
MSK-Mediated Phosphorylation of Histone H3 Ser28 Couples MAPK Signalling with Early Gene Induction and Cardiac Hypertrophy

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animal Experiments
2.3. Echocardiography
2.4. Preparation of Neonatal Rat Ventricular Cardiomyocytes (NRVMs)
2.5. Isolation and Culture of Adult Rat Ventricular Cardiomyocytes (ARVMs)
2.6. Isolation of Human Ventricular Cardiomyocytes
2.7. Isolation of Human Cardiomyocyte Nuclei
2.8. High Content Analysis of NRVM Hypertrophy
2.9. Confocal Imaging of Immunostained NRVM and ARVM
2.10. Confocal Imaging of Immunostained Cardiac Tissue Sections
2.11. Picro Sirius Red Staining for Fibrosis Analysis
2.12. Histone Isolation by Acid Extraction
2.13. Immunoblot Analysis
2.14. Reverse Transcription Quantitative PCR (RT-qPCR)
2.15. Chromatin-Immunoprecipitation (ChIP)
2.16. Jugular Vein Infusion of Endothelin-1/Isoproterenol in Wistar Rat
2.17. Adenoviral Methods
2.18. Analysis of Luciferase Reporter Activity
2.19. Small Interfering RNA (siRNA) Knockdown
2.20. Statistical Analysis
3. Results
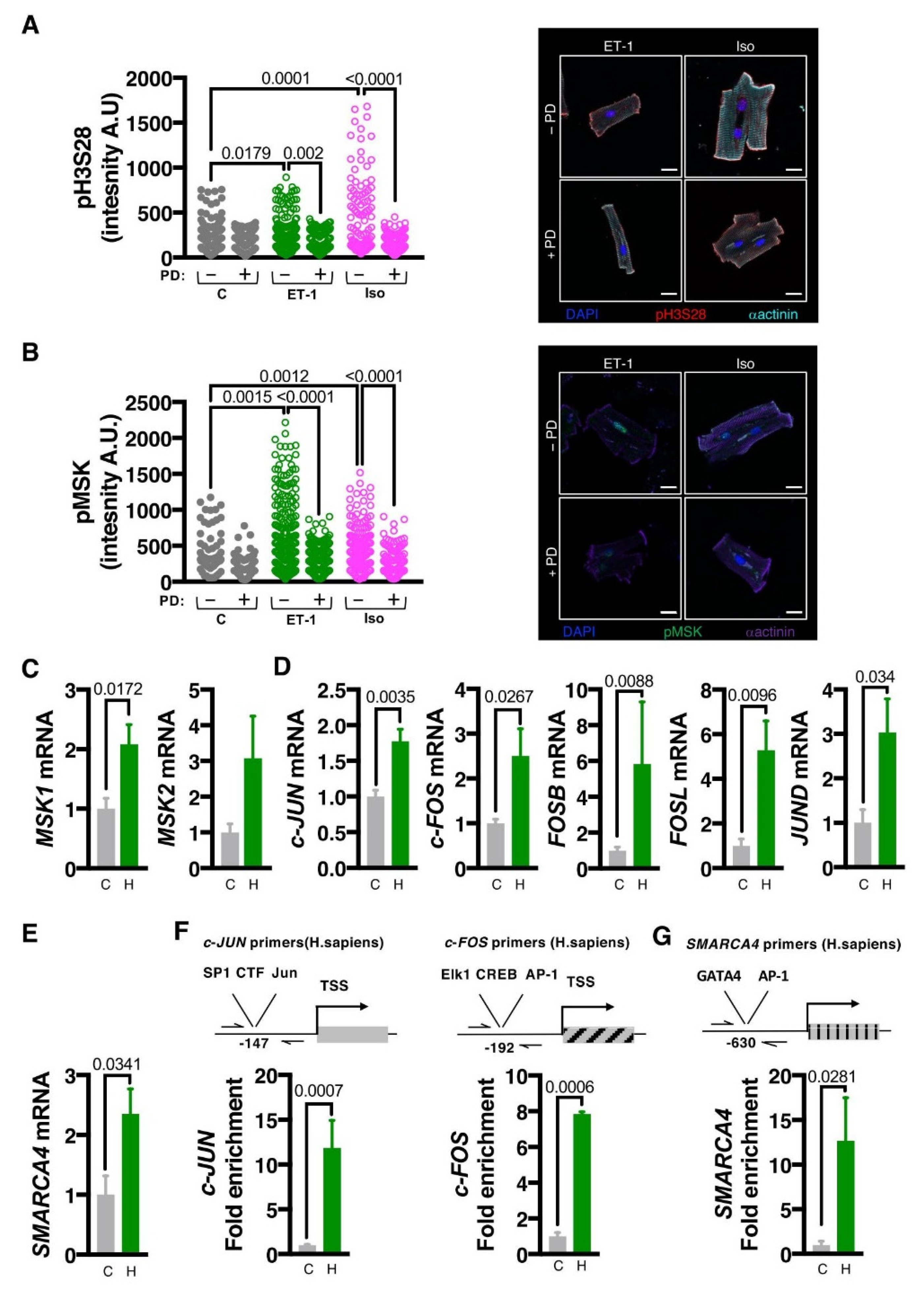
3.1. Endothelin-1 Stimulates ERK-Dependent Phosphorylation of Histone H3 Serine 28
3.2. MSK1/2 Is Activated Following ET-1 Stimulation in an ERK1/2 Dependent Manner
3.3. MSK1/2 Stimulates IEG Induction, Phosphorylation of H3S28 at IEG Promoters and Promotes Induction of Hypertrophic Gene Expression
3.4. MSK1-Mediated Phosphorylation of H3S28 Recruits BRG1, a Component of the BAF60 Chromatin Remodelling Complex to IEG Loci
3.5. MSK1/2 Expression Is Required for the Hypertrophic Response In Vivo
3.6. The MSK1/2/pH3S28/BRG1/IEG Axis Is Engaged in Human Hypertrophic Remodelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [PubMed]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drawnel, F.M.; Archer, C.R.; Roderick, H.L. The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br. J. Pharmacol. 2013, 168, 296–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zi, M.; Jin, J.; Prehar, S.; Oceandy, D.; Kimura, T.E.; Lei, M.; Neyses, L.; Weston, A.H.; Cartwright, E.J.; et al. Cardiac-specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ. Res. 2009, 104, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Bueno, O.F.; De Windt, L.J.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Klevitsky, R.; Hewett, T.E.; Jones, S.P.; Lefer, D.J.; Peng, C.F.; et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000, 19, 6341–6350. [Google Scholar] [CrossRef] [Green Version]
- Garrington, T.P.; Johnson, G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell. Biol. 1999, 11, 211–218. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell. Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Sanna, B.; Bueno, O.F.; Dai, Y.S.; Wilkins, B.J.; Molkentin, J.D. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol. Cell. Biol. 2005, 25, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Archer, C.R.; Robinson, E.L.; Drawnel, F.M.; Roderick, H.L. Endothelin-1 promotes hypertrophic remodelling of cardiac myocytes by activating sustained signalling and transcription downstream of endothelin type A receptors. Cell. Signal. 2017, 36, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, K.; Sukhatme, V.P.; Shubeita, H.E.; Chien, K.R. Alpha- and beta-adrenergic stimulation induces distinct patterns of immediate early gene expression in neonatal rat myocardial cells. fos/jun expression is associated with sarcomere assembly; Egr-1 induction is primarily an alpha 1-mediated response. J. Biol. Chem. 1990, 265, 13809–13817. [Google Scholar] [CrossRef]
- Izumo, S.; Nadal-Ginard, B.; Mahdavi, V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc. Natl. Acad. Sci. USA 1988, 85, 339–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullingford, T.E.; Markou, T.; Fuller, S.J.; Giraldo, A.; Pikkarainen, S.; Zoumpoulidou, G.; Alsafi, A.; Ekere, C.; Kemp, T.J.; Dennis, J.L.; et al. Temporal regulation of expression of immediate early and second phase transcripts by endothelin-1 in cardiomyocytes. Genome Biol. 2008, 9, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gille, H.; Sharrocks, A.D.; Shaw, P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 1992, 358, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell. Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell. Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Glover, J.N.; Harrison, S.C. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 1995, 373, 257–261. [Google Scholar] [CrossRef]
- Chen, L.; Glover, J.N.; Hogan, P.G.; Rao, A.; Harrison, S.C. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature 1998, 392, 42–48. [Google Scholar] [CrossRef]
- Torgerson, T.R.; Colosia, A.D.; Donahue, J.P.; Lin, Y.Z.; Hawiger, J. Regulation of NF-kappa B, AP-1, NFAT, and STAT1 nuclear import in T lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-kappa B p50. J. Immunol. 1998, 161, 6084–6092. [Google Scholar] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Castellazzi, M.; Wollert, K.C.; Trautwein, C.; Schunkert, H.; Drexler, H. JunD attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating AP-1 transcriptional activity. Cardiovasc. Res. 2006, 71, 108–117. [Google Scholar] [CrossRef]
- Omura, T.; Yoshiyama, M.; Yoshida, K.; Nakamura, Y.; Kim, S.; Iwao, H.; Takeuchi, K.; Yoshikawa, J. Dominant negative mutant of c-Jun inhibits cardiomyocyte hypertrophy induced by endothelin 1 and phenylephrine. Hypertension 2002, 39, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Kaminski, K.; Schaefer, A.; Park, J.K.; Michel, K.; Quint, A.; Yaniv, M.; Weitzman, J.B.; Drexler, H. Lack of JunD promotes pressure overload-induced apoptosis, hypertrophic growth, and angiogenesis in the heart. Circulation 2005, 112, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Ricci, R.; Eriksson, U.; Oudit, G.Y.; Eferl, R.; Akhmedov, A.; Sumara, I.; Sumara, G.; Kassiri, Z.; David, J.P.; Bakiri, L.; et al. Distinct functions of junD in cardiac hypertrophy and heart failure. Genes Dev. 2005, 19, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windak, R.; Muller, J.; Felley, A.; Akhmedov, A.; Wagner, E.F.; Pedrazzini, T.; Sumara, G.; Ricci, R. The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS ONE 2013, 8, e73294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirak, E.; Fuller, S.J.; Sugden, P.H.; Clerk, A. p90 ribosomal S6 kinases play a significant role in early gene regulation in the cardiomyocyte response to G(q)-protein-coupled receptor stimuli, endothelin-1 and alpha(1)-adrenergic receptor agonists. Biochem. J. 2013, 450, 351–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, A.L.; Rose, S.; Barratt, M.J.; Mahadevan, L.C. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19, 3714–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, M.H.; Thomson, S.; Inagaki, M.; Goto, H.; Arthur, S.J.; Nightingale, K.; Iborra, F.J.; Mahadevan, L.C. MAP kinase-mediated phosphorylation of distinct pools of histone H3 at S10 or S28 via mitogen- and stress-activated kinase 1/2. J Cell. Sci. 2005, 118, 2247–2259. [Google Scholar] [CrossRef] [Green Version]
- Drobic, B.; Perez-Cadahia, B.; Yu, J.; Kung, S.K.; Davie, J.R. Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res. 2010, 38, 3196–3208. [Google Scholar] [CrossRef]
- Duncan, E.A.; Anest, V.; Cogswell, P.; Baldwin, A.S. The kinases MSK1 and MSK2 are required for epidermal growth factor-induced, but not tumor necrosis factor-induced, histone H3 Ser10 phosphorylation. J. Biol. Chem. 2006, 281, 12521–12525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Shimada, M.; Armache, A.; Li, C.H.; Miller, R.M.; Lin, S.; Yang, A.; Dill, B.D.; Molina, H.; Park, H.S.; et al. Chromatin Kinases Act on Transcription Factors and Histone Tails in Regulation of Inducible Transcription. Mol. Cell. 2016, 64, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soloaga, A.; Thomson, S.; Wiggin, G.R.; Rampersaud, N.; Dyson, M.H.; Hazzalin, C.A.; Mahadevan, L.C.; Arthur, J.S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003, 22, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Malakhova, M.; Kurinov, I.; Liu, K.; Zheng, D.; D’Angelo, I.; Shim, J.H.; Steinman, V.; Bode, A.M.; Dong, Z. Structural diversity of the active N-terminal kinase domain of p90 ribosomal S6 kinase 2. PLoS ONE 2009, 4, e8044. [Google Scholar] [CrossRef]
- McCoy, C.E.; Campbell, D.G.; Deak, M.; Bloomberg, G.B.; Arthur, J.S. MSK1 activity is controlled by multiple phosphorylation sites. Biochem. J. 2005, 387, 507–517. [Google Scholar] [CrossRef]
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [Green Version]
- Markou, T.; Cieslak, D.; Gaitanaki, C.; Lazou, A. Differential roles of MAPKs and MSK1 signalling pathways in the regulation of c-Jun during phenylephrine-induced cardiac myocyte hypertrophy. Mol. Cell. Biochem. 2009, 322, 103–112. [Google Scholar] [CrossRef]
- Markou, T.; Hadzopoulou-Cladaras, M.; Lazou, A. Phenylephrine induces activation of CREB in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J. Mol. Cell. Cardiol. 2004, 37, 1001–1011. [Google Scholar] [CrossRef]
- Alessi, D.R. The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1beta (Rsk-2) and p70 S6 kinase. FEBS Lett. 1997, 402, 121–123. [Google Scholar] [CrossRef] [Green Version]
- Markou, T.; Lazou, A. Phosphorylation and activation of mitogen- and stress-activated protein kinase-1 in adult rat cardiac myocytes by G-protein-coupled receptor agonists requires both extracellular-signal-regulated kinase and p38 mitogen-activated protein kinase. Biochem. J. 2002, 365, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, S.; Macdonald, A.; McCoy, C.E.; Darragh, J.; Reith, A.D.; Arthur, J.S. Characterization of the cellular action of the MSK inhibitor SB-747651A. Biochem. J. 2012, 441, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.S.; Cohen, P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000, 482, 44–48. [Google Scholar] [CrossRef]
- Wiggin, G.R.; Soloaga, A.; Foster, J.M.; Murray-Tait, V.; Cohen, P.; Arthur, J.S. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 2002, 22, 2871–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higazi, D.R.; Fearnley, C.J.; Drawnel, F.M.; Talasila, A.; Corps, E.M.; Ritter, O.; McDonald, F.; Mikoshiba, K.; Bootman, M.D.; Roderick, H.L. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol. Cell. 2009, 33, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Drawnel, F.M.; Wachten, D.; Molkentin, J.D.; Maillet, M.; Aronsen, J.M.; Swift, F.; Sjaastad, I.; Liu, N.; Catalucci, D.; Mikoshiba, K.; et al. Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell. Biol. 2012, 199, 783–798. [Google Scholar] [CrossRef] [Green Version]
- Dries, E.; Santiago, D.J.; Gilbert, G.; Lenaerts, I.; Vandenberk, B.; Nagaraju, C.K.; Johnson, D.M.; Holemans, P.; Roderick, H.L.; Macquaide, N.; et al. Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 2018, 114, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Invest. 2017, 127, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar] [CrossRef]
- Kadel, K.A.; Heistad, D.D.; Faraci, F.M. Effects of endothelin on blood vessels of the brain and choroid plexus. Brain Res. 1990, 518, 78–82. [Google Scholar] [CrossRef]
- Boluyt, M.O.; Long, X.; Eschenhagen, T.; Mende, U.; Schmitz, W.; Crow, M.T.; Lakatta, E.G. Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. Am. J. Physiol. 1995, 269, H638–H647. [Google Scholar] [CrossRef] [PubMed]
- Werhahn, S.M.; Kreusser, J.S.; Hagenmuller, M.; Beckendorf, J.; Diemert, N.; Hoffmann, S.; Schultz, J.H.; Backs, J.; Dewenter, M. Adaptive versus maladaptive cardiac remodelling in response to sustained beta-adrenergic stimulation in a new ‘ISO on/off model’. PLoS ONE 2021, 16, e0248933. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Jansen, C.; She, Q.B.; Goto, H.; Inagaki, M.; Bode, A.M.; Ma, W.Y.; Dong, Z. Ultraviolet B-induced phosphorylation of histone H3 at serine 28 is mediated by MSK1. J. Biol. Chem. 2001, 276, 33213–33219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, C.T.; Yang, J.; Han, P.; Cheng, H.L.; Shang, C.; Ashley, E.; Zhou, B.; Chang, C.P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar] [CrossRef]
- Wang, J.J.; Rau, C.; Avetisyan, R.; Ren, S.; Romay, M.C.; Stolin, G.; Gong, K.W.; Wang, Y.; Lusis, A.J. Genetic Dissection of Cardiac Remodeling in an Isoproterenol-Induced Heart Failure Mouse Model. PLoS Genet. 2016, 12, e1006038. [Google Scholar] [CrossRef]
- Mahadevan, L.C.; Willis, A.C.; Barratt, M.J. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 1991, 65, 775–783. [Google Scholar] [CrossRef]
- Standaert, M.L.; Bandyopadhyay, G.; Antwi, E.K.; Farese, R.V. RO 31-8220 activates c-Jun N-terminal kinase and glycogen synthase in rat adipocytes and L6 myotubes. Comparison to actions of insulin. Endocrinology 1999, 140, 2145–2151. [Google Scholar] [CrossRef]
- Santalucia, T.; Christmann, M.; Yacoub, M.H.; Brand, N.J. Hypertrophic agonists induce the binding of c-Fos to an AP-1 site in cardiac myocytes: Implications for the expression of GLUT1. Cardiovasc. Res. 2003, 59, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Ulm, S.; Liu, W.; Zi, M.; Tsui, H.; Chowdhury, S.K.; Endo, S.; Satoh, Y.; Prehar, S.; Wang, R.; Cartwright, E.J.; et al. Targeted deletion of ERK2 in cardiomyocytes attenuates hypertrophic response but provokes pathological stress induced cardiac dysfunction. J. Mol. Cell. Cardiol. 2014, 72, 104–116. [Google Scholar] [CrossRef]
- Mutlak, M.; Kehat, I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front Pharmacol. 2015, 6, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Yussman, M.G.; Barrett, T.J.; Hahn, H.S.; Osinska, H.; Hilliard, G.M.; Wang, X.; Toyokawa, T.; Yatani, A.; Lynch, R.A.; et al. Increased myocardial Rab GTPase expression: A consequence and cause of cardiomyopathy. Circ. Res. 2001, 89, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2011, 108, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, H.; Perrino, C.; Takaoka, H.; Davis, R.J.; Naga Prasad, S.V.; Rockman, H.A. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem. Biophys. Res. Commun. 2006, 343, 1060–1066. [Google Scholar] [CrossRef]
- Gehani, S.S.; Agrawal-Singh, S.; Dietrich, N.; Christophersen, N.S.; Helin, K.; Hansen, K. Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol. Cell. 2010, 39, 886–900. [Google Scholar] [CrossRef]
- Gilsbach, R.; Preissl, S.; Gruning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Wurch, A.; Bonisch, U.; Gunther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, K.B.; Son, H.J.; Chae, Y.C.; Oh, S.T.; Kim, D.W.; Pak, J.H.; Seo, S.B. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Ibeta. FEBS Lett. 2012, 586, 3159–3165. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Al-Haffar, K.M.; Marashly, Q.; Quijada, P.; Kunhi, M.; Al-Yacoub, N.; Wade, F.S.; Mohammed, S.F.; Al-Dayel, F.; Sutherland, G.; et al. Control of histone H3 phosphorylation by CaMKIIdelta in response to haemodynamic cardiac stress. J. Pathol. 2015, 235, 606–618. [Google Scholar] [CrossRef]
- Saadatmand, A.R.; Sramek, V.; Weber, S.; Finke, D.; Dewenter, M.; Sticht, C.; Gretz, N.; Wustemann, T.; Hagenmueller, M.; Kuenzel, S.R.; et al. CaM kinase II regulates cardiac hemoglobin expression through histone phosphorylation upon sympathetic activation. Proc. Natl. Acad. Sci. USA 2019, 116, 22282–22287. [Google Scholar] [CrossRef]
- Joos, J.P.; Saadatmand, A.R.; Schnabel, C.; Viktorinova, I.; Brand, T.; Kramer, M.; Nattel, S.; Dobrev, D.; Tomancak, P.; Backs, J.; et al. Ectopic expression of S28A-mutated Histone H3 modulates longevity, stress resistance and cardiac function in Drosophila. Sci. Rep. 2018, 8, 2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, S.; Kunhi, M.; Little, G.H.; Bai, Y.; An, W.; Bers, D.; Kedes, L.; Poizat, C. Nuclear CaMKII enhances histone H3 phosphorylation and remodels chromatin during cardiac hypertrophy. Nucleic Acids Res. 2013, 41, 7656–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, M.; Delghandi, M.P.; Moens, U. What turns CREB on? Cell. Signal. 2004, 16, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Thomas, M.C.; Zambetti, G.P.; Brindle, P.K. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle 2011, 10, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.A.; Birdsey, N.; Huggins, G.S.; Svensson, E.; Heppe, D.; Knaub, L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2056–H2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Mahata, B.; Escobar, M.; Goell, J.; Wang, K.; Khemka, P.; Hilton, I.B. Programmable human histone phosphorylation and gene activation using a CRISPR/Cas9-based chromatin kinase. Nat. Commun. 2021, 12, 896. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, E.L.; Drawnel, F.M.; Mehdi, S.; Archer, C.R.; Liu, W.; Okkenhaug, H.; Alkass, K.; Aronsen, J.M.; Nagaraju, C.K.; Sjaastad, I.; et al. MSK-Mediated Phosphorylation of Histone H3 Ser28 Couples MAPK Signalling with Early Gene Induction and Cardiac Hypertrophy. Cells 2022, 11, 604. https://doi.org/10.3390/cells11040604
Robinson EL, Drawnel FM, Mehdi S, Archer CR, Liu W, Okkenhaug H, Alkass K, Aronsen JM, Nagaraju CK, Sjaastad I, et al. MSK-Mediated Phosphorylation of Histone H3 Ser28 Couples MAPK Signalling with Early Gene Induction and Cardiac Hypertrophy. Cells. 2022; 11(4):604. https://doi.org/10.3390/cells11040604
Chicago/Turabian StyleRobinson, Emma L., Faye M. Drawnel, Saher Mehdi, Caroline R. Archer, Wei Liu, Hanneke Okkenhaug, Kanar Alkass, Jan Magnus Aronsen, Chandan K. Nagaraju, Ivar Sjaastad, and et al. 2022. "MSK-Mediated Phosphorylation of Histone H3 Ser28 Couples MAPK Signalling with Early Gene Induction and Cardiac Hypertrophy" Cells 11, no. 4: 604. https://doi.org/10.3390/cells11040604