
Molecular Regulators of Embryonic Diapause and Cancer Diapause-like State

 ,
, 

Abstract
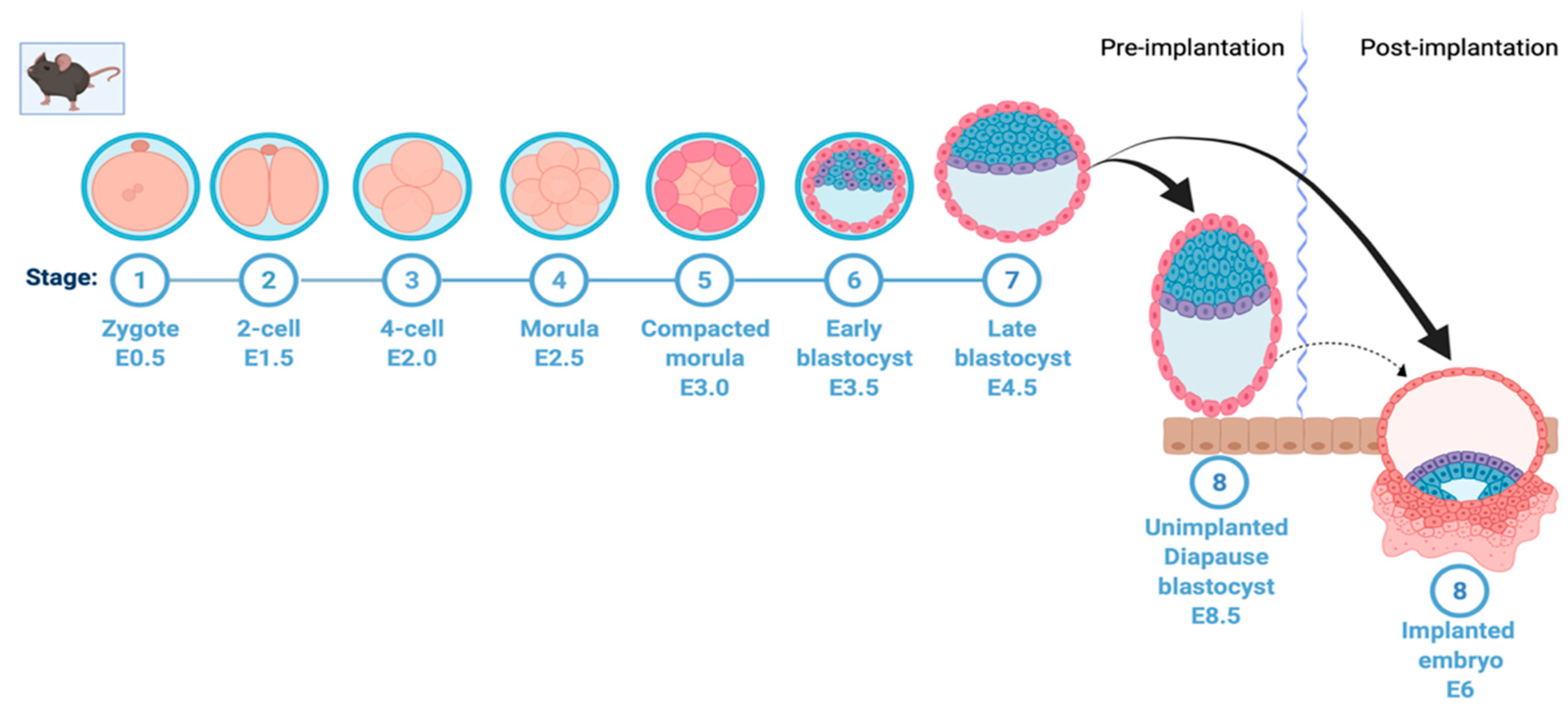
:1. Introduction
2. Diapause Metabolism
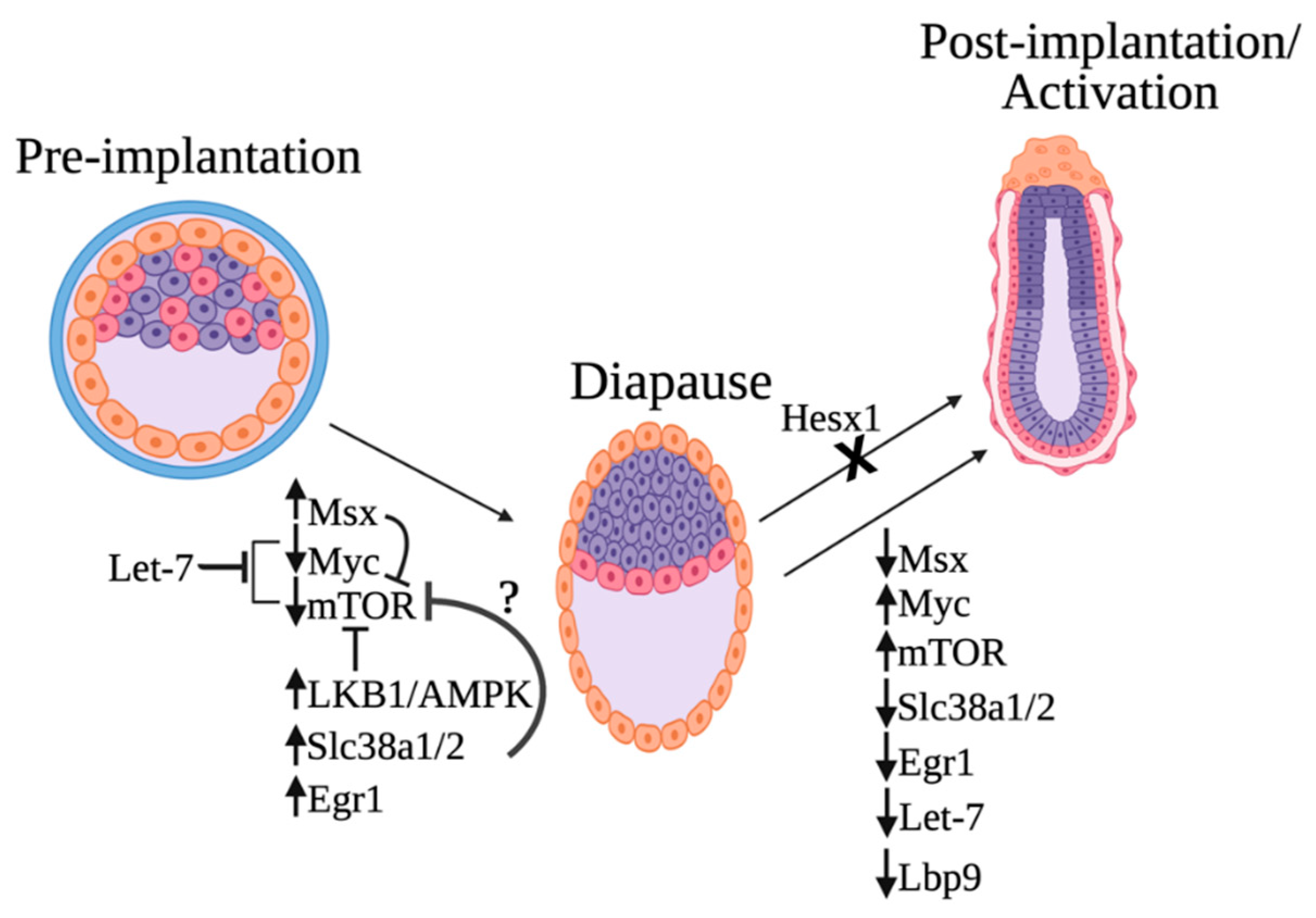
3. Molecular Mechanisms of Diapause Regulation
3.1. The Transcription Factor Hesx1
3.2. Micro-RNA LET-7
3.3. Epigenetic Remodeling in Diapause
3.4. mTOR and the Maternal Control of the Diapause State
4. Embryonic Diapause and Its Relevance in Cancer Stem Cells
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenelon, J.C.; Banerjee, A.; Murphy, B.D. Embryonic diapause: Development on hold. Int. J. Dev. Biol. 2014, 58, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.L.; Desmarais, J.A.; Murphy, B.D. Embryonic diapause and its regulation. Reproduction 2004, 128, 669–678. [Google Scholar] [CrossRef] [PubMed]
- McLaren, A. A study of blastocysts during delay and subsequent implantation in lactating mice. BioScientifica 1968, 42, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, K. A sequence of events in the uterus prior to implantation in the mouse. J. Assist. Reprod. Genet. 2013, 30, 1017–1022. [Google Scholar] [CrossRef]
- Mantalenakis, S.J.; Ketchel, M.M. Frequency and extent of delayed implantation in lactating rats and mice. J. Reprod. Fertil. 1966, 12, 391–394. [Google Scholar] [CrossRef]
- Cha, J.; Fenelon, J.C.; Murphy, B.D.; Shaw, G.; Renfree, M.B.; Dey, S.K. A role for Msx genes in mammalian embryonic diapause. Biosci. Proc. 2020, 10, 44–51. [Google Scholar] [CrossRef]
- Renfree, M.B.; Fenelon, J.C. The enigma of embryonic diapause. Development 2017, 144, 3199–3210. [Google Scholar] [CrossRef]
- Arena, R.; Bisogno, S.; Gąsior, Ł.; Rudnicka, J.; Bernhardt, L.; Haaf, T.; Zacchini, F.; Bochenek, M.; Fic, K.; Bik, E.; et al. Lipid droplets in mammalian eggs are utilized during embryonic diapause. Proc. Natl. Acad. Sci. USA 2021, 118, e2018362118. [Google Scholar] [CrossRef]
- van der Weijden, V.A.; Bick, J.T.; Bauersachs, S.; Rüegg, A.B.; Hildebrandt, T.B.; Goeritz, F.; Jewgenow, K.; Giesbertz, P.; Daniel, H.; Derisoud, E.; et al. Amino acids activate mTORC1 to release roe deer embryos from decelerated proliferation during diapause. Proc. Natl. Acad. Sci. USA. 2021, 118, e2100500118. [Google Scholar] [CrossRef]
- Nichols, J.; Chambers, I.; Taga, T.; Smith, A. Physiological rationale for responsiveness of mouse embryonic stem cells to gp130 cytokines. Development 2001, 128, 2333–2339. [Google Scholar] [CrossRef]
- Rose-John, S. GP130 stimulation and the maintenance of stem cells. Trends Biotechnol. 2002, 20, 417–419. [Google Scholar] [CrossRef]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Menke, T.M.; McLaren, A. Carbon dioxide production by mouse blastocysts during lactational delay of implantation or after ovariectomy. J. Endocrinol. 1970, 47, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Pike, I.L. Comparative studies of embryo metabolism in early pregnancy. J. Reprod. Fertil. Suppl. 1981, 29, 203–213. [Google Scholar]
- Van Blerkom, J.; Chavez, D.J.; Bell, H. Molecular and cellular aspects of facultative delayed implantation in the mouse. Ciba Found. Symp. 1978, 64, 141–172. [Google Scholar]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Mathieu, J.; Ruohola-Baker, H. Metabolic remodeling during the loss and acquisition of pluripotency. Development 2017, 144, 541–551. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Somasundaram, L.; Levy, S.; Hussein, A.M.; Ehnes, D.D.; Mathieu, J.; Ruohola-Baker, H. Epigenetic metabolites license stem cell states. Curr. Top. Dev. Biol. 2020, 138, 209–240. [Google Scholar] [CrossRef]
- Ehnes, D.D.; Hussein, A.M.; Ware, C.B.; Mathieu, J.; Ruohola-Baker, H. Combinatorial metabolism drives the naive to primed pluripotent chromatin landscape. Exp. Cell Res. 2020, 389, 111913. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Malik, N.; Kim, Y.I.; He, Y.; Li, M.; Dubois, W.; Liu, H.; Peat, T.J.; Nguyen, J.T.; Tseng, Y.C.; et al. Fatty acid oxidation is required for embryonic stem cell survival during metabolic stress. EMBO Rep. 2021, 22, e52122. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over mTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250. [Google Scholar] [CrossRef]
- Lee, J.E.; Oh, H.A.; Song, H.; Jun, J.H.; Roh, C.R.; Xie, H.; Dey, S.K.; Lim, H.J. Autophagy regulates embryonic survival during delayed implantation. Endocrinology 2011, 152, 2067–2075. [Google Scholar] [CrossRef]
- Tao, J.; Ma, Y.C.; Yang, Z.S.; Zou, C.G.; Zhang, K.Q. Octopamine connects nutrient cues to lipid metabolism upon nutrient deprivation. Sci. Adv. 2016, 2, e1501372. [Google Scholar] [CrossRef]
- Kouba, D.J.; Nakano, H.; Nishiyama, T.; Kang, J.; Uitto, J.; Mauviel, A. Tumor necrosis factor-alpha induces distinctive NF-kappa B signaling within human dermal fibroblasts. J. Biol. Chem. 2001, 276, 6214–6224. [Google Scholar] [CrossRef]
- Vuong, B.; Hogan-Cann, A.D.; Alano, C.C.; Stevenson, M.; Chan, W.Y.; Anderson, C.M.; Swanson, R.A.; Kauppinen, T.M. NF-κB transcriptional activation by TNFα requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. J. Neuroinflammation 2015, 12, 229. [Google Scholar] [CrossRef]
- Schütze, S.; Potthoff, K.; Machleidt, T.; Berkovic, D.; Wiegmann, K.; Krönke, M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 1992, 71, 765–776. [Google Scholar] [CrossRef]
- Zamorano, J.; Rivas, M.D.; Garcia-Trinidad, A.; Qu, C.K.; Keegan, A.D. Phosphatidylcholine-specific phospholipase C activity is necessary for the activation of STAT6. J. Immunol. 2003, 171, 4203–4209. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF-kappaB signaling: Impact on the aging process. Int. J. Cell Biol. 2012, 2012, 849541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nivon, M.; Richet, E.; Codogno, P.; Arrigo, A.P.; Kretz-Remy, C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 2009, 5, 766–783. [Google Scholar] [CrossRef] [PubMed]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef]
- Molaei, M.; Vandehoef, C.; Karpac, J. NF-κB Shapes Metabolic Adaptation by Attenuating Foxo-Mediated Lipolysis in Drosophila. Dev. Cell 2019, 49, 802–810.e6. [Google Scholar] [CrossRef]
- Tatsumi, T.; Takayama, K.; Ishii, S.; Yamamoto, A.; Hara, T.; Minami, N.; Miyasaka, N.; Kubota, T.; Matsuura, A.; Itakura, E.; et al. Forced lipophagy reveals that lipid droplets are required for early embryonic development in mouse. Development 2018, 145, dev161893. [Google Scholar] [CrossRef]
- Aizawa, R.; Ibayashi, M.; Tatsumi, T.; Yamamoto, A.; Kokubo, T.; Miyasaka, N.; Sato, K.; Ikeda, S.; Minami, N.; Tsukamoto, S. Synthesis and maintenance of lipid droplets are essential for mouse preimplantation embryonic development. Development 2019, 146, dev181925. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, K.; Li, L.; Hao, Z.; Wang, T.; Liu, Y.; Xing, G.; Liu, Z.; Li, H.; Yuan, H.; et al. Plin2-mediated lipid droplet mobilization accelerates exit from pluripotency by lipidomic remodeling and histone acetylation. Cell Death Differ. 2022. [Google Scholar] [CrossRef]
- Pozzi, S.; Bowling, S.; Apps, J.; Brickman, J.M.; Rodriguez, T.A.; Martinez-Barbera, J.P. Genetic Deletion of Hesx1 Promotes Exit from the Pluripotent State and Impairs Developmental Diapause. Stem Cell Rep. 2019, 13, 970–979. [Google Scholar] [CrossRef]
- Dattani, M.T.; Martinez-Barbera, J.P.; Thomas, P.Q.; Brickman, J.M.; Gupta, R.; Mårtensson, I.L.; Toresson, H.; Fox, M.; Wales, J.K.; Hindmarsh, P.C.; et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat. Genet. 1998, 19, 125–133. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Signore, M.; Sajedi, E.; Gaston-Massuet, C.; Kelberman, D.; Burns, A.J.; Itasaki, N.; Dattani, M.; Martinez-Barbera, J.P. Lack of the murine homeobox gene Hesx1 leads to a posterior transformation of the anterior forebrain. Development 2007, 134, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.; Tan, S.P.; Tan, J.H.; Chan, W.K.; Bongso, A. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem Cells 2004, 22, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Z.; Wang, Z.W.; Chen, L.L.; Xue, H.N.; Chen, X.; Guo, Z.K.; Zhang, Y. Hesx1 enhances pluripotency by working downstream of multiple pluripotency-associated signaling pathways. Biochem. Biophys. Res. Commun. 2015, 464, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Renfree, M.B.; Shaw, G. Diapause. Annu. Rev. Physiol. 2000, 62, 353–375. [Google Scholar] [CrossRef]
- Liu, W.M.; Pang, R.T.; Cheong, A.W.; Ng, E.H.; Lao, K.; Lee, K.F.; Yeung, W.S. Involvement of microRNA lethal-7a in the regulation of embryo implantation in mice. PLoS ONE 2012, 7, e37039. [Google Scholar] [CrossRef]
- Cheong, A.W.; Pang, R.T.; Liu, W.M.; Kottawatta, K.S.; Lee, K.F.; Yeung, W.S. MicroRNA Let-7a and dicer are important in the activation and implantation of delayed implanting mouse embryos. Hum. Reprod. 2014, 29, 750–762. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef]
- Hatfield, S.D.; Shcherbata, H.R.; Fischer, K.A.; Nakahara, K.; Carthew, R.W.; Ruohola-Baker, H. Stem cell division is regulated by the microRNA pathway. Nature 2005, 435, 974–978. [Google Scholar] [CrossRef]
- Shcherbata, H.R.; Hatfield, S.; Ward, E.J.; Reynolds, S.; Fischer, K.A.; Ruohola-Baker, H. The MicroRNA pathway plays a regulatory role in stem cell division. Cell Cycle 2006, 5, 172–175. [Google Scholar] [CrossRef]
- Wang, Y.; Hussein, A.M.; Somasundaram, L.; Sankar, R.; Detraux, D.; Mathieu, J.; Ruohola-Baker, H. microRNAs Regulating Human and Mouse Naïve Pluripotency. Int. J. Mol. Sci. 2019, 20, 5864. [Google Scholar] [CrossRef]
- Gurtan, A.M.; Ravi, A.; Rahl, P.B.; Bosson, A.D.; JnBaptiste, C.K.; Bhutkar, A.; Whittaker, C.A.; Young, R.A.; Sharp, P.A. Let-7 represses Nr6a1 and a mid-gestation developmental program in adult fibroblasts. Genes Dev. 2013, 27, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.M.; Cheng, R.R.; Niu, Z.R.; Chen, A.C.; Ma, M.Y.; Li, T.; Chiu, P.C.; Pang, R.T.; Lee, Y.L.; Ou, J.P.; et al. Let-7 derived from endometrial extracellular vesicles is an important inducer of embryonic diapause in mice. Sci. Adv. 2020, 6, eaaz7070. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, Á.; Baumgärtner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- Xu, X.; Ahmed, T.; Wang, L.; Cao, X.; Zhang, Z.; Wang, M.; Lv, Y.; Kanwal, S.; Tariq, M.; Lin, R.; et al. The mTORC1-eIF4F axis controls paused pluripotency. EMBO Rep. 2022, 23, e53081. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Lee, S.L.; Sadovsky, Y.; Swirnoff, A.H.; Polish, J.A.; Goda, P.; Gavrilina, G.; Milbrandt, J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1). Science 1996, 273, 1219–1221. [Google Scholar] [CrossRef]
- Guo, B.; Tian, X.C.; Li, D.D.; Yang, Z.Q.; Cao, H.; Zhang, Q.L.; Liu, J.X.; Yue, Z.P. Expression, regulation and function of Egr1 during implantation and decidualization in mice. Cell Cycle 2014, 13, 2626–2640. [Google Scholar] [CrossRef] [PubMed]
- Battle, S.L.; Doni Jayavelu, N.; Azad, R.N.; Hesson, J.; Ahmed, F.N.; Overbey, E.G.; Zoller, J.A.; Mathieu, J.; Ruohola-Baker, H.; Ware, C.B.; et al. Enhancer Chromatin and 3D Genome Architecture Changes from Naive to Primed Human Embryonic Stem Cell States. Stem Cell Rep. 2019, 12, 1129–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, A.; Torres-Padilla, M.E. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 723–734. [Google Scholar] [CrossRef]
- Wu, J.; Huang, B.; Chen, H.; Yin, Q.; Liu, Y.; Xiang, Y.; Zhang, B.; Liu, B.; Wang, Q.; Xia, W.; et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016, 534, 652–657. [Google Scholar] [CrossRef]
- Cabot, B.; Cabot, R.A. Chromatin remodeling in mammalian embryos. Reproduction 2018, 155, R147–R158. [Google Scholar] [CrossRef]
- Pastor, W.A.; Chen, D.; Liu, W.; Kim, R.; Sahakyan, A.; Lukianchikov, A.; Plath, K.; Jacobsen, S.E.; Clark, A.T. Naive Human Pluripotent Cells Feature a Methylation Landscape Devoid of Blastocyst or Germline Memory. Cell Stem Cell 2016, 8, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Friedli, M.; He, Y.; Planet, E.; O’Neil, R.C.; Markoulaki, S.; Pontis, J.; Wang, H.; Iouranova, A.; Imbeault, M.; et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 2016, 19, 502–515. [Google Scholar] [CrossRef]
- Moody, J.D.; Levy, S.; Mathieu, J.; Xing, Y.; Kim, W.; Dong, C.; Tempel, W.; Robitaille, A.M.; Dang, L.T.; Ferreccio, A.; et al. First critical repressive H3K27me3 marks in embryonic stem cells identified using designed protein inhibitor. Proc. Natl. Acad. Sci. USA 2017, 114, 10125–10130. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, B.; Wang, S.; Wu, W.; Wang, Q.; Chen, Y.; Kong, S.; Lu, J.; Tang, Z.; Ran, H.; et al. Integral proteomic analysis of blastocysts reveals key molecular machinery governing embryonic diapause and reactivation for implantation in mice. Biol. Reprod. 2014, 90, 52. [Google Scholar] [CrossRef]
- He, B.; Zhang, H.; Wang, J.; Liu, M.; Sun, Y.; Guo, C.; Lu, J.; Wang, H.; Kong, S. Blastocyst activation engenders transcriptome reprogram affecting X-chromosome reactivation and inflammatory trigger of implantation. Proc. Natl. Acad. Sci. USA 2019, 116, 16621–16630. [Google Scholar] [CrossRef]
- Schulenburg, H.; Félix, M.A. The Natural Biotic Environment of Caenorhabditis elegans. Genetics 2017, 206, 55–86. [Google Scholar] [CrossRef] [PubMed]
- Kiontke, K.; Sudhaus, W. Ecology of Caenorhabditis species. WormBook 2006, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.E.; Beverly, M.; Russ, C.; Nusbaum, C.; Sengupta, P. A cellular memory of developmental history generates phenotypic diversity in C. elegans. Curr. Biol. 2010, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Sajedi, E.; Gaston-Massuet, C.; Andoniadou, C.L.; Signore, M.; Hurd, P.J.; Dattani, M.; Martinez-Barbera, J.P. DNMT1 interacts with the developmental transcriptional repressor HESX1. Biochim. Biophys. Acta. 2008, 1783, 131–143. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Li, M.; Yu, J.; Tilgner, K.; Ong, S.H.; Koike-Yusa, H.; Yusa, K. Genome-wide CRISPR-KO Screen Uncovers mTORC1-Mediated Gsk3 Regulation in Naive Pluripotency Maintenance and Dissolution. Cell Rep. 2018, 24, 489–502. [Google Scholar] [CrossRef]
- Mathieu, J.; Detraux, D.; Kuppers, D.; Wang, Y.; Cavanaugh, C.; Sidhu, S.; Levy, S.; Robitaille, A.M.; Ferreccio, A.; Bottorff, T.; et al. Folliculin regulates mTORC1/2 and WNT pathways in early human pluripotency. Nat. Commun. 2019, 10, 632. [Google Scholar] [CrossRef]
- Villegas, F.; Lehalle, D.; Mayer, D.; Rittirsch, M.; Stadler, M.B.; Zinner, M.; Olivieri, D.; Vabres, P.; Duplomb-Jego, L.; De Bont, E.S.J.M.; et al. Lysosomal Signaling Licenses Embryonic Stem Cell Differentiation via Inactivation of Tfe3. Cell Stem Cell 2019, 24, 257–270.e8. [Google Scholar] [CrossRef] [PubMed]
- Betschinger, J.; Nichols, J.; Dietmann, S.; Corrin, P.D.; Paddison, P.J.; Smith, A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 2013, 153, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennebeck, G.; Kleymenova, E.V.; Anderson, R.; Yeung, R.S.; Artzt, K.; Walker, C.L. Loss of function of the tuberous sclerosis 2 tumor suppressor gene results in embryonic lethality characterized by disrupted neuroepithelial growth and development. Proc. Natl. Acad. Sci. USA 1998, 95, 15629–15634. [Google Scholar] [CrossRef]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Shiota, C.; Woo, J.T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 2006, 11, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef]
- Goorden, S.M.; Hoogeveen-Westerveld, M.; Cheng, C.; van Woerden, G.M.; Mozaffari, M.; Post, L.; Duckers, H.J.; Nellist, M.; Elgersma, Y. Rheb is essential for murine development. Mol. Cell Biol. 2011, 31, 1672–1678. [Google Scholar] [CrossRef]
- Sousa, M.I.; Correia, B.; Rodrigues, A.S.; Ramalho-Santos, J. Metabolic characterization of a paused-like pluripotent state. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129612. [Google Scholar] [CrossRef]
- Shaw, G.; Renfree, M.B. Uterine and embryonic metabolism after diapause in the tammar wallaby, Macropus eugenii. J. Reprod. Fertil. 1986, 76, 339–347. [Google Scholar] [CrossRef]
- Renfree, M.B.; Shaw, G. Embryo-endometrial interactions during early development after embryonic diapause in the marsupial tammar wallaby. Int. J. Dev. Biol. 2014, 58, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.D. Embryonic diapause: Advances in understanding the enigma of seasonal delayed implantation. Reprod. Domest. Anim. 2012, 47 (Suppl. S6), 121–124. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, H.; Jiang, H.; Ren, Y.; Yu, X.; Qiu, J.; Stablewski, A.B.; Zhang, B.; Buck, M.J.; Feng, J. Transient inhibition of mTOR in human pluripotent stem cells enables robust formation of mouse-human chimeric embryos. Sci. Adv. 2020, 6, eaaz0298. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Sun, X.; Bartos, A.; Fenelon, J.; Lefèvre, P.; Daikoku, T.; Shaw, G.; Maxson, R.; Murphy, B.D.; Renfree, M.B.; et al. A new role for muscle segment homeobox genes in mammalian embryonic diapause. Open Biol. 2013, 3, 130035. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Cha, J.; Sun, X.; Tranguch, S.; Xie, H.; Fujita, T.; Hirota, Y.; Lydon, J.; DeMayo, F.; Maxson, R.; et al. Conditional deletion of Msx homeobox genes in the uterus inhibits blastocyst implantation by altering uterine receptivity. Dev. Cell 2011, 21, 1014–1025. [Google Scholar] [CrossRef]
- Cha, J.; Burnum-Johnson, K.E.; Bartos, A.; Li, Y.; Baker, E.S.; Tilton, S.C.; Webb-Robertson, B.J.; Piehowski, P.D.; Monroe, M.E.; Jegga, A.G.; et al. Muscle Segment Homeobox Genes Direct Embryonic Diapause by Limiting Inflammation in the Uterus. J. Biol. Chem. 2015, 290, 15337–15349. [Google Scholar] [CrossRef]
- Son, M.J.; Rho, S.B.; Kim, K.; Oh, M.; Son, C.; Song, S.Y.; Park, K. Homeoprotein Msx1-PIASy Interaction Inhibits Angiogenesis. Cells 2020, 9, 1854. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Rapp, U.R.; Ceteci, F.; Schreck, R. Oncogene-induced plasticity and cancer stem cells. Cell Cycle 2008, 7, 45–51. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107, 1110. [Google Scholar] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Somasundaram, L.; Raj, I.X.; Ic-Mex, D.; Phal, A.; Schmidt, S.; Ng, W.I.; Mar, D.; Decarreau, J.; Moss, N.; et al. dCas9 fusion to computer-designed PRC2 inhibitor reveals functional TATA box in distal promoter region. Cell Rep. 2022, 38, 110457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| MicroRNA | Target Gene | Context++ Score Percentile |
|---|---|---|
| mmu-miR-199a-3p | Slc38a1 | 75 |
| mmu-miR-199b-3p | Slc38a1 | 75 |
| mmu-miR-199a-3p | Egr1 | 87 |
| mmu-miR-199b-3p | Egr1 | 87 |
| mmu-miR-181-5p | Egr1 | 85 |
| mmu-miR-181-5p | Slc38a2 | 84 |
| mmu-let-7a-5p | Rictor | 83 |
| mmu-let-7b-5p | Rictor | 83 |
| mmu-let-7c-5p | Rictor | 83 |
| mmu-let-7d-5p | Rictor | 85 |
| mmu-let-7e-5p | Rictor | 83 |
| mmu-let-7f-5p | Rictor | 82 |
| mmu-let-7g-5p | Rictor | 84 |
| mmu-let-7i-5p | Rictor | 84 |
| mmu-let-7k | Rictor | 83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussein, A.M.; Balachandar, N.; Mathieu, J.; Ruohola-Baker, H. Molecular Regulators of Embryonic Diapause and Cancer Diapause-like State. Cells 2022, 11, 2929. https://doi.org/10.3390/cells11192929
Hussein AM, Balachandar N, Mathieu J, Ruohola-Baker H. Molecular Regulators of Embryonic Diapause and Cancer Diapause-like State. Cells. 2022; 11(19):2929. https://doi.org/10.3390/cells11192929
Chicago/Turabian StyleHussein, Abdiasis M., Nanditaa Balachandar, Julie Mathieu, and Hannele Ruohola-Baker. 2022. "Molecular Regulators of Embryonic Diapause and Cancer Diapause-like State" Cells 11, no. 19: 2929. https://doi.org/10.3390/cells11192929