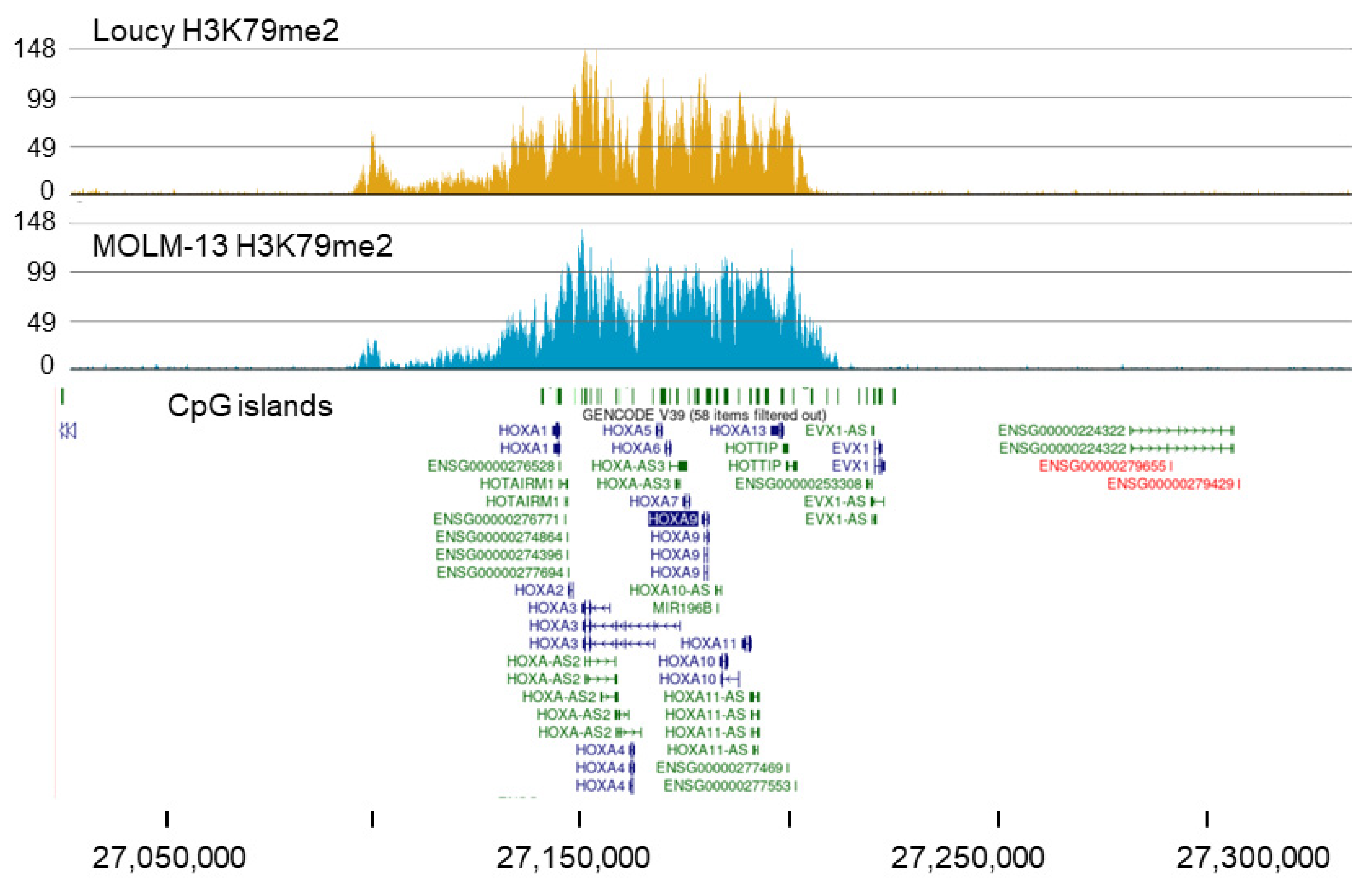
Figure 6.
Distribution of genes with the broad H3K79me2 domain in MVP-11, THP-1, and MOLM-13. Genes unique and in common in the top 5% of genes with the broad H3K79me2 among the three AML cell lines.
Figure 6.
Distribution of genes with the broad H3K79me2 domain in MVP-11, THP-1, and MOLM-13. Genes unique and in common in the top 5% of genes with the broad H3K79me2 among the three AML cell lines.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
GO terms (biological processes) for genes with the broadest H3K79me2 domains in leukemic cells.
Table 1.
GO terms (biological processes) for genes with the broadest H3K79me2 domains in leukemic cells.
| MOLM-13 Top 5% | |||||
|---|---|---|---|---|---|
| Term | Overlap | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
| Phosphorylation (GO:0016310) | 70/400 | 5.35 × 10−12 | 2.09 × 10−8 | 2.785 | 72.295 |
| Positive regulation of transcription, DNA-templated (GO:0045893) | 144/1183 | 4.52 × 10−10 | 8.81 × 10−7 | 1.848 | 39.758 |
| Protein phosphorylation (GO:0006468) | 75/496 | 1.13 × 10−9 | 1.47 × 10−6 | 2.336 | 48.130 |
| Protein dephosphorylation (GO:0006470) | 32/139 | 4.05 × 10−9 | 3.95 × 10−6 | 3.869 | 74.773 |
| DND-41 Top 5% | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Mitotic cell-cycle phase transition (GO:0044772) | 59/209 | 1.06 × 10−14 | 4.49 × 10−11 | 3.812 | 122.675 |
| G1/S transition of mitotic cell cycle (GO:0000082) | 31/85 | 1.65 × 10−11 | 3.50 × 10−8 | 5.510 | 136.810 |
| Ubiquitin-dependent protein catabolic process (GO:0006511) | 71/354 | 1.10 × 10−9 | 1.30 × 10−6 | 2.429 | 49.941 |
| Protein dephosphorylation (GO:0006470) | 38/139 | 1.54 × 10−9 | 1.30 × 10−6 | 3.615 | 73.357 |
| K562 Top 5% | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Negative regulation of translation (GO:0017148) | 26/90 | 7.53 × 10−12 | 2.78 × 10−8 | 6.549 | 167.727 |
| Regulation of translation (GO:0006417) | 36/178 | 7.56 × 10−11 | 1.39 × 10−7 | 4.105 | 95.670 |
| Phosphorylation (GO:0016310) | 57/400 | 7.38 × 10−10 | 9.08 × 10−7 | 2.711 | 57.009 |
| Negative regulation of gene expression (GO:0010629) | 48/322 | 3.72 × 10−9 | 3.43 × 10−6 | 2.846 | 55.243 |
| Loucy Top 5% | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| mRNA splicing, via spliceosome (GO:0000398) | 55/274 | 1.11 × 10−12 | 2.52 × 10−9 | 3.413 | 93.961 |
| RNA splicing via transesterification reactions with bulged adenosine as nucleophile (GO:0000377) | 52/251 | 1.28 × 10−12 | 2.52 × 10−9 | 3.547 | 97.139 |
| mRNA processing (GO:0006397) | 57/300 | 4.58 × 10−12 | 6.01 × 10−9 | 3.188 | 83.248 |
| RNA metabolic process (GO:0016070) | 34/133 | 2.55 × 10−11 | 2.51 × 10−8 | 4.626 | 112.835 |
Table 2.
GO terms (biological processes) for genes with the broadest H3K79me2 domains unique to one of the four cell lines.
Table 2.
GO terms (biological processes) for genes with the broadest H3K79me2 domains unique to one of the four cell lines.
| MOLM-13 | |||||
|---|---|---|---|---|---|
| Term | Overlap | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
| Negative regulation of myeloid-cell differentiation (GO:0045638) | 4/22 | 4.56 × 10−4 | 0.403777 | 13.10978 | 100.8647 |
| Phosphorylation (GO:0016310) | 17/400 | 4.81 × 10−4 | 0.403777 | 2.674282 | 20.42911 |
| Regulation of cellular senescence (GO:2000772) | 4/31 | 0.001739 | 0.573737 | 8.735847 | 55.50902 |
| Negative regulation of NF-kappaB transcription-factor activity (GO:0032088) | 6/79 | 0.002167 | 0.573737 | 4.864462 | 29.83979 |
| DND-41 | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Mitotic-chromosome condensation (GO:0007076) | 6/27 | 1.52 × 10−4 | 0.134681 | 8.94822 | 78.65524 |
| DNA dealkylation (GO:0035510) | 4/10 | 1.70 × 10−4 | 0.134681 | 20.82796 | 180.8268 |
| DNA dealkylation involved in DNA repair (GO:0006307) | 4/10 | 1.70 × 10−4 | 0.134681 | 20.82796 | 180.8268 |
| Mitotic-cell-cycle phase transition (GO:0044772) | 17/209 | 3.14 × 10−4 | 0.18717 | 2.798325 | 22.56889 |
| K562 | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Regulation of organelle organization (GO:0033043) | 7/80 | 2.38 × 10−4 | 0.207771 | 6.229066 | 51.9768 |
| Regulation of cytoskeleton organization (GO:0051493) | 8/112 | 3.51 × 10−4 | 0.207771 | 5.005622 | 39.81809 |
| Negative regulation of stress-fiber assembly (GO:0051497) | 4/24 | 4.65 × 10−4 | 0.207771 | 12.89902 | 98.97629 |
| Establishment of endothelial intestinal barrier (GO:0090557) | 3/11 | 5.50 × 10−4 | 0.207771 | 24.12132 | 181.0597 |
| Loucy | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| RNA splicing, via transesterification reactions with bulged adenosine as nucleophile (GO:0000377) | 31/251 | 6.03 × 10−7 | 0.001095 | 2.982339 | 42.71137 |
| Mitotic-spindle organization (GO:0007052) | 23/157 | 8.91 × 10−7 | 0.001095 | 3.616888 | 50.38532 |
| mRNA splicing via spliceosome (GO:0000398) | 32/274 | 1.39 × 10−6 | 0.001095 | 2.798551 | 37.75151 |
| Gene expression (GO:0010467) | 38/356 | 1.40 × 10−6 | 0.001095 | 2.535929 | 34.17959 |
Table 3.
GO terms (biological processes) for genes with the broadest H3K79me2 domains in AML cell lines.
Table 3.
GO terms (biological processes) for genes with the broadest H3K79me2 domains in AML cell lines.
| MV4-11 Top 5% | |||||
|---|---|---|---|---|---|
| Term | Overlap | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
| mRNA splicing via spliceosome (GO:0000398) | 64/274 | 5.72 × 10−13 | 2.37 × 10−9 | 3.178282 | 89.59665 |
| mRNA processing (GO:0006397) | 66/300 | 4.57 × 10−12 | 9.48 × 10−9 | 2.940925 | 76.79013 |
| RNA processing (GO:0006396) | 46/179 | 3.34 × 10−11 | 4.61 × 10−8 | 3.584974 | 86.48194 |
| RNA splicing via transesterification reactions with bulged adenosine as nucleophile (GO:0000377) | 56/251 | 1.07 × 10−10 | 1.11 × 10−7 | 2.983586 | 68.49088 |
| THP-1 top 5% | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| mRNA splicing via spliceosome (GO:0000398) | 34/274 | 3.18 × 10−11 | 8.11 × 10−8 | 4.32956 | 104.6518 |
| RNA splicing via transesterification reactions with bulged adenosine as nucleophile (GO:0000377) | 32/251 | 5.98 × 10−11 | 8.11 × 10−8 | 4.456271 | 104.9003 |
| mRNA processing (GO:0006397) | 33/300 | 1.47 × 10−9 | 1.33 × 10−6 | 3.765912 | 76.5817 |
| RNA metabolic process (GO:0016070) | 19/133 | 7.67 × 10−8 | 5.20 × 10−5 | 5.007031 | 82.0337 |
Table 4.
GO terms (biological processes) for genes with broadest H3K79me2 domains unique to an AML cell line.
Table 4.
GO terms (biological processes) for genes with broadest H3K79me2 domains unique to an AML cell line.
| MV4-11 | |||||
|---|---|---|---|---|---|
| Term | Overlap | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
| Import into nucleus (GO:0051170) | 14/77 | 6.81 × 10−5 | 0.147625 | 3.858269 | 36.98274 |
| Nucleotide-excision repair (GO:0006289) | 16/105 | 1.92 × 10−4 | 0.147625 | 3.122745 | 26.72998 |
| Positive regulation of establishment of protein localization to mitochondrion (GO:1903749) | 11/56 | 1.99 × 10−4 | 0.147625 | 4.236438 | 36.0943 |
| mRNA splicing via spliceosome (GO:0000398) | 30/274 | 2.59 × 10−4 | 0.147625 | 2.145831 | 17.72484 |
| THP-1 | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Microtubule cytoskeleton organization involved in mitosis (GO:1902850) | 10/128 | 1.17 × 10−4 | 0.097514 | 4.685985 | 42.43785 |
| Mitotic-spindle organization (GO:0007052) | 11/157 | 1.43 × 10−4 | 0.097514 | 4.171875 | 36.93698 |
| Positive regulation of double-strand break repair via nonhomologous end joining (GO:2001034) | 4/16 | 1.63 × 10−4 | 0.097514 | 18.22191 | 158.8791 |
| Nuclear-membrane reassembly (GO:0031468) | 6/54 | 4.25 × 10−4 | 0.164331 | 6.858894 | 53.24899 |
| MOLM-13 | |||||
| Term | Overlap | p-value | Adjusted p-value | Odds Ratio | Combined Score |
| Phosphorylation (GO:0016310) | 45/400 | 9.90 × 10−9 | 2.98 × 10−5 | 2.827469 | 52.11112 |
| Protein phosphorylation (GO:0006468) | 44/496 | 1.02 × 10−5 | 0.015305 | 2.157547 | 24.80196 |
| Regulation of intracellular-signal transduction (GO:1902531) | 38/437 | 6.19 × 10−5 | 0.062065 | 2.101864 | 20.36704 |
| Transmembrane-receptor protein tyrosine kinase-signaling pathway (GO:0007169) | 35/404 | 1.28 × 10−4 | 0.096379 | 2.089284 | 18.72459 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, P.; Sattarifard, H.; Fatemiyan, N.; Lakowski, T.M.; Davie, J.R. Bioinformatic Analyses of Broad H3K79me2 Domains in Different Leukemia Cell Line Data Sets. Cells 2022, 11, 2830. https://doi.org/10.3390/cells11182830
AMA Style
Sharma P, Sattarifard H, Fatemiyan N, Lakowski TM, Davie JR. Bioinformatic Analyses of Broad H3K79me2 Domains in Different Leukemia Cell Line Data Sets. Cells. 2022; 11(18):2830. https://doi.org/10.3390/cells11182830
Chicago/Turabian StyleSharma, Prerna, Hedieh Sattarifard, Narges Fatemiyan, Ted M. Lakowski, and James R. Davie. 2022. "Bioinformatic Analyses of Broad H3K79me2 Domains in Different Leukemia Cell Line Data Sets" Cells 11, no. 18: 2830. https://doi.org/10.3390/cells11182830
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.