

The Extracellular Matrix and Neuroblastoma Cell Communication—A Complex Interplay and Its Therapeutic Implications

Abstract
:1. Introduction
2. Complex Interplay between Tumor Cells and ECM Components in NB
2.1. ECM Constituents Produced by NB Cells
2.2. ECM Content and Its Structural Features Can Aid NB Prognosis
2.3. ECM Components of NB Are Affected by Differentiation
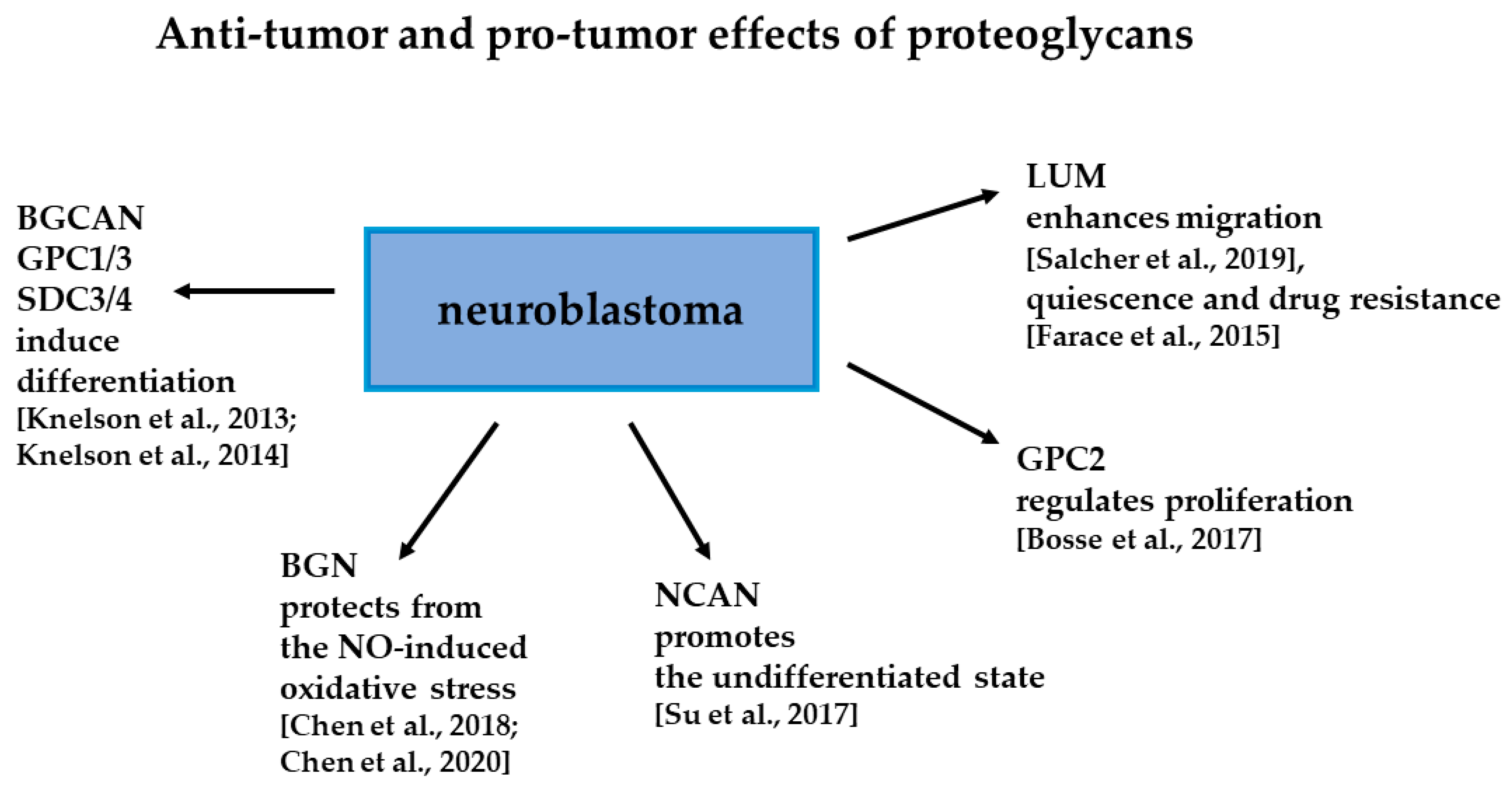
3. Proteoglycans Are Regulating Phenotype of NB Cells
3.1. Small Leucine Rich Repeat Proteoglycans in NB
3.2. Roles of Neurocan in NB
3.3. Cell Surface PGs
3.3.1. Glypicans
3.3.2. Roles of HS-PGs in NB Differentiation
3.4. Enzymes Modifying HS-PGs
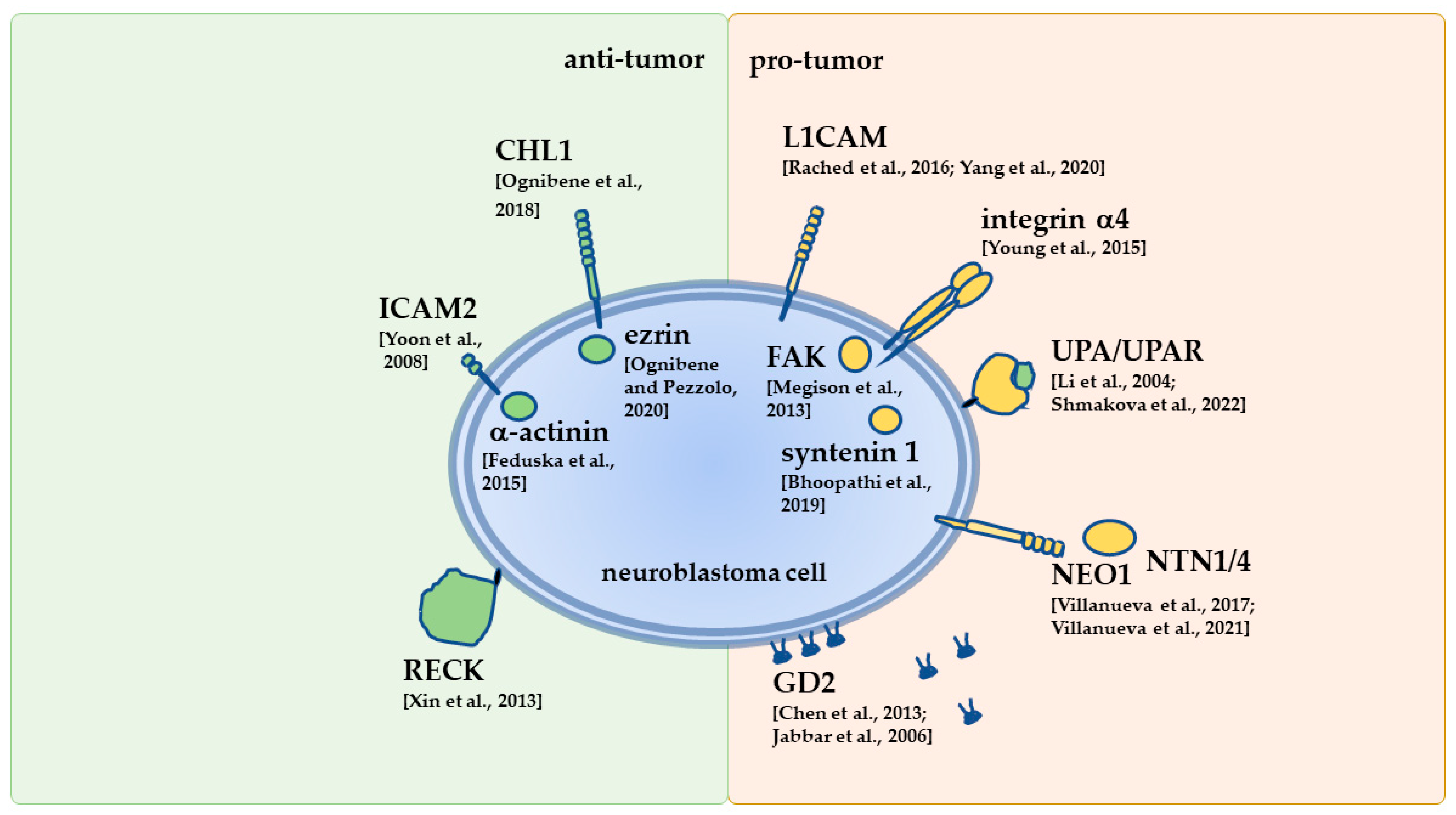
4. Overview of Selected NB Receptors Engaged in Interactions with the ECM
4.1. Integrins Are Major Adhesion Receptors Which Link the Cytoskeleton to ECM
4.1.1. Integrin Expression in NB
4.1.2. Integrins Play Roles in NB Differentiation
4.1.3. Integrins Play Roles in NB Survival
4.1.4. Roles of Integrins in Metastasis of NB Cells
4.1.5. FAK Is Upregulated in MYCN-Amplified NB
4.1.6. Roles of MDA-9/Syntenin 1 in NB
4.1.7. Roles of Caspase 8 in NB
4.2. Laminin Receptor/Ribosomal Protein SA
4.3. Ig-like Cell Adhesion Molecule Family
4.3.1. Intercellular Adhesion Molecule 2
4.3.2. Neural Cell Adhesion Molecule 1
4.3.3. Cell Adhesion Molecule L1-Like
4.3.4. L1 Cell Adhesion Molecule
4.4. Roles of CD44 in Regulation of Phenotype of NB Cells
4.5. Dependence Receptors
5. Glycocalyx Changes Affect Interactions of NB Cells with the ECM
5.1. Roles of Glycosylating Enzymes in Regulation of NB Aggressiveness
5.2. Ganglioside GD2
6. NB and ECM Degradation
6.1. MMPs and Their Inhibitors
6.2. Plasminogen Activator (UPA) and Plasminogen Activator Receptor (UPAR)
7. Targeting Interactions of NB with ECM Components
8. Future Prospects and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kastriti, M.E.; Kameneva, P.; Adameyko, I. Stem cells, evolutionary aspects and pathology of the adrenal medulla: A new developmental paradigm. Mol. Cell. Endocrinol. 2020, 518, 110998. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, H. Linking human sympathoadrenal development and neuroblastoma. Nat. Genet. 2021, 53, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Vo, K.T.; Matthay, K.K.; Neuhaus, J.; London, W.B.; Hero, B.; Ambros, P.F.; Nakagawara, A.; Miniati, D.; Wheeler, K.; Pearson, A.D.; et al. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: A report from the international neuroblastoma risk group project. J. Clin. Oncol. 2014, 32, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.H.; Federico, S.M.; London, W.B.; Naranjo, A.; Irwin, M.S.; Volchenboum, S.L.; Cohn, S.L. Tailoring Therapy for Children with Neuroblastoma on the Basis of Risk Group Classification: Past; Present; and Future. JCO Clin. Cancer. Inform. 2020, 4, 895–905. [Google Scholar] [CrossRef]
- Nakazawa, A. Biological categories of neuroblastoma based on the international neuroblastoma pathology classification for treatment stratification. Pathol. Int. 2021, 71, 232–244. [Google Scholar] [CrossRef]
- Castel, V.; García-Miguel, P.; Cañete, A.; Melero, C.; Navajas, A.; Ruíz-Jiménez, J.I.; Navarro, S.; Badal, M.D. Prospective evaluation of the International Neuroblastoma Staging System (INSS) and the International Neuroblastoma Response Criteria (INRC) in a multicentre setting. Eur. J. Cancer 1999, 35, 606–611. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Brodeur, G.M. Spontaneous regression of neuroblastoma. Cell. Tissue Res. 2018, 372, 277–286. [Google Scholar] [CrossRef]
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book. 2022, 42, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer. Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Mueller, I.; Ehlert, K.; Endres, S.; Pill, L.; Siebert, N.; Kietz, S.; Brock, P.; Garaventa, A.; Valteau-Couanet, D.; Janzek, E.; et al. Tolerability; response and outcome of high-risk neuroblastoma patients treated with long-term infusion of anti-GD2 antibody ch14.18/CHO. MAbs 2018, 10, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuBois, S.G.; Granger, M.M.; Groshen, S.; Tsao-Wei, D.; Ji, L.; Shamirian, A.; Czarnecki, S.; Goodarzian, F.; Berkovich, R.; Shimada, H.; et al. Randomized Phase II Trial of MIBG Versus MIBG; Vincristine; and Irinotecan Versus MIBG and Vorinostat for Patients with Relapsed or Refractory Neuroblastoma: A Report from NANT Consortium. J. Clin. Oncol. 2021, 39, 3506–3514. [Google Scholar] [CrossRef]
- Mody, R.; Yu, A.L.; Naranjo, A.; Zhang, F.F.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.E.; Diccianni, M.B.; Hank, J.A.; et al. Irinotecan; Temozolomide; and Dinutuximab With GM-CSF in Children with Refractory or Relapsed Neuroblastoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2020, 38, 2160–2169. [Google Scholar] [CrossRef]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 5183. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Bagatell, R.; Cohn, S.L.; Hogarty, M.D.; Maris, J.M.; Moreno, L.; Park, J.R.; Pearson, A.D.; Schleiermacher, G.; Valteau-Couanet, D.; et al. The challenge of defining “ultra-high-risk” neuroblastoma. Pediatr. Blood Cancer 2019, 66, e27556. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef]
- Wolpaw, A.J.; Bayliss, R.; Büchel, G.; Dang, C.V.; Eilers, M.; Gustafson, W.C.; Hansen, G.H.; Jura, N.; Knapp, S.; Lemmon, M.A.; et al. Drugging the “Undruggable” MYCN Oncogenic Transcription Factor: Overcoming Previous Obstacles to Impact Childhood Cancers. Cancer Res. 2021, 81, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos Cogo, S.; Gradowski Farias da Costa do Nascimento, T.; de Almeida Brehm Pinhatti, F.; de França Junior, N.; Santos Rodrigues, B.; Cavalli, L.R.; Elifio-Esposito, S. An overview of neuroblastoma cell lineage phenotypes and in vitro models. Exp. Biol. Med. 2020, 245, 1637–1647. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Corallo, D.; Frabetti, S.; Candini, O.; Gregianin, E.; Dominici, M.; Fischer, H.; Aveic, S. Emerging neuroblastoma 3D in vitro models for pre-clinical assessments. Front. Immunol. 2020, 11, 584214:1–584214:12. [Google Scholar] [CrossRef]
- Quinn, C.H.; Beierle, A.M.; Beierle, E.A. Artificial tumor microenvironments in neuroblastoma. Cancers 2021, 13, 1629. [Google Scholar] [CrossRef]
- Braekeveldt, N.; Bexell, D. Patient-derived xenografts as preclinical neuroblastoma models. Cell Tissue Res. 2018, 372, 233–243. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R. The chick embryo chorioallantoic membrane as an in vivo experimental model to study human neuroblastoma. J. Cell. Physiol. 2018, 234, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Casey, M.J.; Stewart, R.A. Zebrafish as a model to study neuroblastoma development. Cell Tissue Res. 2018, 372, 223–232. [Google Scholar] [CrossRef]
- Kamili, A.; Atkinson, C.; Trahair, T.N.; Fletcher, J.I. Mouse models of high-risk neuroblastoma. Cancer Metastasis Rev. 2020, 39, 261–274. [Google Scholar] [CrossRef]
- Ara, T.; DeClerck, Y.A. Mechanisms of invasion and metastasis in human neuroblastoma. Cancer Metastasis Rev. 2006, 25, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players; New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Gladson, C.L.; Dennis, C.; Rotolo, T.C.; Kelly, D.R.; Grammer, J.R. Vitronectin expression in differentiating neuroblastic tumors: Integrin alpha v beta 5 mediates vitronectin-dependent adhesion of retinoic-acid-differentiated neuroblastoma cells. Am. J. Pathol. 1997, 150, 1631–1646. [Google Scholar] [PubMed]
- Burgos-Panadero, R.; Noguera, I.; Cañete, A.; Navarro, S.; Noguera, R. Vitronectin as a molecular player of the tumor microenvironment in neuroblastoma. BMC Cancer 2019, 19, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, S.; Modesti, A.; Triche, T.J. Extracellular matrix synthesis by undifferentiated childhood tumor cell lines. Am. J. Pathol. 1987, 129, 74–85. [Google Scholar] [PubMed]
- Truong, D.Q.; Ho, B.T.; Chau, G.C.; Truong, D.K.; Pham, T.T.T.; Nakagawara, A.; Bui, C.B. Collagen XI Alpha 1 (COL11A1) Expression in the Tumor Microenvironment Drives Neuroblastoma Dissemination. Pediatr. Dev. Pathol. 2022, 25, 91–98. [Google Scholar] [CrossRef]
- Darvishi, B.; Eisavand, M.R.; Majidzadeh-A, K.; Farahmand, L. Matrix stiffening and acquired resistance to chemotherapy: Concepts and clinical significance. Br. J. Cancer 2022, 126, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Tadeo, I.; Berbegall, A.P.; Navarro, S.; Castel, V.; Noguera, R. A stiff extracellular matrix is associated with malignancy in peripheral neuroblastic tumors. Pediatr. Blood Cancer 2017, 64, e26449. [Google Scholar] [CrossRef] [PubMed]
- Tadeo, I.; Berbegall, A.P.; Castel, V.; García-Miguel, P.; Callaghan, R.; Påhlman, S.; Navarro, S.; Noguera, R. Extracellular matrix composition defines an ultra-high-risk group of neuroblastoma within the high-risk patient cohort. Br. J. Cancer 2016, 115, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Carrasco, A.; Martín-Vañó, S.; Burgos-Panadero, R.; Monferrer, E.; Berbegall, A.P.; Fernández-Blanco, B.; Navarro, S.; Noguera, R. Impact of extracellular matrix stiffness on genomic heterogeneity in MYCN-amplified neuroblastoma cell line. J. Exp. Clin. Cancer. Res. 2020, 39, 226. [Google Scholar] [CrossRef]
- Tsokos, M.; Scarpa, S.; Ross, R.A.; Triche, T.J. Differentiation of human neuroblastoma recapitulates neural crest development. Study of morphology; neurotransmitter enzymes; and extracellular matrix proteins. Am. J. Pathol. 1987, 128, 484–496. [Google Scholar]
- Halakos, E.G.; Connell, A.J.; Glazewski, L.; Wei, S.; Mason, R.W. Bottom up proteomics identifies neuronal differentiation pathway networks activated by cathepsin inhibition treatment in neuroblastoma cells that are enhanced by concurrent 13-cis retinoic acid treatment. J. Proteomics 2021, 232, 104068. [Google Scholar] [CrossRef]
- Tan, X.; Gong, W.; Chen, B.; Gong, B.; Hua, Z.; Zhang, S.; Chen, Y.; Li, Q.; Li, Z. Downregulation of fibronectin 1 attenuates ATRA-induced inhibition of cell migration and invasion in neuroblastoma cells. Mol. Cell. Biochem. 2021, 476, 3601–3612. [Google Scholar] [CrossRef]
- Barkovskaya, A.; Buffone, A., Jr.; Žídek, M.; Weaver, V.M. Proteoglycans as Mediators of Cancer Tissue Mechanics. Front. Cell. Dev. Biol. 2020, 8, 569377. [Google Scholar] [CrossRef]
- Appunni, S.; Anand, V.; Khandelwal, M.; Gupta, N.; Rubens, M.; Sharma, A. Small Leucine Rich Proteoglycans (decorin; biglycan and lumican) in cancer. Clin. Chim. Acta 2019, 491, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guo, D.; Zhang, W.; Xie, Y.; Yang, H.; Cheng, B.; Wang, L.; Yang, R.; Bi, J.; Feng, Z. Biglycan, a Nitric Oxide-Downregulated Proteoglycan; Prevents Nitric Oxide-Induced Neuronal Cell Apoptosis via Targeting Erk1/2 and p38 Signaling Pathways. J. Mol. Neurosci. 2018, 66, 68–76. [Google Scholar] [CrossRef]
- Chen, S.; Guo, D.; Lei, B.; Bi, J. Biglycan protects human neuroblastoma cells from nitric oxide-induced death by inhibiting AMPK-mTOR mediated autophagy and intracellular ROS level. Biotechnol. Lett. 2020, 42, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Yang, K.; Ye, L.; You, Z.; Chen, R.; Liu, Y. Decorin gene upregulation mediated by an adeno-associated virus vector increases intratumoral uptake of nab-paclitaxel in neuroblastoma via inhibition of stabilin-1. Investig. New Drugs 2017, 35, 566–575. [Google Scholar] [CrossRef]
- Salcher, S.; Spoden, G.; Huber, J.M.; Golderer, G.; Lindner, H.; Ausserlechner, M.J.; Kiechl-Kohlendorfer, U.; Geiger, K.; Obexer, P. Repaglinide Silences the FOXO3/Lumican Axis and Represses the Associated Metastatic Potential of Neuronal Cancer Cells. Cells 2019, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farace, C.; Oliver, J.A.; Melguizo, C.; Alvarez, P.; Bandiera, P.; Rama, A.R.; Malaguarnera, G.; Ortiz, R.; Madeddu, R.; Prados, J. Microenvironmental Modulation of Decorin and Lumican in Temozolomide-Resistant Glioblastoma and Neuroblastoma Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0134111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Karamanos, N.K. Proteoglycans remodeling in cancer: Underlying molecular mechanisms. Matrix Biol. 2019, 75–76, 220–259. [Google Scholar] [CrossRef]
- Su, Z.; Kishida, S.; Tsubota, S.; Sakamoto, K.; Cao, D.; Kiyonari, S.; Ohira, M.; Kamijo, T.; Narita, A.; Xu, Y.; et al. Neurocan, an extracellular chondroitin sulfate proteoglycan; stimulates neuroblastoma cells to promote malignant phenotypes. Oncotarget 2017, 8, 106296–106310. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, N.; Chen, G.Y.; Kadomatsu, K.; Ikematsu, S.; Sakuma, S.; Muramatsu, T. Glypican-2 binds to midkine: The role of glypican-2 in neuronal cell adhesion and neurite outgrowth. Glycoconj. J. 2001, 18, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, M.; Zheng, C.; Zhong, Q.; Shi, Y.; Han, X. Diagnostic value of serum glypican-3 alone and in combination with AFP as an aid in the diagnosis of liver cancer. Clin. Biochem. 2020, 79, 54–60. [Google Scholar] [CrossRef]
- Tenorio, J.; Arias, P.; Martínez-Glez, V.; Santos, F.; García-Miñaur, S.; Nevado, J.; Lapunzina, P. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J. Rare Dis. 2014, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.S.; Pawel, B.R.; Corao, D.A.; Venneti, S.; Russo, P.; Santi, M.; Sullivan, L.M. Immunohistochemical expression of glypican-3 in pediatric tumors: An analysis of 414 cases. Pediatr. Dev. Pathol. 2013, 16, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Shibui, Y.; Miyoshi, K.; Kohashi, K.; Kinoshita, Y.; Kuda, M.; Yamamoto, H.; Taguchi, T.; Oda, Y. Glypican-3 expression in malignant small round cell tumors. Oncol. Lett. 2019, 17, 3523–3528. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Raman, P.; Zhu, Z.; Lane, M.; Martinez, D.; Heitzeneder, S.; Rathi, K.S.; Kendsersky, N.M.; Randall, M.; Donovan, L.; et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell 2017, 32, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Lee, C.; Sabharwal, P.; Zhang, M.; Meyers, C.L.; Sockanathan, S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 2013, 339, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Matas-Rico, E.; van Veen, M.; Leyton-Puig, D.; van den Berg, J.; Koster, J.; Kedziora, K.M.; Molenaar, B.; Weerts, M.J.; de Rink, I.; Medema, R.H.; et al. Glycerophosphodiesterase GDE2 Promotes Neuroblastoma Differentiation through Glypican Release and Is a Marker of Clinical Outcome. Cancer Cell 2016, 30, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Knelson, E.H.; Gaviglio, A.L.; Tewari, A.K.; Armstrong, M.B.; Mythreye, K.; Blobe, G.C. Type III TGF-β receptor promotes FGF2-mediated neuronal differentiation in neuroblastoma. J. Clin. Investig. 2013, 123, 4786–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knelson, E.H.; Gaviglio, A.L.; Nee, J.C.; Starr, M.D.; Nixon, A.B.; Marcus, S.G.; Blobe, G.C. Stromal heparan sulfate differentiates neuroblasts to suppress neuroblastoma growth. J. Clin. Investig. 2014, 124, 3016–3031. [Google Scholar] [CrossRef] [Green Version]
- Masola, V.; Zaza, G.; Gambaro, G.; Franchi, M.; Onisto, M. Role of heparanase in tumor progression: Molecular aspects and therapeutic options. Semin. Cancer Biol. 2020, 62, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.D.; Tong, Q.S.; Tang, S.T.; Du, Z.Y.; Liu, Y.; Jiang, G.S.; Cai, J.B. Expression and clinical significance of heparanase in neuroblastoma. World J. Pediatr. 2009, 5, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Zheng, L.; Pu, J.; Mei, H.; Xiang, X.; Zhao, X.; Li, D.; Li, S.; Mao, L.; Huang, K.; et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Hum. Mol. Genet. 2015, 24, 2539–5251. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Zheng, L.; Jiao, W.; Mei, H.; Li, D.; Song, H.; Fang, E.; Wang, X.; Li, S.; Huang, K.; et al. Smad4 suppresses the tumorigenesis and aggressiveness of neuroblastoma through repressing the expression of heparanase. Sci. Rep. 2016, 6, 32628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell. Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction; Stemness; Epithelial Plasticity; and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favrot, M.C.; Combaret, V.; Goillot, E.; Lutz, P.; Frappaz, D.; Thiesse, P.; Thyss, A.; Dolbeau, D.; Bouffet, E.; Tabone, E.; et al. Expression of integrin receptors on 45 clinical neuroblastoma specimens. Int. J. Cancer. 1991, 49, 347–355. [Google Scholar] [CrossRef]
- Gladson, C.L.; Hancock, S.; Arnold, M.M.; Faye-Petersen, O.M.; Castleberry, R.P.; Kelly, D.R. Stage-specific expression of integrin alphaVbeta3 in neuroblastic tumors. Am. J. Pathol. 1996, 48, 1423–1434. [Google Scholar]
- Rossino, P.; Defilippi, P.; Silengo, L.; Tarone, G. Up-regulation of the integrin alpha 1/beta 1 in human neuroblastoma cells differentiated by retinoic acid: Correlation with increased neurite outgrowth response to laminin. Cell Regul. 1991, 2, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Rozzo, C.; Ratti, P.; Ponzoni, M.; Cornaglia-Ferraris, P. Modulation of alpha 1 beta 1; alpha 2 beta 1 and alpha 3 beta 1 integrin heterodimers during human neuroblastoma cell differentiation. FEBS Lett. 1993, 332, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Bozzo, C.; Bellomo, G.; Silengo, L.; Tarone, G.; Altruda, F. Soluble integrin ligands and growth factors independently rescue neuroblastoma cells from apoptosis under nonadherent conditions. Exp. Cell Res. 1997, 237, 326–337. [Google Scholar] [CrossRef]
- Bonfoco, E.; Chen, W.; Paul, R.; Cheresh, D.A.; Cooper, N.R. beta1 integrin antagonism on adherent, differentiated human neuroblastoma cells triggers an apoptotic signaling pathway. Neuroscience 2000, 101, 1145–1152. [Google Scholar] [CrossRef]
- Young, S.A.; McCabe, K.E.; Bartakova, A.; Delaney, J.; Pizzo, D.P.; Newbury, R.O.; Varner, J.A.; Schlaepfer, D.D.; Stupack, D.G. Integrin α4 Enhances Metastasis and May Be Associated with Poor Prognosis in MYCN-low Neuroblastoma. PLoS ONE 2015, 10, e0120815. [Google Scholar] [CrossRef] [Green Version]
- Tzinia, A.K.; Kitsiou, P.V.; Talamagas, A.A.; Georgopoulos, A.; Tsilibary, E.C. Effects of collagen IV on neuroblastoma cell matrix-related functions. Exp. Cell Res. 2002, 274, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bownes, L.V.; Marayati, R.; Quinn, C.H.; Hutchins, S.C.; Stewart, J.E.; Anderson, J.C.; Willey, C.D.; Datta, P.K.; Beierle, E.A. Serine-Threonine Kinase Receptor Associate Protein (STRAP) confers an aggressive phenotype in neuroblastoma via regulation of Focal Adhesion Kinase (FAK). J. Pediatr. Surg. 2022, 57, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Beierle, E.A.; Massoll, N.A.; Hartwich, J.; Kurenova, E.V.; Golubovskaya, V.M.; Cance, W.G.; McGrady, P.; London, W.B. Focal adhesion kinase expression in human neuroblastoma: Immunohistochemical and real-time PCR analyses. Clin. Cancer Res. 2008, 14, 3299–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beierle, E.A.; Trujillo, A.; Nagaram, A.; Kurenova, E.V.; Finch, R.; Ma, X.; Vella, J.; Cance, W.G.; Golubovskaya, V.M. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J. Biol. Chem. 2007, 282, 12503–12516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megison, M.L.; Stewart, J.E.; Nabers, H.C.; Gillory, L.A.; Beierle, E.A. FAK inhibition decreases cell invasion; migration and metastasis in MYCN amplified neuroblastoma. Clin. Exp. Metastasis 2013, 30, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhoopathi, P.; Pradhan, A.K.; Bacolod, M.D.; Emdad, L.; Sarkar, D.; Das, S.K.; Fisher, P.B. Regulation of neuroblastoma migration, invasion, and in vivo metastasis by genetic and pharmacological manipulation of MDA-9/Syntenin. Oncogene 2019, 38, 6781–6793. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.K.; Maji, S.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin/SDCBP: New insights into a unique multifunctional scaffold protein. Cancer Metastasis Rev. 2020, 39, 769–781. [Google Scholar] [CrossRef]
- Hassan, W.M.; Bakry, M.S.; Siepmann, T.; Illigens, B. Association of RASSF1A; DCR2; and CASP8 Methylation with Survival in Neuroblastoma: A Pooled Analysis Using Reconstructed Individual Patient Data. Biomed. Res. Int. 2020, 2020, 7390473. [Google Scholar] [CrossRef]
- Teitz, T.; Stupack, D.G.; Lahti, J.M. Halting neuroblastoma metastasis by controlling integrin-mediated death. Cell Cycle 2006, 5, 681–685. [Google Scholar] [CrossRef]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barilà, D.; Lahti, J.M.; Schlaepfer, D.; et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teitz, T.; Inoue, M.; Valentine, M.B.; Zhu, K.; Rehg, J.E.; Zhao, W.; Finkelstein, D.; Wang, Y.D.; Johnson, M.D.; Calabrese, C.; et al. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer Res. 2013, 73, 4086–4097. [Google Scholar] [CrossRef] [PubMed]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57–58, 213–243. [Google Scholar] [CrossRef] [PubMed]
- DiGiacomo, V.; Meruelo, D. Looking into laminin receptor: Critical discussion regarding the non-integrin 37/67-kDa laminin receptor/RPSA protein. Biol. Rev. Camb. Philos. Soc. 2016, 91, 288–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escamilla, J.M.; Bäuerl, C.; López, C.M.R.; Pekkala, S.P.; Navarro, S.; Barettino, D. Retinoic-Acid-Induced Downregulation of the 67 KDa Laminin Receptor Correlates with Reduced Biological Aggressiveness of Human Neuroblastoma Cells. In Neuroblastoma—Present and Future; Shimada, H., Ed.; InTech: Rijeka, Chroatia, 2012; pp. 217–232. ISBN 978-953-307-016-2. [Google Scholar]
- Chetty, C.J.; Ferreira, E.; Jovanovic, K.; Weiss, S.F.T. Knockdown of LRP/LR induces apoptosis in pancreatic cancer and neuroblastoma cells through activation of caspases. Exp. Cell. Res. 2017, 360, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Feduska, J.M.; Aller, S.G.; Garcia, P.L.; Cramer, S.L.; Council, L.N.; van Waardenburg, R.C.; Yoon, K.J. ICAM-2 confers a non-metastatic phenotype in neuroblastoma cells by interaction with α-actinin. Oncogene 2015, 34, 1553–1562. [Google Scholar] [CrossRef]
- Yoon, K.J.; Phelps, D.A.; Bush, R.A.; Remack, J.S.; Billups, C.A.; Khoury, J.D. ICAM-2 expression mediates a membrane-actin link; confers a nonmetastatic phenotype and reflects favorable tumor stage or histology in neuroblastoma. PLoS ONE 2008, 3, e3629. [Google Scholar] [CrossRef] [Green Version]
- Feduska, J.M.; Garcia, P.L.; Brennan, S.B.; Bu, S.; Council, L.N.; Yoon, K.J. N-glycosylation of ICAM-2 is required for ICAM-2-mediated complete suppression of metastatic potential of SK-N-AS neuroblastoma cells. BMC Cancer 2013, 13, 261. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Pawel, B.; Seiser, E.; Zhao, H.; Raabe, E.; Wang, Q.; Judkins, A.R.; Attiyeh, E.; Maris, J.M. Neural cell adhesion molecule (NCAM) isoform expression is associated with neuroblastoma differentiation status. Pediatr. Blood Cancer 2008, 51, 10–16. [Google Scholar] [CrossRef]
- Ognibene, M.; Pagnan, G.; Marimpietri, D.; Cangelosi, D.; Cilli, M.; Benedetti, M.C.; Boldrini, R.; Garaventa, A.; Frassoni, F.; Eva, A.; et al. CHL1 gene acts as a tumor suppressor in human neuroblastoma. Oncotarget 2018, 9, 25903–25921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ognibene, M.; Pezzolo, A. Ezrin interacts with the tumor suppressor CHL1 and promotes neuronal differentiation of human neuroblastoma. PLoS ONE 2020, 15, e0244069. [Google Scholar] [CrossRef] [PubMed]
- Zaatiti, H.; Abdallah, J.; Nasr, Z.; Khazen, G.; Sandler, A.; Abou-Antoun, T.J. Tumorigenic proteins upregulated in the MYCN-amplified IMR-32 human neuroblastoma cells promote proliferation and migration. Int. J. Oncol. 2018, 52, 787–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhang, X.; Zhao, Y.; Sun, G.; Zhang, J.; Gao, Y.; Liu, Q.; Zhang, W.; Zhu, H. Downregulation of lncRNA XIST Represses Tumor Growth and Boosts Radiosensitivity of Neuroblastoma via Modulation of the miR-375/L1CAM Axis. Neurochem. Res. 2020, 45, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Rached, J.; Nasr, Z.; Abdallah, J.; Abou-Antoun, T. L1-CAM knock-down radiosensitizes neuroblastoma IMR-32 cells by simultaneously decreasing MycN; but increasing PTEN protein expression. Int. J. Oncol. 2016, 49, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.; Cheung, N.K.; Gerald, W.L.; LaQuaglia, M.; Kushner, B.H.; LeClerc, J.M.; LeSauter, L.; Saragovi, H.U. Correlation of MYCN amplification; Trk-A and CD44 expression with clinical stage in 250 patients with neuroblastoma. Eur. J. Cancer 1997, 33, 2098–2100. [Google Scholar] [CrossRef]
- Gross, N.; Beck, D.; Beretta, C.; Jackson, D.; Perruisseau, G. CD44 expression and modulation on human neuroblastoma tumours and cell lines. Eur. J. Cancer 1995, 31, 471–475. [Google Scholar] [CrossRef]
- Gross, N.; Balmas, K.; Beretta Brognara, C. Role of CD44H carbohydrate structure in neuroblastoma adhesive properties. Med. Pediatr. Oncol. 2001, 36, 139–141. [Google Scholar] [CrossRef]
- Fichter, M.; Hinrichs, R.; Eissner, G.; Scheffer, B.; Classen, S.; Ueffing, M. Expression of CD44 isoforms in neuroblastoma cells is regulated by PI 3-kinase and protein kinase C. Oncogene 1997, 14, 2817–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siapati, E.K.; Rouka, E.; Kyriakou, D.; Vassilopoulos, G. Neuroblastoma cells negative for CD44 possess tumor-initiating properties. Cell. Oncol. 2011, 34, 189–197. [Google Scholar] [CrossRef]
- Valentiner, U.; Valentiner, F.U.; Schumacher, U. Expression of CD44 is associated with a metastatic pattern of human neuroblastoma cells in a SCID mouse xenograft model. Tumour Biol. 2008, 29, 152–160. [Google Scholar] [CrossRef]
- Vega, F.M.; Colmenero-Repiso, A.; Gómez-Muñoz, M.A.; Rodríguez-Prieto, I.; Aguilar-Morante, D.; Ramírez, G.; Márquez, C.; Cabello, R.; Pardal, R. CD44-high neural crest stem-like cells are associated with tumour aggressiveness and poor survival in neuroblastoma tumours. EBioMedicine 2019, 49, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; Pezzolo, A. Roniciclib down-regulates stemness and inhibits cell growth by inducing nucleolar stress in neuroblastoma. Sci. Rep. 2020, 10, 12902. [Google Scholar] [CrossRef]
- Gibert, B.; Mehlen, P. Dependence Receptors and Cancer: Addiction to Trophic Ligands. Cancer Res. 2015, 75, 5171–5175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisset, M.; Grandin, M.; Bernet, A.; Mehlen, P.; Hollande, F. Dependence receptors: New targets for cancer therapy. EMBO Mol. Med. 2021, 13, e14495. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.H.; Key, B. Neogenin: One receptor; many functions. Int. J. Biochem. Cell Biol. 2007, 39, 874–878. [Google Scholar] [CrossRef]
- Li, X.; Saint-Cyr-Proulx, E.; Aktories, K.; Lamarche-Vane, N. Rac1 and Cdc42 but not RhoA or Rho kinase activities are required for neurite outgrowth induced by the Netrin-1 receptor DCC (deleted in colorectal cancer) in N1E-115 neuroblastoma cells. J. Biol. Chem. 2002, 277, 15207–15214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.T.; Choi, S.H.; Bessho, F.; Kobayashi, M.; Hanada, R.; Yamamoto, K.; Hayashi, Y. Codon 201(Gly) polymorphic type of the DCC gene is related to disseminated neuroblastoma. Neoplasia 2001, 3, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.A.; Sanchez-Gomez, P.; Muñoz-Palma, E.; Puvogel, S.; Casas, B.S.; Arriagada, C.; Peña-Villalobos, I.; Lois, P.; Ramírez Orellana, M.; Lubieniecki, F.; et al. The Netrin-1-Neogenin-1 signaling axis controls neuroblastoma cell migration via integrin-β1 and focal adhesion kinase activation. Cell. Adh. Migr. 2021, 15, 58–73. [Google Scholar] [CrossRef]
- Delloye-Bourgeois, C.; Fitamant, J.; Paradisi, A.; Cappellen, D.; Douc-Rasy, S.; Raquin, M.A.; Stupack, D.; Nakagawara, A.; Rousseau, R.; Combaret, V.; et al. Netrin-1 acts as a survival factor for aggressive neuroblastoma. J. Exp. Med. 2009, 206, 833–847. [Google Scholar] [CrossRef]
- Villanueva, A.A.; Falcón, P.; Espinoza, N.; Luis, S.R.; Milla, L.A.; Hernandez-SanMiguel, E.; Torres, V.A.; Sanchez-Gomez, P.; Palma, V. The Netrin-4/Neogenin-1 axis promotes neuroblastoma cell survival and migration. Oncotarget 2017, 8, 9767–9782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.A.; Puvogel, S.; Lois, P.; Muñoz-Palma, E.; Ramírez Orellana, M.; Lubieniecki, F.; Casco Claro, F.; Gallegos, I.; García-Castro, J.; Sanchez-Gomez, P.; et al. The Netrin-4/Laminin γ1/Neogenin-1 complex mediates migration in SK-N-SH neuroblastoma cells. Cell. Adh Migr. 2019, 13, 33–40. [Google Scholar] [CrossRef]
- Buffone, A.; Weaver, V.M. Don’t sugarcoat it: How glycocalyx composition influences cancer progression. J. Cell. Biol. 2020, 219, e201910070. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell. 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Inamori, K.; Gu, J.; Ohira, M.; Kawasaki, A.; Nakamura, Y.; Nakagawa, T.; Kondo, A.; Miyoshi, E.; Nakagawara, A.; Taniguchi, N. High expression of N-acetylglucosaminyltransferase V in favorable neuroblastomas: Involvement of its effect on apoptosis. FEBS Lett. 2006, 580, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.K.; Burch, A.P.; Schwalbe, R.A. Functional analysis of N-acetylglucosaminyltransferase-I knockdown in 2D and 3D neuroblastoma cell cultures. PLoS ONE 2021, 16, e0259743. [Google Scholar] [CrossRef] [PubMed]
- Abbott, K.L.; Troupe, K.; Lee, I.; Pierce, M. Integrin-dependent neuroblastoma cell adhesion and migration on laminin is regulated by expression levels of two enzymes in the O-mannosyl-linked glycosylation pathway; PomGnT1 and GnT-Vb. Exp. Cell. Res. 2006, 312, 2837–2850. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.M.; Che, M.I.; Liao, Y.F.; Chang, H.H.; Chen, C.H.; Huang, Y.M.; Jeng, Y.M.; Huang, J.; Quon, M.J.; Lee, H.; et al. B4GALNT3 expression predicts a favorable prognosis and suppresses cell migration and invasion via β₁ integrin signaling in neuroblastoma. Am. J. Pathol. 2011, 179, 1394–1404. [Google Scholar] [CrossRef]
- Ho, W.L.; Che, M.I.; Chou, C.H.; Chang, H.H.; Jeng, Y.M.; Hsu, W.M.; Lin, K.H.; Huang, M.C. B3GNT3 expression suppresses cell migration and invasion and predicts favorable outcomes in neuroblastoma. Cancer Sci. 2013, 104, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Chen, C.H.; Chou, C.H.; Liao, Y.F.; Huang, M.J.; Chen, Y.H.; Wang, W.J.; Huang, J.; Hung, J.S.; Ho, W.L.; et al. β-1;4-Galactosyltransferase III enhances invasive phenotypes via β1-integrin and predicts poor prognosis in neuroblastoma. Clin. Cancer Res. 2013, 19, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, A.M.; Gabri, M.R.; Hernández, A.M.; Alonso, D.F.; Beausoleil, I.; Gomez, D.E.; Pérez, R. Antitumor properties of an anti-idiotypic monoclonal antibody in relation to N-glycolyl-containing gangliosides. Oncol. Rep. 2000, 7, 751–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleurence, J.; Fougeray, S.; Bahri, M.; Cochonneau, D.; Clémenceau, B.; Paris, F.; Heczey, A.; Birklé, S. Targeting O-Acetyl-GD2 Ganglioside for Cancer Immunotherapy. J. Immunol. Res. 2017, 2017, 604891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, S.; Bobowski, M.; Steenackers, A.; Le Bourhis, X.; Delannoy, P. How Do Gangliosides Regulate RTKs Signaling? Cells 2013, 2, 751–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniotti, J.L.; Vilcaes, A.A.; Torres Demichelis, V.; Ruggiero, F.M.; Rodriguez-Walker, M. Glycosylation of glycolipids in cancer: Basis for development of novel therapeutic approaches. Front. Oncol. 2013, 3, 306. [Google Scholar] [CrossRef] [Green Version]
- Beiske, K.; Burchill, S.A.; Cheung, I.Y.; Hiyama, E.; Seeger, R.C.; Cohn, S.L.; Pearson, A.D.; Matthay, K.K.; International neuroblastoma Risk Group Task Force. Consensus criteria for sensitive detection of minimal neuroblastoma cells in bone marrow, blood and stem cell preparations by immunocytology and QRT-PCR: Recommendations by the International Neuroblastoma Risk Group Task Force. Br. J. Cancer 2009, 100, 1627–1637. [Google Scholar] [CrossRef]
- Horwacik, I.; Rokita, H. Targeting of tumor-associated gangliosides with antibodies affects signaling pathways and leads to cell death including apoptosis. Apoptosis 2015, 20, 679–688. [Google Scholar] [CrossRef]
- Horwacik, I.; Durbas, M.; Boratyn, E.; Węgrzyn, P.; Rokita, H. Targeting GD2 ganglioside and aurora A kinase as a dual strategy leading to cell death in cultures of human neuroblastoma cells. Cancer Lett. 2013, 341, 248–264. [Google Scholar] [CrossRef]
- Durbas, M.; Horwacik, I.; Boratyn, E.; Kamycka, E.; Rokita, H. GD2 ganglioside specific antibody treatment downregulates PI3K/Akt/mTOR signaling network in human neuroblastoma cell lines. Int. J. Oncol. 2015, 47, 1143–1159. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; Gil, M.; Horwacik, I.; Odrowaz, Z.; Kozbor, D.; Rokita, H. The GD2-specific 14G2a monoclonal antibody induces apoptosis and enhances cytotoxicity of chemotherapeutic drugs in IMR-32 human neuroblastoma cells. Cancer Lett. 2009, 281, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Durbas, M.; Rokita, H.; Horwacik, I.; Wiśniewska, A.; Nowak, I. Apoptosis is responsible for the cytotoxic effects of anti-GD2 ganglioside antibodies and aurora A kinase inhibitors on human neuroblastoma cells. Acta Biochim. Pol. 2022, 69, 485–494. [Google Scholar] [CrossRef]
- Horwacik, I.; Rokita, H. Modulation of interactions of neuroblastoma cell lines with extracellular matrix proteins affects their sensitivity to treatment with the anti-GD2 ganglioside antibody 14G2a. Int. J. Oncol. 2017, 50, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Kazarian, T.; Jabbar, A.A.; Wen, F.Q.; Patel, D.A.; Valentino, L.A. Gangliosides regulate tumor cell adhesion to collagen. Clin. Exp. Metastasis 2003, 20, 311–319. [Google Scholar] [CrossRef]
- Czaplicki, D.; Horwacik, I.; Kowalczyk, A.; Wieczorek, A.; Bolek-Marzec, K.; Balwierz, W.; Kozik, A.; Rokita, H. New method for quantitative analysis of GD2 ganglioside in plasma of neuroblastoma patients. Acta Biochim. Pol. 2009, 56, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A.; Ladisch, S. Tumor gangliosides enhance alpha2 beta1 integrin-dependent platelet activation. Biochim. Biophys. Acta. 1996, 1316, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.X.; Chen, X.W.; Li, C.G.; Yue, L.J.; Mai, H.R.; Wen, F.Q. Effect of tumor gangliosides on tyrosine phosphorylation of p125FAK in platelet adhesion to collagen. Oncol. Rep. 2013, 29, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Jabbar, A.A.; Kazarian, T.; Hakobyan, N.; Valentino, L.A. Gangliosides promote platelet adhesion and facilitate neuroblastoma cell adhesion under dynamic conditions simulating blood flow. Pediatr. Blood Cancer 2006, 46, 292–299. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Ara, T.; Fukuzawa, M.; Kusafuka, T.; Komoto, Y.; Oue, T.; Inoue, M.; Okada, A. Immunohistochemical expression of MMP-2; MMP-9; and TIMP-2 in neuroblastoma: Association with tumor progression and clinical outcome. J. Pediatr. Surg. 1998, 33, 1272–1278. [Google Scholar] [CrossRef]
- Zhang, H.; Qi, M.; Li, S.; Qi, T.; Mei, H.; Huang, K.; Zheng, L.; Tong, Q. microRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol. Cancer Ther. 2012, 11, 1454–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.B.; O’Neill, G.M. Cooperative cell invasion: Matrix metalloproteinase-mediated incorporation between cells. Mol. Biol. Cell. 2016, 27, 3284–3292. [Google Scholar] [CrossRef] [PubMed]
- Chantrain, C.F.; Shimada, H.; Jodele, S.; Groshen, S.; Ye, W.; Shalinsky, D.R.; Werb, Z.; Coussens, L.M.; DeClerck, Y.A. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004, 64, 1675–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, D.B.; Aravindan, S.; Major, R.; Natarajan, M.; Aravindan, N. MMP-9 reinforces radiation-induced delayed invasion and metastasis of neuroblastoma cells through second-signaling positive feedback with NFκB via both ERK and IKK activation. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, F.; Ma, T.; Zeng, Z.; Zhang, N. miR-338-3p inhibits cell growth, invasion, and EMT process in neuroblastoma through targeting MMP-2. Open Life Sci. 2021, 16, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Rellinger, E.J.; Qiao, J.; Lee, S.; Volny, N.; Padmanabhan, C.; Romain, C.V.; Mobley, B.; Correa, H.; Chung, D.H. Elevated TIMP-1 expression is associated with a prometastatic phenotype, disease relapse, and poor survival in neuroblastoma. Oncotarget 2017, 8, 82609–82620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, D.M.; Pérez-Martínez, L. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression is regulated by multiple neural differentiation signals. J. Neurochem. 2006, 98, 234–247. [Google Scholar] [CrossRef]
- Spurbeck, W.W.; Ng, C.Y.; Strom, T.S.; Vanin, E.F.; Davidoff, A.M. Enforced expression of tissue inhibitor of matrix metalloproteinase-3 affects functional capillary morphogenesis and inhibits tumor growth in a murine tumor model. Blood 2002, 100, 3361–3368. [Google Scholar] [CrossRef] [Green Version]
- Xin, C.; Buhe, B.; Hongting, L.; Chuanmin, Y.; Xiwei, H.; Hong, Z.; Lulu, H.; Qian, D.; Renjie, W. MicroRNA-15a promotes neuroblastoma migration by targeting reversion-inducing cysteine-rich protein with Kazal motifs (RECK) and regulating matrix metalloproteinase-9 expression. FEBS J. 2013, 280, 855–866. [Google Scholar] [CrossRef]
- Lv, T.; Zhao, Y.; Jiang, X.; Yuan, H.; Wang, H.; Cui, X.; Xu, J.; Zhao, J.; Wang, J. uPAR: An Essential Factor for Tumor Development. J. Cancer 2021, 12, 7026–7040. [Google Scholar] [CrossRef]
- Zhai, B.T.; Tian, H.; Sun, J.; Zou, J.B.; Zhang, X.F.; Cheng, J.X.; Shi, Y.J.; Fan, Y.; Guo, D.Y. Urokinase-type plasminogen activator receptor (uPAR) as a therapeutic target in cancer. J. Transl. Med. 2022, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gao, Y.; Ji, Z.; Zhang, X.; Xu, Q.; Li, G.; Guo, Z.; Zheng, B.; Guo, X. Role of urokinase plasminogen activator and its receptor in metastasis and invasion of neuroblastoma. J. Pediatr. Surg. 2004, 39, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.A.; Klimovich, P.S.; Rysenkova, K.D.; Popov, V.S.; Gorbunova, A.S.; Karpukhina, A.A.; Karagyaur, M.N.; Rubina, K.A.; Tkachuk, V.A.; Semina, E.V. Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis. Cancers 2022, 14, 994. [Google Scholar] [CrossRef]
- Rysenkova, K.D.; Klimovich, P.S.; Shmakova, A.A.; Karagyaur, M.N.; Ivanova, K.A.; Aleksandrushkina, N.A.; Tkachuk, V.A.; Rubina, K.A.; Semina, E.V. Urokinase receptor deficiency results in EGFR-mediated failure to transmit signals for cell survival and neurite formation in mouse neuroblastoma cells. Cell Signal. 2020, 75, 109741. [Google Scholar] [CrossRef]
- Gutova, M.; Najbauer, J.; Frank, R.T.; Kendall, S.E.; Gevorgyan, A.; Metz, M.Z.; Guevorkian, M.; Edmiston, M.; Zhao, D.; Glackin, C.A.; et al. Urokinase plasminogen activator and urokinase plasminogen activator receptor mediate human stem cell tropism to malignant solid tumors. Stem Cells 2008, 26, 1406–1413. [Google Scholar] [CrossRef]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef]
- Otsuka, K.; Sasada, M.; Iyoda, T.; Nohara, Y.; Sakai, S.; Asayama, T.; Suenaga, Y.; Yokoi, S.; Higami, Y.; Kodama, H.; et al. Combining peptide TNIIIA2 with all-trans retinoic acid accelerates N-Myc protein degradation and neuronal differentiation in MYCN-amplified neuroblastoma cells. Am. J. Cancer Res. 2019, 9, 434–448. [Google Scholar]
- Otsuka, K.; Sasada, M.; Hirano, Y.U.; Nohara, Y.; Iyoda, T.; Higami, Y.; Kodama, H.; Fukai, F. Acyclic Retinoid Combined with Tenascin-C-derived Peptide Reduces the Malignant Phenotype of Neuroblastoma Cells Through N-Myc Degradation. Anticancer Res. 2019, 39, 3487–3492. [Google Scholar] [CrossRef]
- Bergh, J.J.; Lin, H.Y.; Lansing, L.; Mohamed, S.N.; Davis, F.B.; Mousa, S.; Davis, P.J. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 2005, 146, 2864–2871. [Google Scholar] [CrossRef]
- Ozen Karakus, O.; Godugu, K.; Mousa, S.A. Discovery of dual targeting PEGylated BG-P1600-TAT to norepinephrine transporter (NET) and thyrointegrin αvβ3 in the treatment of neuroblastoma. Bioorg. Med. Chem. 2021, 43, 116278. [Google Scholar] [CrossRef]
- Burgos-Panadero, R.; El Moukhtari, S.H.; Noguera, I.; Rodríguez-Nogales, C.; Martín-Vañó, S.; Vicente-Munuera, P.; Cañete, A.; Navarro, S.; Blanco-Prieto, M.J.; Noguera, R. Unraveling the extracellular matrix-tumor cell interactions to aid better targeted therapies for neuroblastoma. Int. J. Pharm. 2021, 608, 121058. [Google Scholar] [CrossRef] [PubMed]
- Stafman, L.L.; Williams, A.P.; Marayati, R.; Aye, J.M.; Markert, H.R.; Garner, E.F.; Quinn, C.H.; Lallani, S.B.; Stewart, J.E.; Yoon, K.J.; et al. Focal Adhesion Kinase Inhibition Contributes to Tumor Cell Survival and Motility in Neuroblastoma Patient-Derived Xenografts. Sci. Rep. 2019, 9, 13259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillory, L.A.; Stewart, J.E.; Megison, M.L.; Waters, A.M.; Beierle, E.A. Focal adhesion kinase and p53 synergistically decrease neuroblastoma cell survival. J. Surg. Res. 2015, 196, 339–349. [Google Scholar] [CrossRef]
- Markovsky, E.; Eldar-Boock, A.; Ben-Shushan, D.; Baabur-Cohen, H.; Yeini, E.; Pisarevsky, E.; Many, A.; Aviel-Ronen, S.; Barshack, I.; Satchi-Fainaro, R. Targeting NCAM-expressing neuroblastoma with polymeric precision nanomedicine. J. Control. Release. 2017, 249, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, T.M.; Chetty, C.J.; Ferreira, E.; Weiss, S.F. Anti-LRP/LR-specific antibody IgG1-iS18 impedes adhesion and invasion of pancreatic cancer and neuroblastoma cells. BMC Cancer 2016, 16, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, J.I.; Pressey, J.G.; Smith, M.A.; Kudgus, R.A.; Cajaiba, M.; Reid, J.M.; Hall, D.; Barkauskas, D.A.; Voss, S.D.; Cho, S.Y.; et al. ADVL1522: A phase 2 study of lorvotuzumab mertansine (IMGN901) in children with relapsed or refractory wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, or synovial sarcoma-A Children’s Oncology Group study. Cancer 2020, 126, 5303–5310. [Google Scholar] [CrossRef] [PubMed]
- Künkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin. Cancer Res. 2017, 23, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, S.; Buongervino, S.N.; Lane, M.V.; Zhelev, D.V.; Zhu, Z.; Cui, H.; Martinez, B.; Martinez, D.; Wang, Y.; Upton, K.; et al. A GPC2 antibody-drug conjugate is efficacious against neuroblastoma and small-cell lung cancer via binding a conformational epitope. Cell. Rep. Med. 2021, 2, 100344:1–100344:17. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, H.; Hewitt, S.M.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc. Natl. Acad. Sci. USA 2017, 114, E6623–E6631. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Torres, M.B.; Spetz, M.R.; Wang, R.; Peng, L.; Tian, M.; Dower, C.M.; Nguyen, R.; Sun, M.; Tai, C.H.; et al. CAR T cells targeting tumor-associated exons of glypican 2 regress neuroblastoma in mice. Cell Rep. Med. 2021, 2, 100297. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Cheuk, A.T.; Wei, J.S.; Abdelmaksoud, A.; Chou, H.C.; Milewski, D.; Kelly, M.C.; Song, Y.K.; Dower, C.M.; Li, N.; et al. An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. J. Clin. Investig. 2022, 132, e155621. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Cheung, I.Y.; Cheung, N.V.; Modak, S.; Mauguen, A.; Feng, Y.; Basu, E.; Roberts, S.S.; Ragupathi, G.; Kushner, B.H. Survival Impact of Anti-GD2 Antibody Response in a Phase II Ganglioside Vaccine Trial Among Patients with High-Risk Neuroblastoma with Prior Disease Progression. J. Clin. Oncol. 2021, 39, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Voeller, J.; Sondel, P.M. Advances in Anti-GD2 Immunotherapy for Treatment of High-risk Neuroblastoma. J. Pediatr. Hematol. Oncol. 2019, 41, 163–169. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapy | Description | References |
|---|---|---|
| Tenascin C-derived peptide (TNIIIA2) | To activate integrin β1, tested in a combination with ATRA or acyclic retinoid | [168] [169] |
| BG-PEG1600-TAT | An antagonist of αvβ3 integrin linked via poly(ethylene glycol) to benzylguanidine | [171] |
| SB273005 Cilengitide | Inhibitors of αvβ3 and αvβ5 integrins combined with anti-GD2 14G2a mAb or etoposide | [144] [172] |
| PF-573,288 | An inhibitor of FAK combined with nutlin 3 (to activate P53) | [174] |
| PGA–PTX–NTP | Targeting NCAM-positive cells with a peptide (NTP) to deliver paclitaxel (PTX) | [175] |
| IgG1-iS18 | Anti-laminin receptor mAb | [176] |
| Lorvotuzumab mertansine (IMGN901) | Anti-NCAM mAb huN-901 conjugated to an anti-tubule agent | [177] |
| D3-GPC2-IgG1 combined with pyrrolobenzadiazepine | Anti-GPC2 mAb recognizing a tumor-specific epitope for the targeted delivery of a DNA-damaging agent | [179] |
| Immunotoxin of LH7 and PE38 | The anti-GPC2 single domain Ab fragment LH7 linked to the Pseudomonas exotoxin (PE38) | [180] |
| CAR to modify T cells for adoptive immunotherapy | Contains the scFv of anti-L1CAM mAb CE7. Contains the anti-GPC2 Ab fragment LH7. Contains the scFv of anti-GPC2 mAb CT3. | [178] [180] [181] |
| GD2/GD3 vaccine | Keyhole limpet hemocyanin (KLH) conjugated to deliver active immunotherapy | [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horwacik, I. The Extracellular Matrix and Neuroblastoma Cell Communication—A Complex Interplay and Its Therapeutic Implications. Cells 2022, 11, 3172. https://doi.org/10.3390/cells11193172
Horwacik I. The Extracellular Matrix and Neuroblastoma Cell Communication—A Complex Interplay and Its Therapeutic Implications. Cells. 2022; 11(19):3172. https://doi.org/10.3390/cells11193172
Chicago/Turabian StyleHorwacik, Irena. 2022. "The Extracellular Matrix and Neuroblastoma Cell Communication—A Complex Interplay and Its Therapeutic Implications" Cells 11, no. 19: 3172. https://doi.org/10.3390/cells11193172