

Cardiovascular Protective Effects of NP-6A4, a Drug with the FDA Designation for Pediatric Cardiomyopathy, in Female Rats with Obesity and Pre-Diabetes
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Metabolic and Cardiac Phenotyping of 5-Month-Old ZDF-F Rats
3.2. Effects of NP-6A4 and NP-6A4 + PD123319 Treatments on Cardiac Functions of Obese and Hyperglycemic ZDF-F Rats
3.3. Effects of NP-6A4 and NP-6A4 + PD123319 Treatments on Cardiac Capillary Density and Coronary Microvascular Damage in ZDF-F Rats
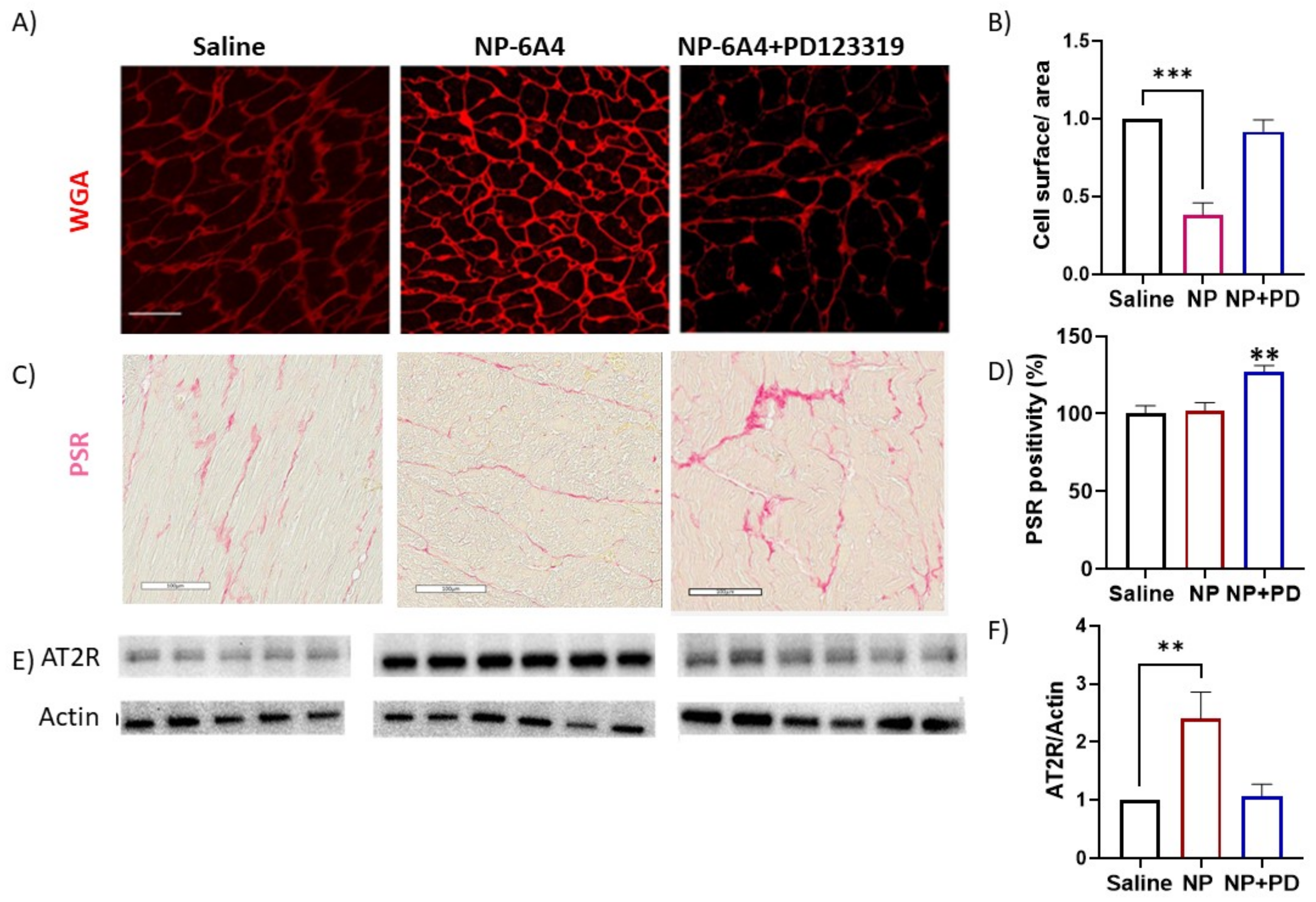
3.4. Effects of NP-6A4 and NP-6A4 + PD123319 Treatments on Cardiomyocyte Hypertrophy, Fibrosis and Cardiac AT2R Expression in ZDF-F Rats
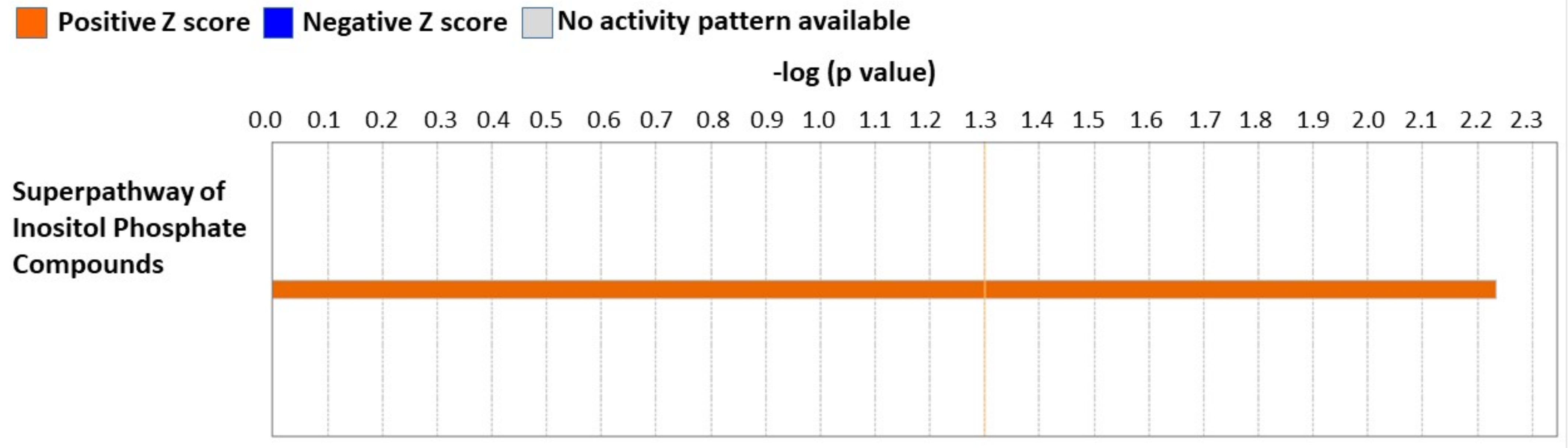
3.5. Cardiac Proteome Analysis Shows Activation of Superpathway of Inositol Compounds by NP-6A4-AT2R Signaling in ZDF-F Rat Hearts
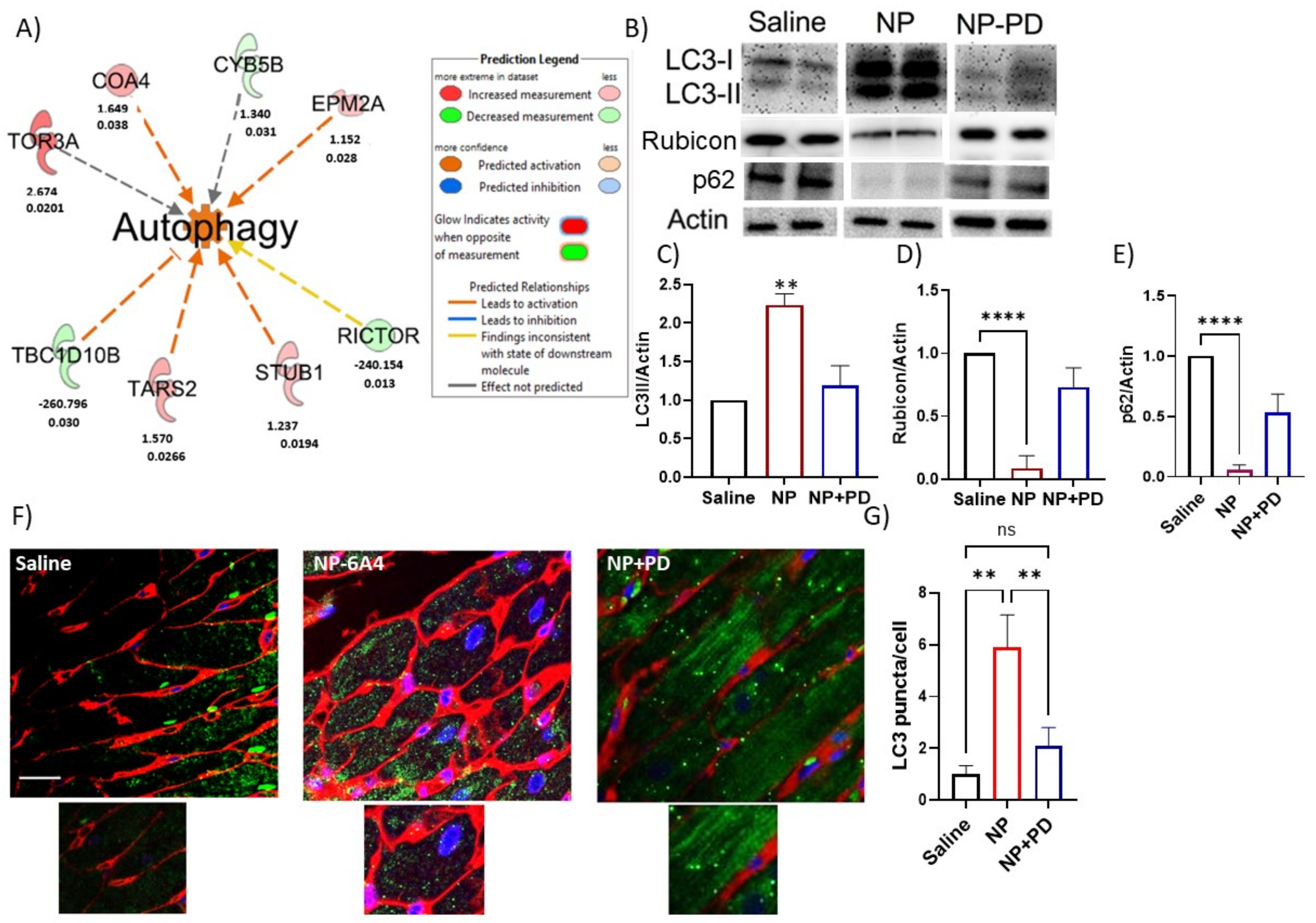
3.6. NP-6A4 Induced Cardiac Autophagy Activation in Obese ZDF-F Rats and PD123319 Suppressed This Effect
3.7. Inhibition of AT2R by Co-Treatment with PD123319 Induced Inflammatory Reelin Signaling Pathway in the Heart
3.8. Inhibition of NP-6A4-AT2R Signaling in ZDF-F Rat Heart by Co-Treatment with PD123319 Induces Signaling Networks That Decrease ATP Concentration, and Increase Muscle Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Animals
Appendix A.2. Drug Treatments
Appendix A.3. Body Weight, Fasting Plasma Profile and Blood Pressure
Appendix A.4. Echocardiography
Appendix A.5. Evaluation of the Effect of NP-6A4 Treatment on Healthy Rats
Appendix A.6. Histopathology and NT-ProBNP
Appendix A.7. Quantification of Microvascular Damage in ZDF-F Rat Heart
Appendix A.8. Capillary Density and Cardiomyocyte Hypertrophy
Appendix A.9. Quantification of LC3, RUBICON and AT2R by Immunohistochemistry and Immunoblotting
Appendix A.10. Cardiac Proteome Analysis
References
- Nowbar, A.N.; Gitto, M.; Howard, J.P.; Francis, D.P.; Al-Lamee, R. Mortality From Ischemic Heart Disease. Circ Cardiovasc. Qual. Outcomes 2019, 12, e005375. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. American Heart Association Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Epidemiology and Prevention; and Stroke Council. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [PubMed]
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- McPherson, R. Obesity and ischemic heart disease: Defining the link. Circ. Res. 2015, 116, 570–571. [Google Scholar] [CrossRef]
- Savji, N.; Meijers, W.C.; Bartz, T.M.; Bhambhani, V.; Cushman, M.; Nayor, M.; Kizer, J.R.; Sarma, A.; Blaha, M.J.; Gansevoort, R.T.; et al. The Association of Obesity and Cardiometabolic Traits with Incident HFpEF and HFrEF. JACC Hear. Fail. 2018, 6, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Somaratne, J.B.; Prior, D.L.; Yii, M.; Kenny, J.F.; Newcomb, A.E.; Kelly, D.J.; Black, M.J. Obesity Is Associated with Lower Coronary Microvascular Density. PLoS ONE 2013, 8, e81798. [Google Scholar] [CrossRef]
- Paavonsalo, S.; Hariharan, S.; Lackman, M.H.; Karaman, S. Capillary Rarefaction in Obesity and Metabolic Diseases—Organ-Specificity and Possible Mechanisms. Cells 2020, 9, 2683. [Google Scholar] [CrossRef]
- Levy, B.I.; Ambrosio, G.; Pries, A.R.; Struijker-Boudier, H.A. Microcirculation in hypertension: A new target for treatment? Circulation 2001, 104, 735–740. [Google Scholar] [CrossRef]
- Wilson, P.W.; D’Agostino, R.B.; Sullivan, L.; Parise, H.; Kannel, W.B. Overweight and obesity as determinants of cardiovascular risk: The Framingham experience. Arch. Intern. Med. 2002, 162, 1867–1872. [Google Scholar] [CrossRef]
- Song, X.; for the DECODE Study Group; Tabák, A.G.; Zethelius, B.; Yudkin, J.S.; Söderberg, S.; Laatikainen, T.; Da Stehouwer, C.; Dankner, R.; Jousilahti, P.; et al. Obesity attenuates gender differences in cardiovascular mortality. Cardiovasc. Diabetol. 2014, 13, 144. [Google Scholar] [CrossRef]
- Dikaiou, P.; Björck, L.; Adiels, M.; Lundberg, C.E.; Mandalenakis, Z.; Manhem, K.; Rosengren, A. Obesity, overweight and risk for cardiovascular disease and mortality in young women. Eur. J. Prev. Cardiol. 2020, 28, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.B.; Buring, J.E.; Manson, J.E.; Whisnant, J.P.; Winston, M. Cardiovascular Disease in Women. Circ. Res. 2016, 118, 1273–1293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; O’neil, A.; Jiao, Y.; Wang, L.; Huang, J.; Lan, Y.; Zhu, Y.; Yu, C. Sex differences in the association between diabetes and risk of cardiovascular disease, cancer, and all-cause and cause-specific mortality: A systematic review and meta-analysis of 5,162,654 participants. BMC Med. 2019, 17, 136. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Bullo, M.; Tschumi, S.; Bucher, B.S.; Bianchetti, M.G.; Simonetti, G.D. Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: A systematic review. Hypertension 2012, 60, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Koutouroushis, C.; Sarkar, O. Role of Autophagy in Cardiovascular Disease and Aging. Cureus 2021, 13, e20042. [Google Scholar] [CrossRef]
- Nah, J.; Zablocki, D.; Sadoshima, J. The role of autophagic cell death in cardiac disease. J. Mol. Cell. Cardiol. 2022, 173, 16–24. [Google Scholar] [CrossRef]
- Ren, J.; Wu, N.N.; Wang, S.; Sowers, J.R.; Zhang, Y. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev. 2021, 101, 1745–1807. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Therapeutic targeting of autophagy in cardiovascular disease. J. Mol. Cell. Cardiol. 2015, 95, 86–93. [Google Scholar] [CrossRef]
- Castañeda, D.; Gabani, M.; Choi, S.; Nguyen, Q.M.; Chen, C.; Mapara, A.; Kassan, A.; Gonzalez, A.A.; Ait-Aissa, K.; Kassan, M. Targeting Autophagy in Obesity-Associated Heart Disease. Obesity 2019, 27, 1050–1058. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Reineke, D.C.; Müller-Schweinitzer, E.; Winkler, B.; Kunz, D.; Konerding, M.A.; Grussenmeyer, T.; Carrel, T.P.; Eckstein, F.S.; Grapow, M.T. Rapamycin impairs endothelial cell function in human internal thoracic arteries. Eur. J. Med. Res. 2015, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Zahr, E.; Molano, R.D.; Pileggi, A.; Ichii, H.; Jose, S.S.; Bocca, N.; An, W.; Gonzalez-Quintana, J.; Fraker, C.; Ricordi, C.; et al. Rapamycin Impairs In Vivo Proliferation of Islet Beta-Cells. Transplantation 2007, 84, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Lum-Naihe, K.; Toedebusch, R.; Mahmood, A.; Bajwa, J.; Carmack, T.; Kumar, S.A.; Ardhanari, S.; DeMarco, V.G.; Emter, C.A.; Pulakat, L. Cardiovascular disease progression in female Zucker Diabetic Fatty rats occurs via unique mechanisms compared to males. Sci. Rep. 2017, 7, 17823. [Google Scholar] [CrossRef]
- Tramunt, B.; Smati, S.; Grandgeorge, N.; Lenfant, F.; Arnal, J.-F.; Montagner, A.; Gourdy, P. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2019, 63, 453–461. [Google Scholar] [CrossRef]
- Mahmood, A.; Pulakat, L. Differential Effects of β-Blockers, Angiotensin II Receptor Blockers, and a Novel AT2R Agonist NP-6A4 on Stress Response of Nutrient-Starved Cardiovascular Cells. PLoS ONE 2015, 10, e0144824. [Google Scholar] [CrossRef]
- Gavini, M.P.; Mahmood, A.; Belenchia, A.M.; Beauparlant, P.; Kumar, S.A.; Ardhanari, S.; DeMarco, V.G.; Pulakat, L. Suppression of Inflammatory Cardiac Cytokine Network in Rats with Untreated Obesity and Pre-Diabetes by AT2 Receptor Agonist NP-6A4. Front. Pharmacol. 2021, 12, 693167. [Google Scholar] [CrossRef]
- Toedebusch, R.; Belenchia, A.; Pulakat, L. Cell-Specific Protective Signaling Induced by the Novel AT2R-Agonist NP-6A4 on Human Endothelial and Smooth Muscle Cells. Front. Pharmacol. 2018, 9, 928. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]
- Tanase, D.M.; Radu, S.; Al Shurbaji, S.; Baroi, G.L.; Costea, C.F.; Turliuc, M.D.; Ouatu, A.; Floria, M. Natriuretic Peptides in Heart Failure with Preserved Left Ventricular Ejection Fraction: From Molecular Evidences to Clinical Implications. Int. J. Mol. Sci. 2019, 20, 2629. [Google Scholar] [CrossRef] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Healio Learn The Heart: Congestive Heart Failure–Diastolic Topic Review. Available online: https://www.healio.com/cardiology/learn-the-heart/cardiology-review/topic-reviews/congestive-heart-failure-diastolic (accessed on 15 September 2022).
- Dieseldorff Jones, K.M.; Vied, C.; Valera, I.C.; Chase, P.B.; Parvatiyar, M.S.; Pinto, J.R. Sexual dimorphism in cardiac transcriptome associated with a troponin C murine model of hypertrophic cardiomyopathy. Physiol. Rep. 2020, 8, e14396. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Álvarez, J.; Andersen, J.K. mTORC2: The other mTOR in autophagy regulation. Aging Cell 2021, 20, e13431. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Chung, K.-P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. J. Clin. Investig. 2019, 4, e132826. [Google Scholar] [CrossRef]
- Popovic, D.; Akutsu, M.; Novak, I.; Harper, J.W.; Behrends, C.; Dikic, I. Rab GTPase-Activating Proteins in Autophagy: Regulation of Endocytic and Autophagy Pathways by Direct Binding to Human ATG8 Modifiers. Mol. Cell. Biol. 2012, 32, 1733–1744. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.-I.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Mizushima, N.; Murphy, L.O. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends Biochem. Sci. 2020, 45, 1080–1093. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P. Untangling autophagy measurements: All fluxed up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nature 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Calvier, L.; Xian, X.; Lee, R.G.; Sacharidou, A.; Mineo, C.; Shaul, P.W.; Kounnas, M.Z.; Tsai, S.; Herz, J. Reelin Depletion Protects Against Atherosclerosis by Decreasing Vascular Adhesion of Leukocytes. Arter. Thromb. Vasc. Biol. 2021, 41, 1309–1318. [Google Scholar] [CrossRef]
- Ding, Y.; Huang, L.; Xian, X.; Yuhanna, I.S.; Wasser, C.R.; Frotscher, M.; Mineo, C.; Shaul, P.W.; Herz, J. Loss of Reelin protects against atherosclerosis by reducing leukocyte–endothelial cell adhesion and lesion macrophage accumulation. Sci. Signal. 2016, 9, ra29. [Google Scholar] [CrossRef]
- Kitzman, D.W.; Lam, C.S.P. Obese Heart Failure With Preserved Ejection Fraction Phenotype: From Pariah to Central Player. Circulation 2017, 136, 20–23. [Google Scholar] [CrossRef]
- Pfaller, B.; Siu, S.C.; D’Souza, R.; Wichert-Schmitt, B.; Nair, G.K.K.; Haberer, K.; Maxwell, C.; Silversides, C.K. Impact of Obesity on Outcomes of Pregnancy in Women With Heart Disease. J. Am. Coll. Cardiol. 2021, 77, 1317–1326. [Google Scholar] [CrossRef]
- Samuel, P.; Khan, M.A.; Nag, S.; Inagami, T.; Hussain, T. Angiotensin AT2 Receptor Contributes towards Gender Bias in Weight Gain. PLoS ONE 2013, 8, e48425. [Google Scholar] [CrossRef]
- Sampson, A.K.; Moritz, K.M.; Jones, E.S.; Flower, R.L.; Widdop, R.E.; Denton, K.M. Enhanced Angiotensin II Type 2 Receptor Mechanisms Mediate Decreases in Arterial Pressure Attributable to Chronic Low-Dose Angiotensin II in Female Rats. Hypertension 2008, 52, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, L.M.; Jones, E.S.; Steckelings, U.M.; Unger, T.; Widdop, R.E.; Denton, K.M. Sex-specific influence of angiotensin type 2 receptor stimulation on renal function: A novel therapeutic target for hypertension. Hypertension 2012, 59, 409–414. [Google Scholar] [CrossRef]
- Rehman, A.; Leibowitz, A.; Yamamoto, N.; Rautureau, Y.; Paradis, P.; Schiffrin, E.L. Angiotensin Type 2 Receptor Agonist Compound 21 Reduces Vascular Injury and Myocardial Fibrosis in Stroke-Prone Spontaneously Hypertensive Rats. Hypertension 2012, 59, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Patel, S.N.; Hussain, T. Angiotensin II Type 2 Receptor: A Target for Protection Against Hypertension, Metabolic Dysfunction, and Organ Remodeling. Hypertension 2021, 77, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, Y.; Carretero, O.A.; Zhu, L.; Harding, P.; Shesely, E.G.; Dai, X.; Rhaleb, N.-E.; Peterson, E.; Yang, X.-P. Effects of Cardiac Overexpression of the Angiotensin II Type 2 Receptor on Remodeling and Dysfunction in Mice Post–Myocardial Infarction. Hypertension 2014, 63, 1251–1259. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Matsubara, H.; Masaki, H.; Kurihara, H.; Murasawa, S.; Takai, S.; Miyazaki, M.; Nozawa, Y.; Ozono, R.; Nakagawa, K.; et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Investig. 1999, 104, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Masaki, H.; Kurihara, T.; Yamaki, A.; Inomata, N.; Nozawa, Y.; Mori, Y.; Murasawa, S.; Kizima, K.; Maruyama, K.; Horiuchi, M.; et al. Cardiac-specific overexpression of angiotensin II AT2 receptor causes attenuated response to AT1 receptor-mediated pressor and chronotropic effects. J. Clin. Investig. 1998, 101, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Pettersson-Fernholm, K.; Fröjdö, S.; Fagerudd, J.; Thomas, M.; Forsblom, C.; Wessman, M.; Groop, P.-H. The AT2 gene may have a gender-specific effect on kidney function and pulse pressure in type I diabetic patients. Kidney Int. 2006, 69, 1880–1884. [Google Scholar] [CrossRef]
- Cwynar, M.; Gąsowski, J.; Głuszewska, A.; Królczyk, J.; Bartoń, H.; Słowik, A.; Grodzicki, T. Blood pressure, arterial stiffness and endogenous lithium clearance in relation to AGTR1 A1166C and AGTR2 G1675A gene polymorphisms. J. Renin. Angiotensin Aldosterone Syst. 2016, 17, 1470320316655669. [Google Scholar] [CrossRef]
- Tornling, G.; Batta, R.; Porter, J.C.; Williams, B.; Bengtsson, T.; Parmar, K.; Kashiva, R.; Hallberg, A.; Cohrt, A.K.; Westergaard, K.; et al. Seven days treatment with the angiotensin II type 2 receptor agonist C21 in hospitalized COVID-19 patients; a placebo-controlled randomised multi-centre double-blind phase 2 trial. Eclinicalmedicine 2021, 41, 101152. [Google Scholar] [CrossRef]
- Safety, Efficacy and Pharmacokinetics of C21 in Subjects with IPF. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04533022 (accessed on 15 September 2022).
- Porrello, E.R.; D’Amore, A.; Curl, C.L.; Allen, A.M.; Harrap, S.B.; Thomas, W.; Delbridge, L.M. Angiotensin II Type 2 Receptor Antagonizes Angiotensin II Type 1 Receptor–Mediated Cardiomyocyte Autophagy. Hypertension 2009, 53, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Delbridge, L.M. Cardiomyocyte autophagy is regulated by angiotensin II type 1 and type 2 receptors. Autophagy 2009, 5, 1215–1216. [Google Scholar] [CrossRef] [PubMed]
- Luck, C.; DeMarco, V.G.; Mahmood, A.; Gavini, M.P.; Pulakat, L. Differential Regulation of Cardiac Function and Intracardiac Cytokines by Rapamycin in Healthy and Diabetic Rats. Oxidative Med. Cell. Longev. 2017, 2017, 5724046. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Koppula, P.R.; Gul, R.; Luck, C.; Pulakat, L. Regulation of Cardiac Expression of the Diabetic Marker MicroRNA miR-29. PLoS ONE 2014, 9, e103284. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Belenchia, A.M.; Toedebusch, R.; Pulakat, L.; Hans, C.P. AT2R agonist NP-6A4 mitigates aortic stiffness and proteolytic activity in mouse model of aneurysm. J. Cell. Mol. Med. 2020, 24, 7393–7404. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.A.; Powell, P.C.; Agarwal, A.; Dell’Italia, L.J.; Chatham, J.C. Cardiovascular dysfunction in Zucker obese and Zucker diabetic fatty rats: Role of hydronephrosis. Am. J. Physiol. Circ. Physiol. 2007, 293, H292–H298. [Google Scholar] [CrossRef]
- Yang, B.; Li, R.; Liu, P.N.; Geng, X.; Mooney, B.P.; Chen, C.; Cheng, J.; Fritsche, K.L.; Beversdorf, D.Q.; Lee, J.C.; et al. Quantitative Proteomics Reveals Docosahexaenoic Acid-Mediated Neuroprotective Effects in Lipopolysaccharide-Stimulated Microglial Cells. J. Proteome Res. 2020, 19, 2236–2246. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Parameters | Saline (N = 5) | NP-6A4 (N = 6) | NP-6A4 + PD123319 (N = 6) | Saline vs. NP-6A4 (p Value) | NP-6A4 vs. NP-6A4 + PD123319 (p Value) |
|---|---|---|---|---|---|
| Heart rate | 247 ± 9 | 310 ± 23 | 269 ± 10 | 0.034 | 0.15 |
| E/A | 2.11 ± 0.048 | 1.49 ± 0.14 | 1.84 ± 0.09 | 0.006 | 0.08 |
| Stroke Volume (SV) (µL) | 185 ± 9 | 253 ± 31 | 165 ± 20 | 0.034 | 0.035 |
| Ejection Fraction (EF) | 56.4 ± 2.1 | 63 ± 3 | 56.3 ± 3.2 | 0.059 | 0.167 |
| Cardiac Output (CO) | 50 ± 3 | 80 ± 10 | 47 ± 5 | 0.012 | 0.011 |
| Radial Strain (Pk%) | 22.8 ± 2.77 | 38.0 ± 5.28 | 20.0 ± 3.67 | 0.019 | 0.018 |
| Radial Strain Rate (Pk1/s) | 3.69 ± 0.43 | 6.44 ± 1.15 | 3.55 ± 0.51 | 0.033 | 0.037 |
| Isovolumic Relaxation time (IVRT (ms) | 22.3 ± 0.617 | 18.6 ± 1.01 | 24.5 ± 0.935 | 0.022 | 0.002 |
| Isovolumic Contraction time (IVCT) (ms) | 13.6 ± 0.835 | 9.78 ± 0.45 | 14.1 ± 1.12 | 0.004 | 0.009 |
| Systolic Time (Syst T) (ms) | 117 ± 3.065 | 102 ± 5.33 | 120 ± 3.329 | 0.056 | 0.016 |
| Myocardial Performance Index (MPI) | 0.44 ± 0.027 | 0.39 ± 0.01 | 0.48 ± 0.02 | 0.06 | 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belenchia, A.M.; Boukhalfa, A.; DeMarco, V.G.; Mehm, A.; Mahmood, A.; Liu, P.; Tang, Y.; Gavini, M.P.; Mooney, B.; Chen, H.H.; et al. Cardiovascular Protective Effects of NP-6A4, a Drug with the FDA Designation for Pediatric Cardiomyopathy, in Female Rats with Obesity and Pre-Diabetes. Cells 2023, 12, 1373. https://doi.org/10.3390/cells12101373
Belenchia AM, Boukhalfa A, DeMarco VG, Mehm A, Mahmood A, Liu P, Tang Y, Gavini MP, Mooney B, Chen HH, et al. Cardiovascular Protective Effects of NP-6A4, a Drug with the FDA Designation for Pediatric Cardiomyopathy, in Female Rats with Obesity and Pre-Diabetes. Cells. 2023; 12(10):1373. https://doi.org/10.3390/cells12101373
Chicago/Turabian StyleBelenchia, Anthony M., Asma Boukhalfa, Vincent G. DeMarco, Alexander Mehm, Abuzar Mahmood, Pei Liu, Yinian Tang, Madhavi P. Gavini, Brian Mooney, Howard H. Chen, and et al. 2023. "Cardiovascular Protective Effects of NP-6A4, a Drug with the FDA Designation for Pediatric Cardiomyopathy, in Female Rats with Obesity and Pre-Diabetes" Cells 12, no. 10: 1373. https://doi.org/10.3390/cells12101373