
Loss of Mature Lamin A/C Triggers a Shift in Intracellular Metabolic Homeostasis via AMPKα Activation
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, LMNA-KO or LMNA-KD Plasmids and Transfection
2.2. Construction of the LMNA cDNA and Pre–LMNA and Mutant Plasmids
2.3. Immunohistochemistry (IHC)
2.4. RNA Extraction and qRT–PCR
2.5. Detection of the Intracellular TG and Lactic Acid Contents
2.6. Endogenous or Exogenous Immunoprecipitation Assay
2.7. Tissue and Cell Protein Extraction, BCA Assay and Western Blot Analysis
2.8. Extraction, Culture, and Induction of AD-MSCs
2.9. High Glucose-Mediated Increase in Cellular Lipid Synthesis and Oil Red O Staining
2.10. Mass Spectrometry
2.11. CCK-8 Assay
2.12. Statistical Analysis
3. Results
3.1. pLamin A/C Levels Increased around Lipid Droplets in Human Liver Tissue
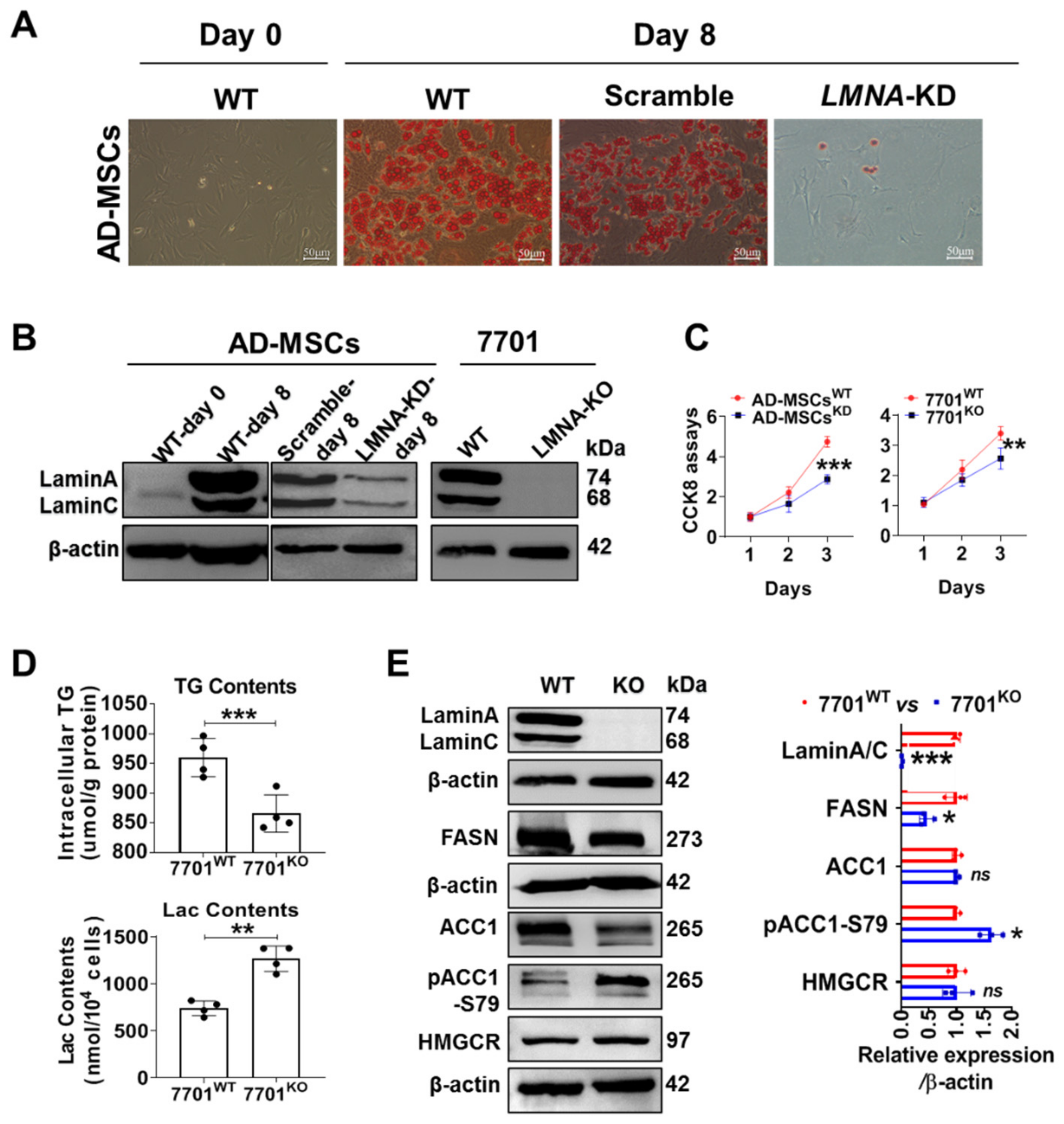
3.2. LMNA KD Blocked AD-MSC Maturation, and LMNA Deletion Decreased Lipid Synthesis in 7701 Cells
3.3. Lamin C and Phosphorylation-Activated AMPKα Directly Interacted in 7701 Cells
3.4. AMPKα Activation Is Closely Associated with Abnormal Lamin A/C Function
3.5. LMNA Deletion Activates AMPKα, Possibly via a Change in the ADP/ATP Ratio
3.6. pLamin A/C Was Upregulated and pAMPKα-T172 Levels Were Decreased during High Glucose-Induced Lipid Synthesis
3.7. Combined Targeting of AMPKα and Lamin A/C Effectively Inhibited the Growth of HCC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FPLD | familial partial lipodystrophy of Dunnigan |
| AD-MSCs | adipose-derived mesenchymal stem cells |
| pLamin A/C | phosphorylated lamin A/C |
| ACC1 | acetyl-CoA-carboxylase 1 |
| FASN | fatty acid synthase |
| PC | prostate cancer |
| HCC | hepatocellular carcinoma |
| CRC | metastatic colorectal cancer |
| HGPS | Hutchinson–Gilford syndrome |
| FTIs | farnesyltransferase inhibitors |
| AMPK | adenosine 5′-monophosphate-activated protein kinase |
| TG | triglyceride |
| Acetyl-CoA | acetyl coenzyme A |
| MPC | mitochondrial pyruvate carrier |
| WT | wild type |
| PTM | post-translational modification |
References
- Gonzalez, J.M.; Pla, D.; Perez-Sala, D.; Andres, V. A-type lamins and Hutchinson-Gilford progeria syndrome: Pathogenesis and therapy. Front Biosci. 2011, 3, 1133–1146. [Google Scholar] [CrossRef] [Green Version]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 2000, 151, 1155–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.B.; Mastaglia, F.L.; Wilton, S.D. Normal and aberrant splicing of LMNA. J. Med. Genet. 2014, 51, 215–223. [Google Scholar] [CrossRef]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinensky, M.; Fantle, K.; Trujillo, M.; McLain, T.; Kupfer, A.; Dalton, M. The processing pathway of prelamin A. J. Cell Sci. 1994, 107, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S.; Cremer, J.; Faucher, O.; Reynes, J.; Dellamonica, P.; Micallef, J.; Solas, C.; Lacarelle, B.; Stretti, C.; Kaspi, E.; et al. HIV protease inhibitors do not cause the accumulation of prelamin A in PBMCs from patients receiving first line therapy: The ANRS EP45 “aging” study. PLoS ONE 2012, 7, e53035. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Comai, L. Lamin A, farnesylation and aging. Exp. Cell Res. 2012, 318, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, V.L.; Broers, J.L.; Ramaekers, F.C.; van Steensel, M.A. The nuclear envelope, a key structure in cellular integrity and gene expression. Curr. Med. Chem. 2007, 14, 1231–1248. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [Green Version]
- Torvaldson, E.; Kochin, V.; Eriksson, J.E. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 2015, 6, 166–171. [Google Scholar] [CrossRef]
- Foisner, R.; Gerace, L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 1993, 73, 1267–1279. [Google Scholar] [CrossRef]
- Wang, A.S.; Kozlov, S.V.; Stewart, C.L.; Horn, H.F. Tissue specific loss of A-type lamins in the gastrointestinal epithelium can enhance polyp size. Differentiation 2015, 89, 11–21. [Google Scholar] [CrossRef]
- Zuo, L.; Zhao, H.; Yang, R.; Wang, L.; Ma, H.; Xu, X.; Zhou, P.; Kong, L. Lamin A/C might be involved in the EMT signalling pathway. Gene 2018, 663, 51–64. [Google Scholar] [CrossRef]
- Liu, H.; Li, D.; Zhou, L.; Kan, S.; He, G.; Zhou, K.; Wang, L.; Chen, M.; Shu, W. LMNA functions as an oncogene in hepatocellular carcinoma by regulating the proliferation and migration ability. J. Cell. Mol. Med. 2020, 24, 12008–12019. [Google Scholar] [CrossRef]
- Lin, Y.T.; Chien, K.Y.; Wu, C.C.; Chang, W.Y.; Chu, L.J.; Chen, M.C.; Yeh, C.T.; Yu, J.S. Super-SILAC mix coupled with SIM/AIMS assays for targeted verification of phosphopeptides discovered in a large-scale phosphoproteome analysis of hepatocellular carcinoma. J. Proteom. 2017, 157, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Ardini, E.; Bosotti, R.; Amatu, A.; Valtorta, E.; Somaschini, A.; Raddrizzani, L.; Palmeri, L.; Banfi, P.; Bonazzina, E.; et al. Sensitivity to Entrectinib Associated with a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2016, 108, djv306. [Google Scholar] [CrossRef]
- Russo, M.; Misale, S.; Wei, G.; Siravegna, G.; Crisafulli, G.; Lazzari, L.; Corti, G.; Rospo, G.; Novara, L.; Mussolin, B.; et al. Acquired Resistance to the TRK Inhibitor Entrectinib in Colorectal Cancer. Cancer Discov. 2016, 6, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilanczyk, E.; Graczyk, A.; Ostrowska, H.; Kasacka, I.; Lesniak, W.; Filipek, A. S100A6 is transcriptionally regulated by beta-catenin and interacts with a novel target, lamin A/C, in colorectal cancer cells. Cell Calcium 2012, 51, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Neilan, E.G. Laminopathies, other progeroid disorders, and aging: Common pathogenic themes and possible treatments. Am. J. Med. Genet. A 2009, 149A, 563–566. [Google Scholar] [CrossRef]
- Decaudain, A.; Vantyghem, M.C.; Guerci, B.; Hecart, A.C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.H.; Donadille, B.; Lebbe, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef]
- Haithcock, E.; Dayani, Y.; Neufeld, E.; Zahand, A.J.; Feinstein, N.; Mattout, A.; Gruenbaum, Y.; Liu, J. Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 16690–16695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piekarowicz, K.; Machowska, M.; Dzianisava, V.; Rzepecki, R. Hutchinson-Gilford Progeria Syndrome-Current Status and Prospects for Gene Therapy Treatment. Cells 2019, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenburg, A.R.; Delbarre, E.; Thiede, B.; Vigouroux, C.; Collas, P. Deregulation of Fragile X-related protein 1 by the lipodystrophic lamin A p.R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum. Mol. Genet. 2014, 23, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacene, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Youssef, S.J.; Macielak, R.J.; Schimmenti, L.A.; Chatzopoulos, K.; Price, D.L. Hypopharyngeal Squamous Cell Carcinoma in Sisters with LMNA Associated Familial Partial Lipodystrophy: A Case Report and Review of the Literature. Ann. Otol. Rhinol. Laryngol. 2020, 129, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Vasandani, C.; Li, X.; Sekizkardes, H.; Adams-Huet, B.; Brown, R.J.; Garg, A. Diagnostic Value of Anthropometric Measurements for Familial Partial Lipodystrophy, Dunnigan Variety. J. Clin. Endocrinol. Metab. 2020, 105, 2132–2141. [Google Scholar] [CrossRef]
- Miranda, M.; Chacon, M.R.; Gutierrez, C.; Vilarrasa, N.; Gomez, J.M.; Caubet, E.; Megia, A.; Vendrell, J. LMNA mRNA expression is altered in human obesity and type 2 diabetes. Obesity 2008, 16, 1742–1748. [Google Scholar] [CrossRef]
- Wegner, L.; Andersen, G.; Sparso, T.; Grarup, N.; Glumer, C.; Borch-Johnsen, K.; Jorgensen, T.; Hansen, T.; Pedersen, O. Common variation in LMNA increases susceptibility to type 2 diabetes and associates with elevated fasting glycemia and estimates of body fat and height in the general population: Studies of 7495 Danish whites. Diabetes 2007, 56, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. NAFLD Related-HCC: The Relationship with Metabolic Disorders. Adv. Exp. Med. Biol. 2018, 1061, 55–62. [Google Scholar] [CrossRef]
- Tadros, S.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Yu, F.; Lazenby, A.J.; Berim, L.; et al. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.; Martinou, J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lepropre, S.; Kautbally, S.; Octave, M.; Ginion, A.; Onselaer, M.B.; Steinberg, G.R.; Kemp, B.E.; Hego, A.; Wera, O.; Brouns, S.; et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood 2018, 132, 1180–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.B.; Wei, Y.H. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: Implication of the cell survival in mitochondrial diseases. Biochim. Biophys. Acta 2012, 1822, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A Potent and Selective AMPK Activator That Inhibits de Novo Lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, S.; Lloyd, D.J.; Jackson, S.N.; Evans, R.; Niermeijer, M.F.; Singh, B.M.; Schmidt, H.; Brabant, G.; Kumar, S.; Durrington, P.N.; et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 2000, 24, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.; Cao, H.; Rabkin, S.W.; Hanna, A.; Hegele, R.A. Novel LMNA mutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660). Clin. Genet. 2007, 71, 183–186. [Google Scholar] [CrossRef]
- Speckman, R.A.; Garg, A.; Du, F.; Bennett, L.; Veile, R.; Arioglu, E.; Taylor, S.I.; Lovett, M.; Bowcock, A.M. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet. 2000, 66, 1192–1198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shen, H.; Zhao, Z.; Bing, Q.; Hu, J. Cardiac effects of the c.1583 C-->G LMNA mutation in two families with Emery-Dreifuss muscular dystrophy. Mol. Med. Rep. 2015, 12, 5065–5071. [Google Scholar] [CrossRef]
- Young, S.G.; Meta, M.; Yang, S.H.; Fong, L.G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 2006, 281, 39741–39745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopietz, F.; Alshuweishi, Y.; Bijland, S.; Alghamdi, F.; Degerman, E.; Sakamoto, K.; Salt, I.P.; Goransson, O. A-769662 inhibits adipocyte glucose uptake in an AMPK-independent manner. Biochem. J. 2021, 478, 633–646. [Google Scholar] [CrossRef]
- Alannan, M.; Fayyad-Kazan, H.; Trezeguet, V.; Merched, A. Targeting Lipid Metabolism in Liver Cancer. Biochemistry 2020, 59, 3951–3964. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yang, Y.; Keyimu, R.; Hao, J.; Zhao, Z.; Ye, R. The role of lamin A/C in mesenchymal stem cell differentiation. J. Physiol. Biochem. 2019, 75, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Patni, N.; Garg, A. Lipodystrophy for the Diabetologist-What to Look For. Curr. Diab. Rep. 2022, 22, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Ding, H.; Dong, J.; Zhang, R.; Huang, D.; Lei, L.; Wang, Z.; Liu, G.; Li, X. Insulin suppresses the AMPK signaling pathway to regulate lipid metabolism in primary cultured hepatocytes of dairy cows. J. Dairy Res. 2018, 85, 157–162. [Google Scholar] [CrossRef]
- Finley, J. Alteration of splice site selection in the LMNA gene and inhibition of progerin production via AMPK activation. Med. Hypotheses 2014, 83, 580–587. [Google Scholar] [CrossRef]
- Kazgan, N.; Williams, T.; Forsberg, L.J.; Brenman, J.E. Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase. Mol. Biol. Cell 2010, 21, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Wu, M.; Braunschweig, U.; Dyakov, B.J.A.; Luo, Z.; Turner, K.M.; Durbic, T.; Lin, Z.Y.; Weatheritt, R.J.; Maass, P.G.; et al. Systematic mapping of nuclear domain-associated transcripts reveals speckles and lamina as hubs of functionally distinct retained introns. Mol. Cell 2022, 82, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.S.; Morse, M.A. Lonafarnib for cancer and progeria. Expert Opin. Investig. Drugs 2012, 21, 1043–1055. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | PSMs 1 | Group Description |
|---|---|---|
| 32 | 279 | Prelamin-A/C (LMNA) |
| 10 | 61 | Lamin-B1 (LMNB1) |
| 19 | 36 | Keratin, type II cytoskeletal 1 (KRT1) |
| 12 | 20 | Keratin, type II cytoskeletal 2 epidermal (KRT2) |
| 7 | 20 | Tubulin alpha-1B chain (TUBA1B) |
| 9 | 19 | Keratin, type I cytoskeletal 9 (KRT9) |
| 6 | 16 | Tubulin alpha-1C chain (TUBA1C) |
| 4 | 16 | Lamin-B2 (LMNB2) |
| 8 | 13 | Heat shock protein HSP 90-beta (HSP90AB1) |
| 6 | 13 | Tubulin beta chain (TUBB) |
| 7 | 12 | Heat shock 70 kDa protein 1B (HSPA1B) |
| 7 | 12 | Actin, cytoplasmic 1 (ACTB) |
| 8 | 11 | Keratin, type I cytoskeletal 10 (KRT10) |
| 7 | 11 | Keratin, type II cytoskeletal 6B (KRT6B) |
| 5 | 10 | Elongation factor 1-alpha 1 (EEF1A1) |
| 1 | 10 | Serum albumin (ALB) |
| 8 | 9 | Keratin, type II cytoskeletal 5 (KRT5) |
| 7 | 8 | Keratin, type II cytoskeletal 6C (KRT6C) |
| 4 | 8 | Heterogeneous nuclear ribonucleoprotein H (HNRNPH1) |
| 6 | 7 | Nucleolar RNA helicase 2 (DDX21) |
| 7 | 7 | Keratin, type I cytoskeletal 16 (KRT16) |
| 4 | 7 | Alpha-enolase (ENO1) |
| 4 | 7 | Heat shock cognate 71 kDa protein (HSPA8) |
| 5 | 6 | Pyruvate kinase (PKM) |
| 4 | 6 | Elongation factor 2 (EEF2) |
| 5 | 5 | T-complex protein 1 subunit alpha (TCP1) |
| 3 | 3 | Transitional endoplasmic reticulum ATPase (VCP) |
| 2 | 2 | ADP/ATP translocase 2 (SLC25A5) |
| 1 | 1 | Fatty acid-binding protein 5 (FABP5) |
| 1 | 1 | Proliferation-associated protein 2G4 (PA2G4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Yang, J.-J.; Cheng, Y.; Feng, G.-X.; Yang, R.-H.; Yuan, Y.; Wang, L.-Y.; Wang, M.; Kong, L. Loss of Mature Lamin A/C Triggers a Shift in Intracellular Metabolic Homeostasis via AMPKα Activation. Cells 2022, 11, 3988. https://doi.org/10.3390/cells11243988
Zhou Y, Yang J-J, Cheng Y, Feng G-X, Yang R-H, Yuan Y, Wang L-Y, Wang M, Kong L. Loss of Mature Lamin A/C Triggers a Shift in Intracellular Metabolic Homeostasis via AMPKα Activation. Cells. 2022; 11(24):3988. https://doi.org/10.3390/cells11243988
Chicago/Turabian StyleZhou, Ying, Jia-Jie Yang, Yuan Cheng, Ge-Xuan Feng, Rong-Hui Yang, Yuan Yuan, Li-Yong Wang, Miao Wang, and Lu Kong. 2022. "Loss of Mature Lamin A/C Triggers a Shift in Intracellular Metabolic Homeostasis via AMPKα Activation" Cells 11, no. 24: 3988. https://doi.org/10.3390/cells11243988