
Astragaloside IV Blunts Epithelial–Mesenchymal Transition and G2/M Arrest to Alleviate Renal Fibrosis via Regulating ALDH2-Mediated Autophagy
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Drug Preparation
2.3. Animal Procedures
2.4. Biochemical Measurements
2.5. Hematoxylin and Eosin (H&E) Staining
2.6. Masson Staining and Sirius Red Staining
2.7. Immunohistochemistry
2.8. Immunofluorescence
2.9. RNA-Seq Analysis
2.10. RNA Extraction and RT-PCR
2.11. Western Blotting
2.12. Cell Culture and Treatment
2.13. Cell Viability Determination
2.14. siRNA Transfectiony
2.15. Dual Luciferase Reporter Assay
2.16. Molecular Docking Analysis
2.17. Statistical Analysis
3. Results
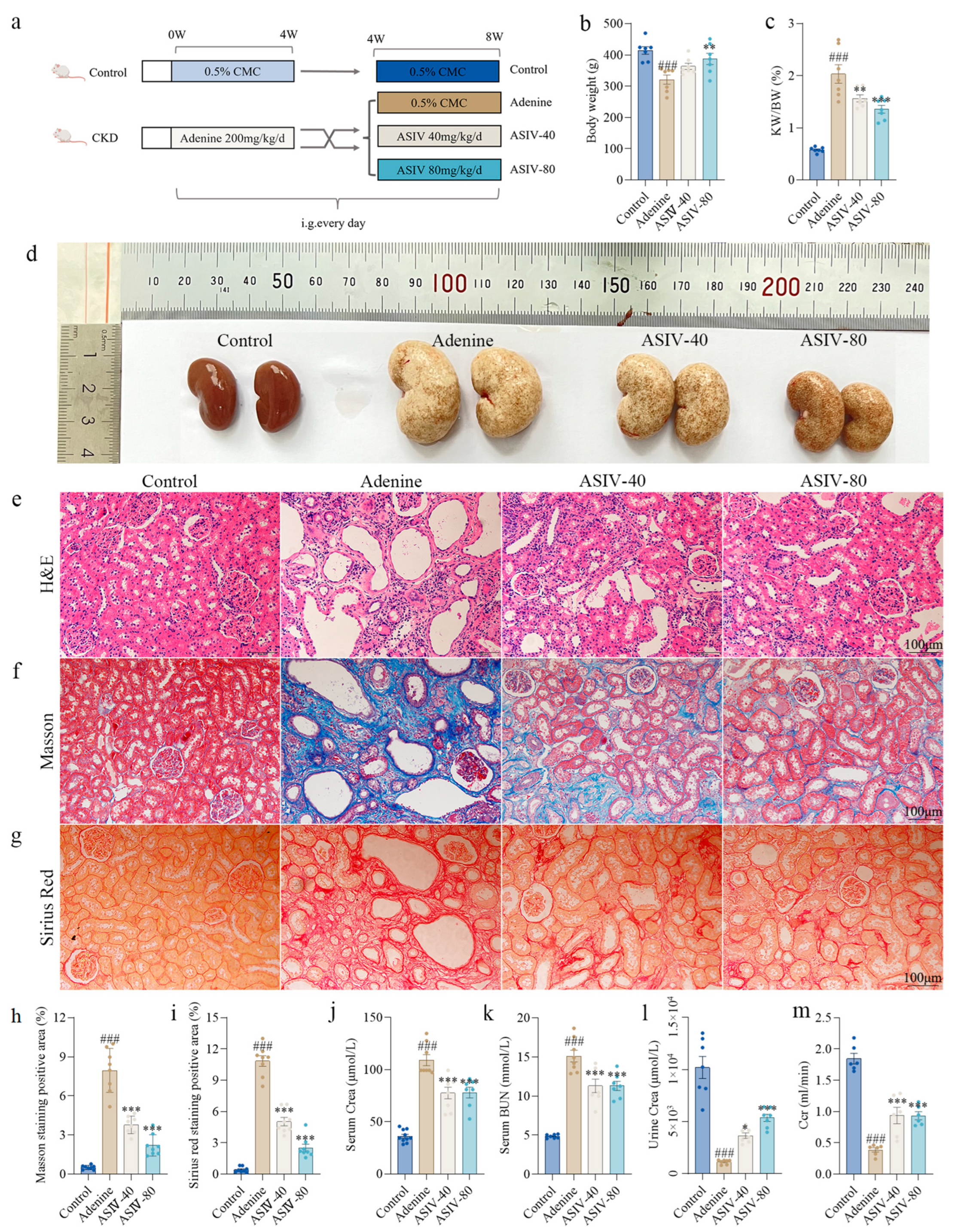
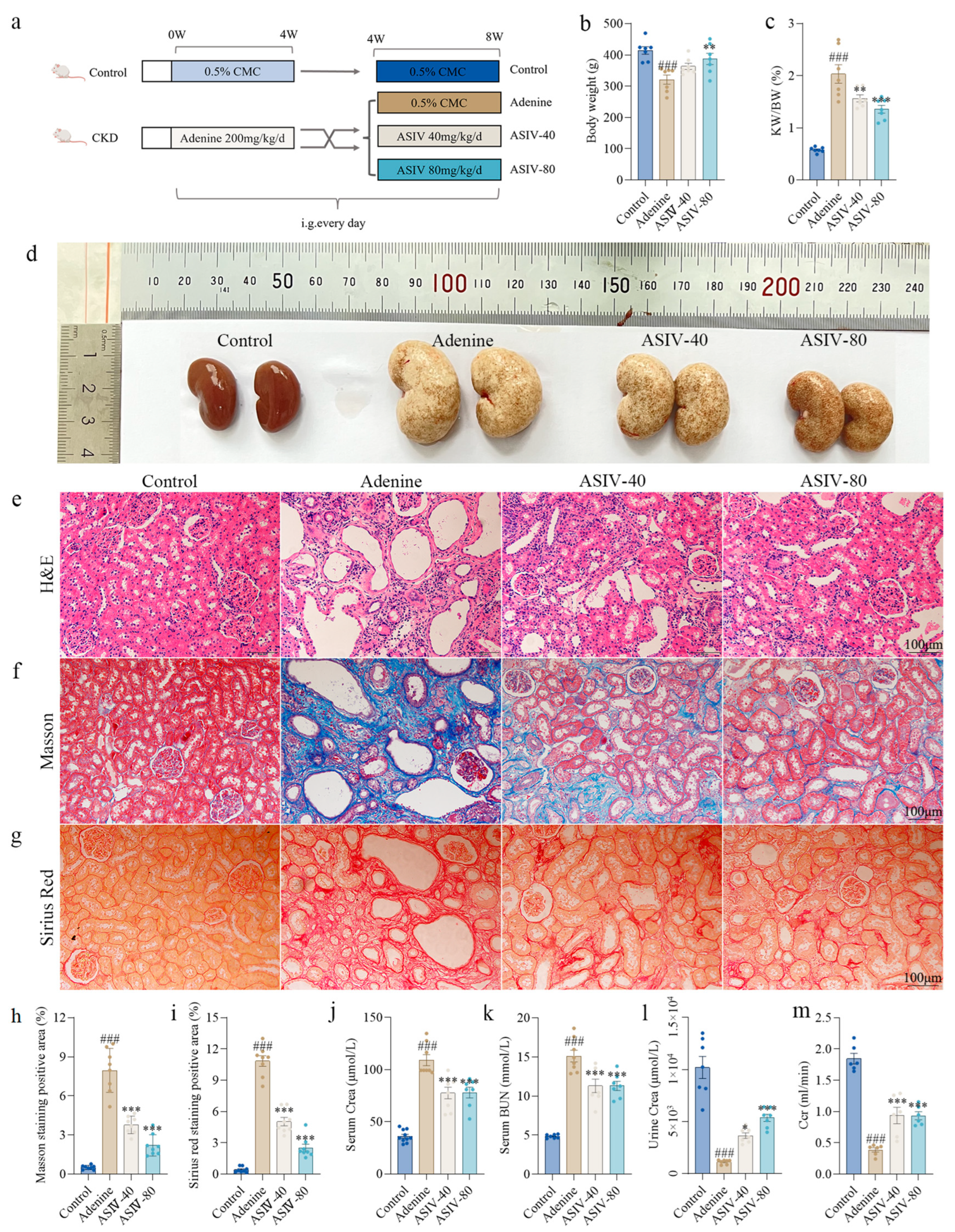
3.1. ASIV Alleviated Renal Pathological Changes and Improved Renal Function in Adenine-Induced CKD Rats
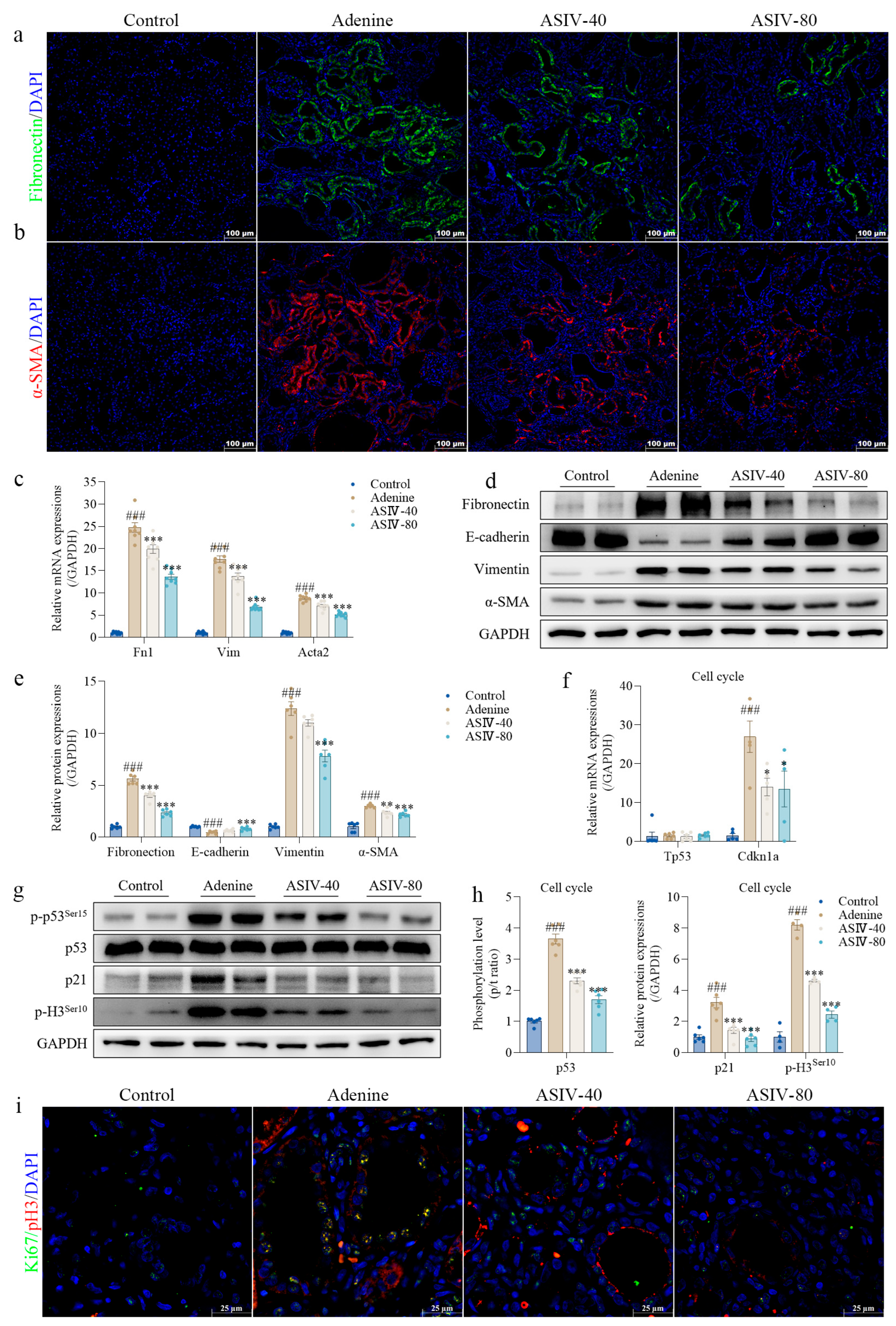
3.2. ASIV Alleviates EMT and G2/M Arrest in the Renal Tissues of CKD Rats
3.3. RNA-Seq Analysis Reveals the Mechanisms of ASIV on the Kidney in Adenine-Induced CKD Rats
3.4. ASIV Promotes ALDH2 Expression, Inhibits Autophagy, and Activates AKT/mTOR Signaling Pathway In Vivo
3.5. ASIV Mitigates EMT and G2/M Arrest, Promotes ALDH2 Expression, Inhibits Autophagy, and Activates AKT/mTOR Signaling Pathway in TGF-β1-Stimulated HK-2 Cells
3.6. ALDH2 siRNA Blocks the Beneficial Effects of ASIV on Renal Fibrosis In Vitro
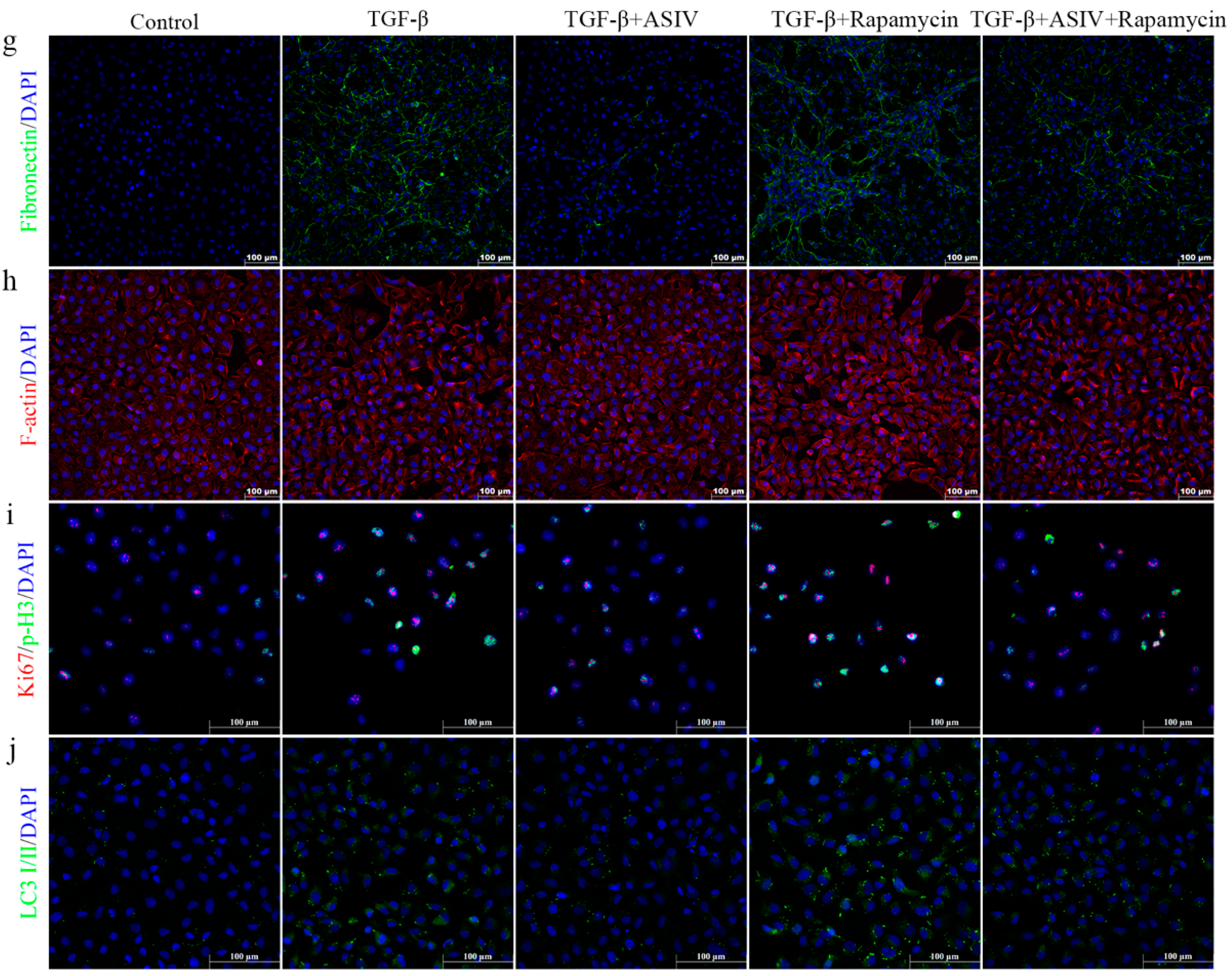
3.7. mTOR Signaling Pathway-Mediated Autophagy Is Required for the ASIV-Induced Alleviation of EMT and G2/M Arrest In Vitro
3.8. ASIV Upregulates the Transcriptional Activity of ALDH2 Promoter, and Molecular Docking Predicts the Potential Interaction between ASIV and ALDH2 Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hewitson, T.D. Renal tubulointerstitial fibrosis: Common but never simple. Am. J. Physiol. Ren. Physiol. 2009, 296, F1239–F1244. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Murphy, D.; McCulloch, C.E.; Lin, F.; Banerjee, T.; Bragg-Gresham, J.L.; Eberhardt, M.S.; Morgenstern, H.; Pavkov, M.E.; Saran, R.; Powe, N.R. Trends in prevalence of chronic kidney disease in the United States. Ann. Intern. Med. 2016, 165, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Savira, F.; Ademi, Z.; Wang, B.H.; Kompa, A.R.; Owen, A.J.; Liew, D.; Zomer, E. The preventable productivity burden of kidney disease in Australia. J. Am. Soc. Nephrol. 2021, 32, 938–949. [Google Scholar] [CrossRef]
- Haritha, C.; Lingaraju, M.C.; Mathesh, K.; Jadhav, S.E.; Shyamkumar, T.; Aneesha, V.; Parida, S.; Singh, T.U.; Kumar, D. PRE-084 ameliorates adenine-induced renal fibrosis in rats. Tissue Cell 2022, 79, 101905. [Google Scholar] [CrossRef]
- Collins, A.J.; Foley, R.N.; Herzog, C.; Chavers, B.; Gilbertson, D.; Herzog, C.; Ishani, A.; Johansen, K.; Kasiske, B.; Kutner, N. US Renal Data System 2012 annual data report. Am. J. Kidney Dis. 2013, 61, E1–E459. [Google Scholar] [CrossRef]
- Honeycutt, A.A.; Segel, J.E.; Zhuo, X.; Hoerger, T.J.; Imai, K.; Williams, D. Medical costs of CKD in the Medicare population. J. Am. Soc. Nephrol. 2013, 24, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Neovius, M.; Jacobson, S.H.; Eriksson, J.K.; Elinder, C.-G.; Hylander, B. Mortality in chronic kidney disease and renal replacement therapy: A population-based cohort study. BMJ Open 2014, 4, e004251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L. How tubular epithelial cell injury contributes to renal fibrosis. In Renal Fibrosis: Mechanisms and Therapies; Springer: Singapore, 2019; pp. 233–252. [Google Scholar]
- Tan, R.J.; Bastacky, S.I.; Liu, Y. Molecular Basis of Kidney Disease. In Molecular Pathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 531–553. [Google Scholar]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Levin, A.; Tonelli, M.; Bonventre, J.; Coresh, J.; Donner, J.-A.; Fogo, A.B.; Fox, C.S.; Gansevoort, R.T.; Heerspink, H.J.; Jardine, M. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 2017, 390, 1888–1917. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Zhang, J.; Cui, L.-Y.; Yang, S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp. Biol. Med. 2015, 240, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Wang, M.; Song, S.; Miao, Y.; Zhang, Q. Autophagy activation promotes removal of damaged mitochondria and protects against renal tubular injury induced by albumin overload. Histol. Histopathol. 2018, 33, 681–690. [Google Scholar]
- Livingston, M.J.; Ding, H.-F.; Huang, S.; Hill, J.A.; Yin, X.-M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, D.; He, Z.; Liu, Q.; Wu, J.; Guan, X.; Tao, Z.; Deng, Y. Inhibition of autophagy-attenuated calcium oxalate crystal-induced renal tubular epithelial cell injury in vivo and in vitro. Oncotarget 2018, 9, 4571. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Zhou, Y.; Huang, Y.; Chen, B.; Wu, M.; Xie, Y.; Chen, X.; Sun, M.; Liu, Y.; Chen, C. Effect of ATM on inflammatory response and autophagy in renal tubular epithelial cells in LPS-induced septic AKI. Exp. Ther. Med. 2019, 18, 4707–4717. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tao, M.; Ma, X.; Hu, Y.; Huang, G.; Qiu, A.; Zhuang, S.; Liu, N. Delayed treatment with an autophagy inhibitor 3-MA alleviates the progression of hyperuricemic nephropathy. Cell Death Dis. 2020, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Babcock, S.A.; Hu, N.; Maris, J.R.; Wang, H.; Ren, J. RETRACTED ARTICLE: Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: Role of GSK3β and mitochondrial function. BMC Med. 2022, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.-n.; Yue, S.-s.; Wang, A.-y.; Xu, L.; Hu, Y.-t.; Qiao, X.; Wu, T.-Y.; Ye, M.; Wu, Y.-C.; Qi, R. Antrodia cinnamomea and its compound dehydroeburicoic acid attenuate nonalcoholic fatty liver disease by upregulating ALDH2 activity. J. Ethnopharmacol. 2022, 292, 115146. [Google Scholar] [CrossRef]
- Kang, P.; Wang, J.; Fang, D.; Fang, T.; Yu, Y.; Zhang, W.; Shen, L.; Li, Z.; Wang, H.; Ye, H.; et al. Activation of ALDH2 attenuates high glucose induced rat cardiomyocyte fibrosis and necroptosis. Free Radic. Biol. Med. 2020, 146, 198–210. [Google Scholar] [CrossRef]
- Li, X.; Weng, X.; Shi, H.; Gao, R.; Wang, P.; Jia, D.; Zhang, S.; Dong, Z.; Sun, X.; Yang, J.; et al. Acetaldehyde dehydrogenase 2 deficiency exacerbates cardiac fibrosis by promoting mobilization and homing of bone marrow fibroblast progenitor cells. J. Mol. Cell. Cardiol. 2019, 137, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Shi, X.; Chen, Z.; Xu, J.; Zhao, R.; Liu, Y.; Wen, Y.; Chen, L. Aldehyde dehydrogenase 2 alleviates mitochondrial dysfunction by promoting PGC-1α-mediated biogenesis in acute kidney injury. Cell Death Dis. 2023, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Peng, H.; Wang, S.; Xu, X.; Xu, F.; Wang, Q.; Chen, Y.; Barton, L.A.; Chen, Y.; Zhang, Y.; et al. Mitochondrial ALDH2 protects against lipopolysaccharide-induced myocardial contractile dysfunction by suppression of ER stress and autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1627–1641. [Google Scholar] [CrossRef]
- Lin, D.; Xiang, T.; Qiu, Q.; Leung, J.; Xu, J.; Zhou, W.; Hu, Q.; Lan, J.; Liu, Z.; Zhong, Z.; et al. Aldehyde dehydrogenase 2 regulates autophagy via the Akt-mTOR pathway to mitigate renal ischemia-reperfusion injury in hypothermic machine perfusion. Life Sci. 2020, 253, 117705. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, C.; Gao, L.; Du, G.; Qin, X. Astragaloside IV derived from Astragalus membranaceus: A research review on the pharmacological effects. Adv. Pharmacol. 2020, 87, 89–112. [Google Scholar] [CrossRef]
- Xu, W.; Shao, X.; Tian, L.; Gu, L.; Zhang, M.; Wang, Q.; Wu, B.; Wang, L.; Yao, J.; Xu, X.; et al. Astragaloside IV ameliorates renal fibrosis via the inhibition of mitogen-activated protein kinases and antiapoptosis in vivo and in vitro. J. Pharmacol. Exp. Ther. 2014, 350, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Sun, X.; Gong, X.; Yang, Y.; Chen, C.; Shan, G.; Yao, Q. Astragaloside IV from Astragalus membranaceus ameliorates renal interstitial fibrosis by inhibiting inflammation via TLR4/NF-kB in vivo and in vitro. Int. Immunopharmacol. 2017, 42, 18–24. [Google Scholar] [CrossRef]
- Jin, D.; Lin, L.; Xie, Y.; Jia, M.; Qiu, H.; Xun, K. NRF2-suppressed vascular calcification by regulating the antioxidant pathway in chronic kidney disease. FASEB J. 2022, 36, e22098. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Peng, R.; Ni, Y.; Chen, L.; He, Q.; Li, Q.; Zhou, Q. Tetratricopeptide repeat domain 36 deficiency mitigates renal tubular injury by inhibiting TGF-β1-induced epithelial-mesenchymal transition in a mouse model of chronic kidney disease. Genes Dis. 2022, 9, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Takiyama, Y.; Harumi, T.; Watanabe, J.; Fujita, Y.; Honjo, J.; Shimizu, N.; Makino, Y.; Haneda, M. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: A possible role of HIF-1α expression and oxygen metabolism. Diabetes 2011, 60, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Huang, Z.; Peng, Y.; Li, H.; Lin, T.; Zhao, Y.; Hu, Z.; Zhou, Z.; Zhou, W.; Liu, Y.; et al. Circular RNA circBNC2 inhibits epithelial cell G2-M arrest to prevent fibrotic maladaptive repair. Nat. Commun. 2022, 13, 6502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Lu, Y.; Yang, Y.; Chen, W.; Shen, B.; Hu, J.; Jia, P.; Xu, S.; Shi, Y.; et al. TWIK-related acid-sensitive K(+) channel 2 promotes renal fibrosis by inducing cell-cycle arrest. iScience 2022, 25, 105620. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Sherman, J.; Wang, R. Rapid profiling of G2 phase to mitosis progression by flow cytometry in asynchronous cells. Cell Cycle 2020, 19, 2897–2905. [Google Scholar] [CrossRef]
- Yu, C.C.; Woods, A.L.; Levison, D.A. The assessment of cellular proliferation by immunohistochemistry: A review of currently available methods and their applications. Histochem. J. 1992, 24, 121–131. [Google Scholar] [CrossRef]
- Crosio, C.; Fimia, G.M.; Loury, R.; Kimura, M.; Okano, Y.; Zhou, H.; Sen, S.; Allis, C.D.; Sassone-Corsi, P. Mitotic phosphorylation of histone H3: Spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 2002, 22, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Lemos, D.R.; McMurdo, M.; Karaca, G.; Wilflingseder, J.; Leaf, I.A.; Gupta, N.; Miyoshi, T.; Susa, K.; Johnson, B.G.; Soliman, K.; et al. Interleukin-1β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wan, T.; Zhang, F.; Zhu, X.; Guo, S.; Mei, X. Aldehyde Dehydrogenase 2 Protects Against Lipopolysaccharide-Induced Myocardial Injury by Suppressing Mitophagy. Front. Pharmacol. 2021, 12, 641058. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Li, J.; Leung, C.K.; Shen, B.; Wang, C.; Xu, Y.; Lin, S.; Zhang, S.; Tan, Y.; Zhang, H.; et al. Methamphetamine and HIV-1 Tat proteins synergistically induce microglial autophagy via activation of the Nrf2/NQO1/HO-1 signal pathway. Neuropharmacology 2022, 220, 109256. [Google Scholar] [CrossRef]
- Wei, H.; Chen, L.; Li, Q.; Liang, X.; Wang, K.; Zhang, Y.; Li, Y.; Liu, Y.; Xu, G. CD137L-macrophage induce lymphatic endothelial cells autophagy to promote lymphangiogenesis in renal fibrosis. Int. J. Biol. Sci. 2022, 18, 1171–1187. [Google Scholar] [CrossRef]
- Sun, Y.; Dai, S.; Tao, J.; Li, Y.; He, Z.; Liu, Q.; Zhao, J.; Deng, Y.; Kang, J.; Zhang, X.; et al. Taurine suppresses ROS-dependent autophagy via activating Akt/mTOR signaling pathway in calcium oxalate crystals-induced renal tubular epithelial cell injury. Aging 2020, 12, 17353–17366. [Google Scholar] [CrossRef]
- Chen, L.; Sha, M.L.; Chen, F.T.; Jiang, C.Y.; Li, D.; Xu, C.L.; Pan, D.S.; Xu, Z.J.; Tang, Q.L.; Xia, S.J.; et al. Upregulation of KLF14 expression attenuates kidney fibrosis by inducing PPARα-mediated fatty acid oxidation. Free. Radic. Biol. Med. 2022, 195, 132–144. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Fox, C.H.; Vassalotti, J.; Choi, M. Complications of progression of CKD. Adv. Chronic Kidney Dis. 2011, 18, 400–405. [Google Scholar] [CrossRef]
- Koppe, L.; Fouque, D. The Role for Protein Restriction in Addition to Renin-Angiotensin-Aldosterone System Inhibitors in the Management of CKD. Am. J. Kidney Dis. 2019, 73, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.V.; Patel, Z.M. Role of low protein diet in management of different stages of chronic kidney disease—Practical aspects. BMC Nephrol. 2016, 17, 156. [Google Scholar] [CrossRef] [Green Version]
- Cupisti, A.; Brunori, G.; Di Iorio, B.R.; D’Alessandro, C.; Pasticci, F.; Cosola, C.; Bellizzi, V.; Bolasco, P.; Capitanini, A.; Fantuzzi, A.L.; et al. Nutritional treatment of advanced CKD: Twenty consensus statements. J. Nephrol. 2018, 31, 457–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Menon, M.C.; Deng, Y.; Chen, Y.; He, J.C. Recent Advances in Traditional Chinese Medicine for Kidney Disease. Am. J. Kidney Dis. 2015, 66, 513–522. [Google Scholar] [CrossRef]
- Wang, Y.J.; He, L.Q.; Sun, W.; Lu, Y.; Wang, X.Q.; Zhang, P.Q.; Wei, L.B.; Cao, S.L.; Yang, N.Z.; Ma, H.Z.; et al. Optimized project of traditional Chinese medicine in treating chronic kidney disease stage 3: A multicenter double-blinded randomized controlled trial. J. Ethnopharmacol. 2012, 139, 757–764. [Google Scholar] [CrossRef]
- Gong, F.; Qu, R.; Li, Y.; Lv, Y.; Dai, J. Astragalus Mongholicus: A review of its anti-fibrosis properties. Front. Pharmacol. 2022, 13, 976561. [Google Scholar] [CrossRef]
- Lu, M.; Wang, H.; Wang, J.; Zhang, J.; Yang, J.; Liang, L.; Maslov, L.N. Astragaloside IV protects against cardiac hypertrophy via inhibiting the Ca2+/CaN signaling pathway. Planta Med. 2014, 80, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Han, J.; Chu, Z.G.; Song, H.P.; Zhang, D.X.; Zhang, Q.; Huang, Y.S. Astragaloside IV attenuates hypoxia-induced cardiomyocyte damage in rats by upregulating superoxide dismutase-1 levels. Clin. Exp. Pharmacol. Physiol. 2009, 36, 351–357. [Google Scholar] [CrossRef]
- Liu, G.; Song, J.; Guo, Y.; Wang, T.; Zhou, Z. Astragalus injection protects cerebral ischemic injury by inhibiting neuronal apoptosis and the expression of JNK3 after cerebral ischemia reperfusion in rats. Behav. Brain Funct. 2013, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.N.; Sun, L.F.; Yang, H. Inhibitory Effects of Astragaloside IV on Bleomycin-Induced Pulmonary Fibrosis in Rats Via Attenuation of Oxidative Stress and Inflammation. Inflammation 2016, 39, 1835–1841. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2020, 82, 297–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, O.; Tharaux, P.L.; Huber, T.B. Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int. 2016, 90, 950–964. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhou, R.; Ma, Z. Autophagy and energy metabolism. In Autophagy: Biology and Diseases; Springer: Singapore, 2019; pp. 329–357. [Google Scholar]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, Y.M.; Bima, A.I.; Elsamanoudy, A.Z. An overview of the perspective of cellular autophagy: Mechanism, regulation, and the role of autophagy dysregulation in the pathogenesis of diseases. J. Microsc. Ultrastruct. 2021, 9, 47. [Google Scholar] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Shi, Y.; Zhuang, S. Autophagy in Chronic Kidney Diseases. Kidney Dis. 2016, 2, 37–45. [Google Scholar] [CrossRef]
- Li, L.; Wang, Z.V.; Hill, J.A.; Lin, F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J. Am. Soc. Nephrol. 2014, 25, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Hu, R.; Wang, Y.; Liu, L.; You, H.; Zhang, J.; Wu, X.; Pei, T.; Wang, F.; Lu, L. Atractylenolide III attenuates muscle wasting in chronic kidney disease via the oxidative stress-mediated PI3K/AKT/mTOR pathway. Oxidative Med. Cell. Longev. 2019, 2019, 1875471. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Zuo, Z.; Yang, Z.; Guo, H.; Fang, J.; Cui, H.; Ouyang, P.; Chen, X.; Chen, J.; Geng, Y.; et al. Nickel induces autophagy via PI3K/AKT/mTOR and AMPK pathways in mouse kidney. Ecotoxicol. Environ. Saf. 2021, 223, 112583. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Q.; Link, P.A.; Meridew, J.A.; Pham, T.X.; Caporarello, N.; Ligresti, G.; Tschumperlin, D.J. Spontaneous Lung Fibrosis Resolution Reveals Novel Antifibrotic Regulators. Am. J. Respir. Cell Mol. Biol. 2021, 64, 453–464. [Google Scholar] [CrossRef]
- Seo, W.; Gao, Y.; He, Y.; Sun, J.; Xu, H.; Feng, D.; Park, S.H.; Cho, Y.E.; Guillot, A.; Ren, T.; et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J. Hepatol. 2019, 71, 1000–1011. [Google Scholar] [CrossRef]
- Yuan, Q.; Yang, F.; Dai, S.; Wang, Z.; Xu, Y.; Xu, B.C.; Sun, Y.; Zheng, B.; Zhao, Y.; Wang, W.; et al. Aldehyde dehydrogenase 2 protects against sympathetic excitation-induced cardiac fibrosis. Biochem. Biophys. Res. Commun. 2020, 533, 1427–1434. [Google Scholar] [CrossRef]
- Wimborne, H.J.; Takemoto, K.; Woster, P.M.; Rockey, D.C.; Lemasters, J.J.; Zhong, Z. Aldehyde dehydrogenase-2 activation by Alda-1 decreases necrosis and fibrosis after bile duct ligation in mice. Free. Radic. Biol. Med. 2019, 145, 136–145. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Chiong, M.; Lavandero, S.; Klionsky, D.J.; Ren, J. Mitophagy in cardiovascular diseases: Molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med. 2022, 28, 836–849. [Google Scholar] [CrossRef]
- Li, W.; Yin, L.; Sun, X.; Wu, J.; Dong, Z.; Hu, K.; Sun, A.; Ge, J. Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis. 2020, 11, 599. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Qin, X.; Turdi, S.; Sun, D.; Culver, B.; Reiter, R.J.; Wang, X.; Zhou, H.; Ren, J. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduct. Target. Ther. 2020, 5, 119. [Google Scholar] [CrossRef]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ren, J. Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: Slayer or savior? Autophagy 2010, 6, 1212–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Sun, A.; Xie, Y.; Isse, T.; Kawamoto, T.; Zou, Y.; Ge, J. Aldehyde dehydrogenase-2 deficiency aggravates cardiac dysfunction elicited by endoplasmic reticulum stress induction. Mol. Med. 2012, 18, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Diwan, V.; Brown, L.; Gobe, G.C. Adenine-induced chronic kidney disease in rats. Nephrology 2018, 23, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Liu, Y.; Zhan, Q.; Zeng, Y.; Peng, Z.; He, Q.; Tan, Q.; Cao, W.; Wang, S.; Wang, J. Astragaloside IV Blunts Epithelial–Mesenchymal Transition and G2/M Arrest to Alleviate Renal Fibrosis via Regulating ALDH2-Mediated Autophagy. Cells 2023, 12, 1777. https://doi.org/10.3390/cells12131777
Li D, Liu Y, Zhan Q, Zeng Y, Peng Z, He Q, Tan Q, Cao W, Wang S, Wang J. Astragaloside IV Blunts Epithelial–Mesenchymal Transition and G2/M Arrest to Alleviate Renal Fibrosis via Regulating ALDH2-Mediated Autophagy. Cells. 2023; 12(13):1777. https://doi.org/10.3390/cells12131777
Chicago/Turabian StyleLi, Dong, Yuzhe Liu, Quancao Zhan, Yan Zeng, Ze Peng, Qifeng He, Qi Tan, Wenfu Cao, Shang Wang, and Jianwei Wang. 2023. "Astragaloside IV Blunts Epithelial–Mesenchymal Transition and G2/M Arrest to Alleviate Renal Fibrosis via Regulating ALDH2-Mediated Autophagy" Cells 12, no. 13: 1777. https://doi.org/10.3390/cells12131777