
Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells
 , , , and
, , , and
Abstract
:
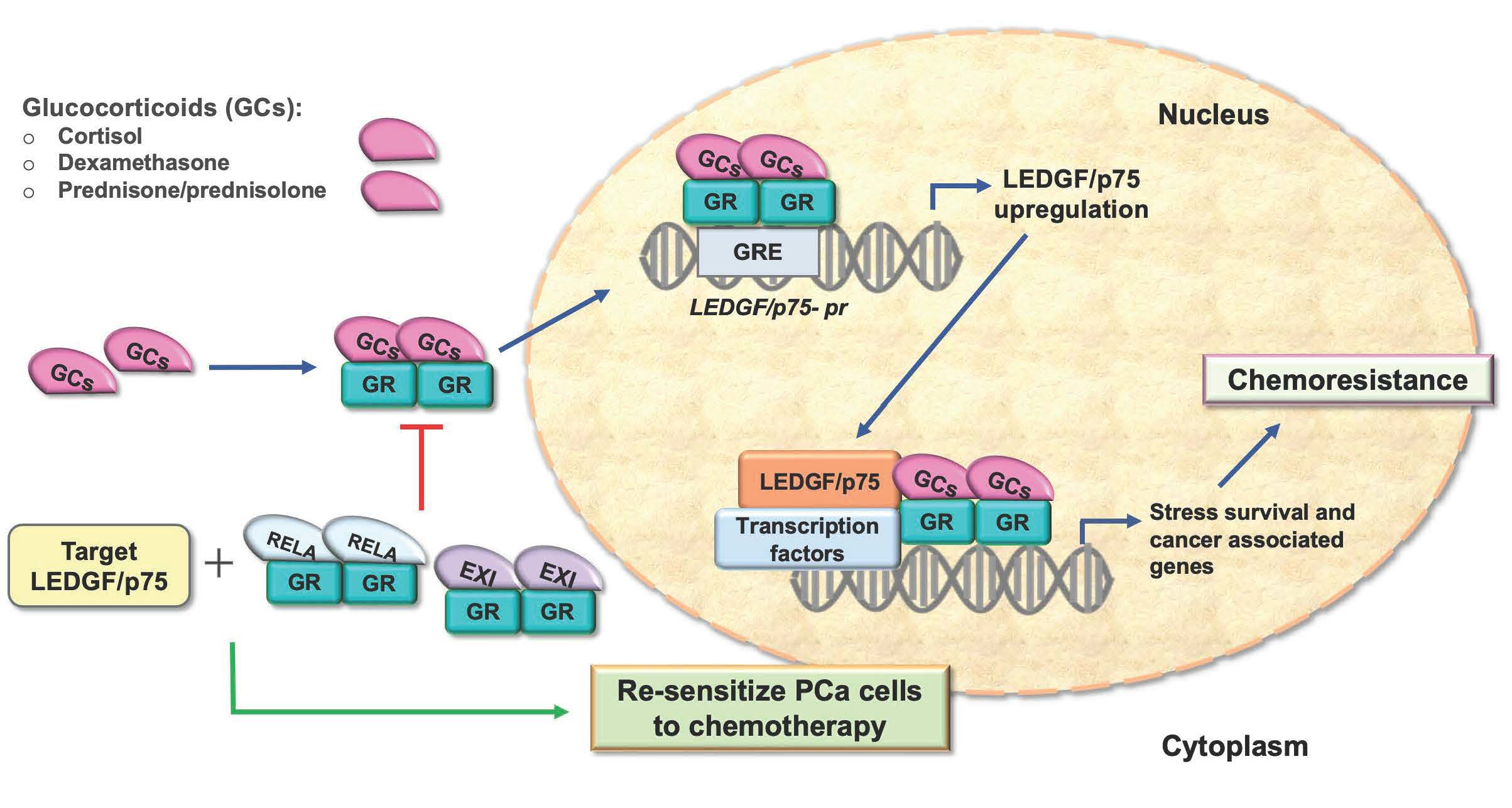{kind=link}
{kind=link}
{kind=link}
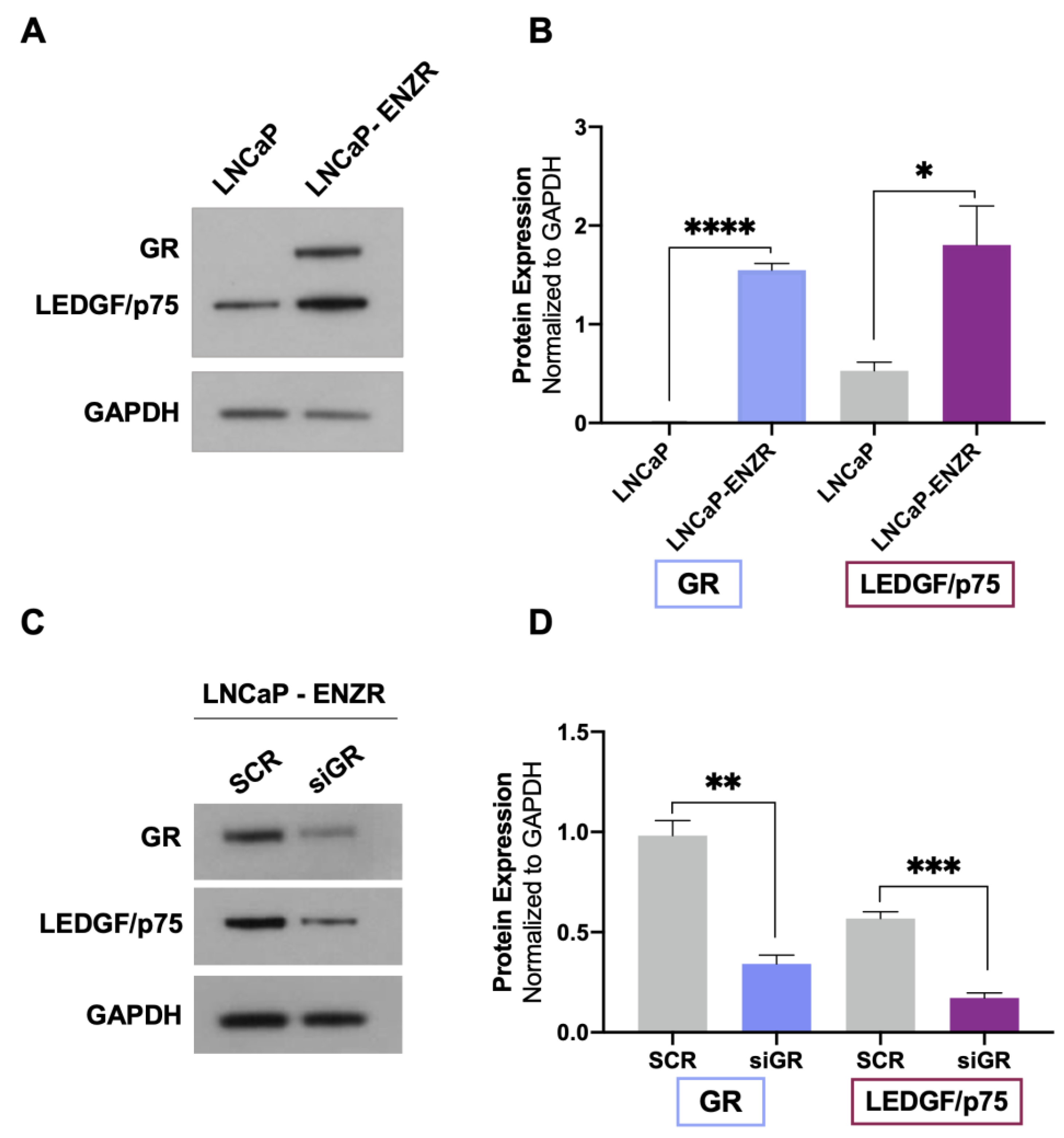{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
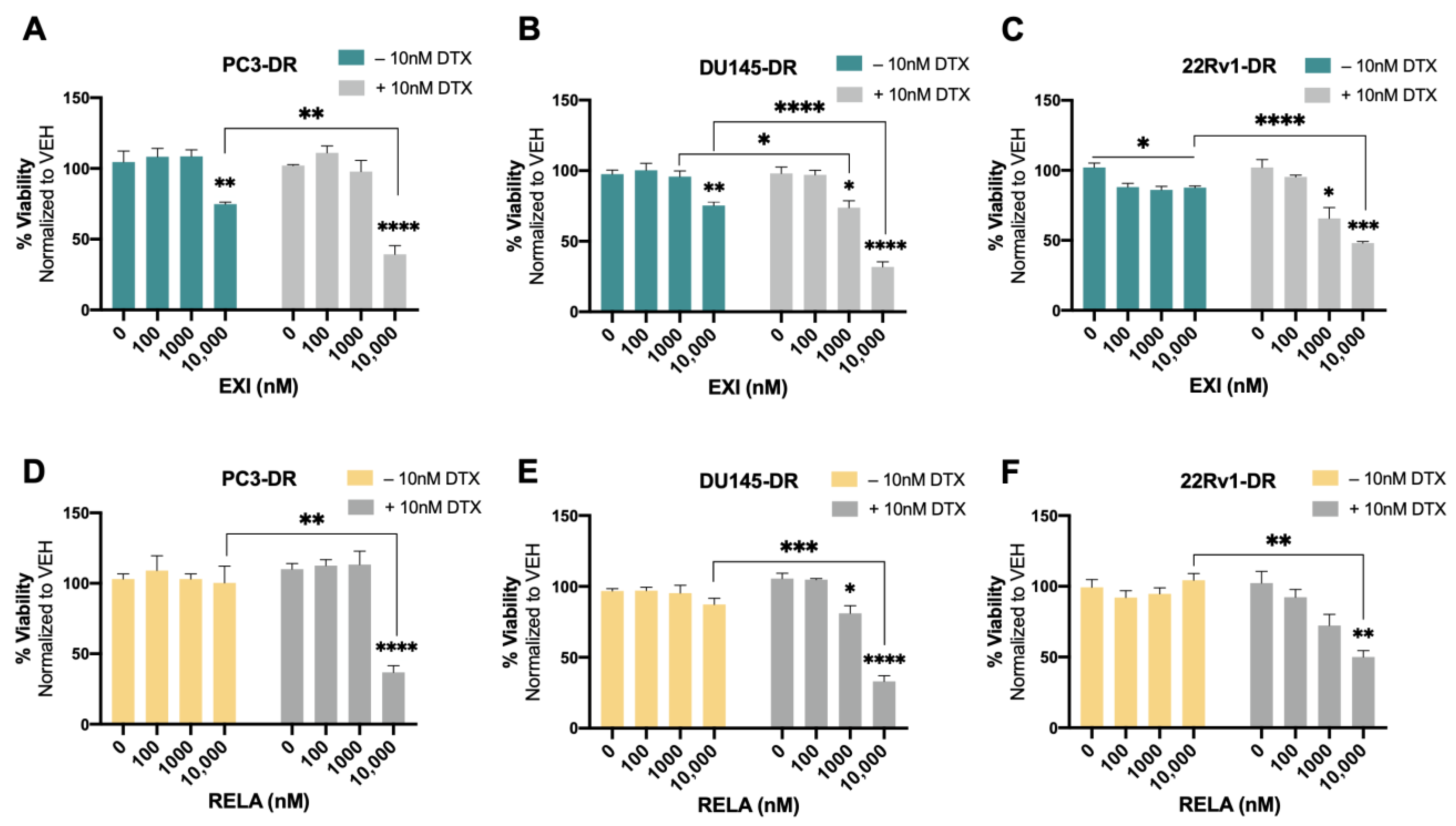{kind=link}
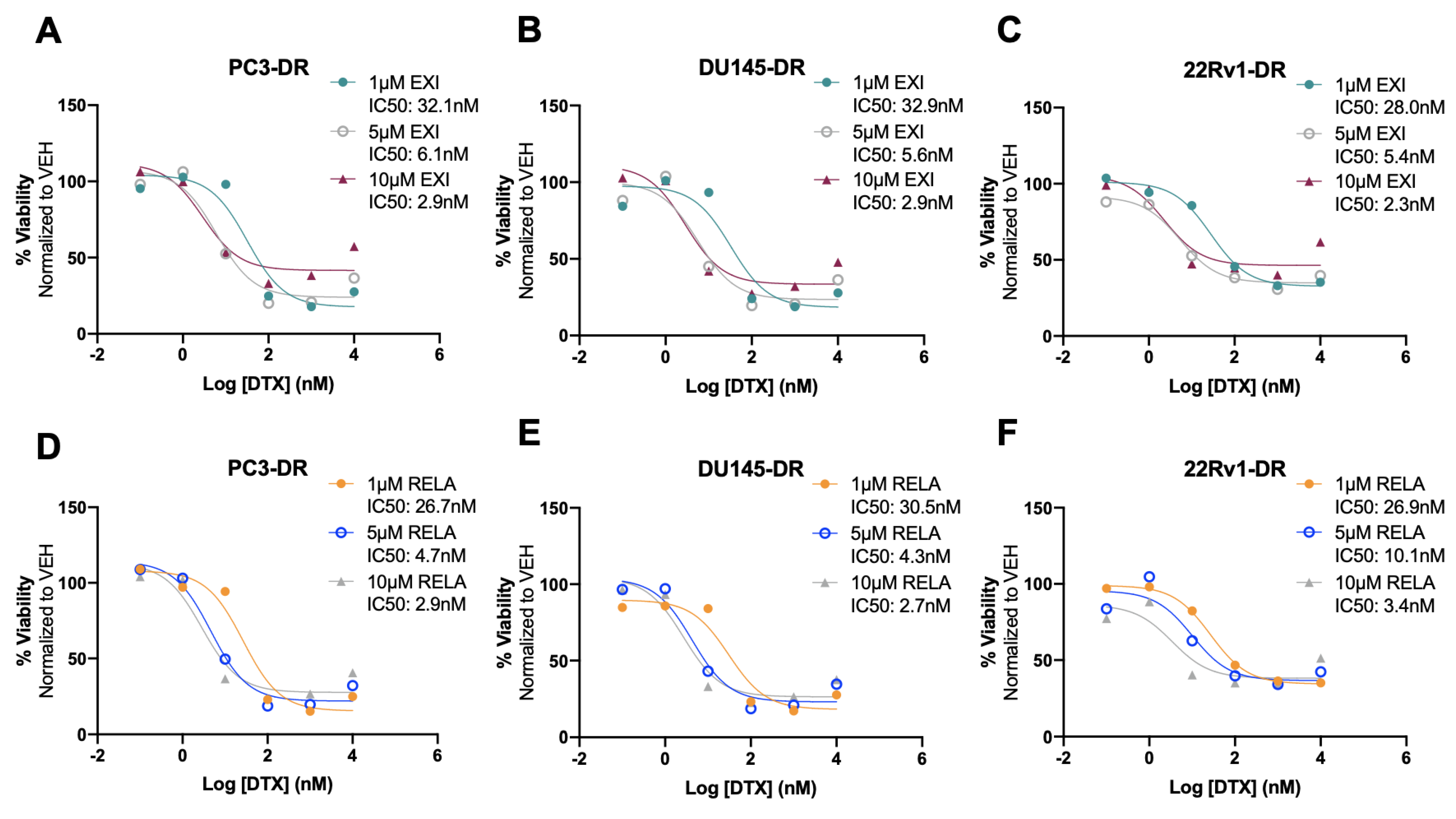{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Small Interfering RNA (siRNA)-Mediated Knockdown
2.3. Quantitative Reverse Transcription PCR (RT-qPCR)
2.4. Immunoblotting
2.5. Immunoprecipitation
2.6. Confocal Microscopy
2.7. Cell Viability and Apoptosis Assays
2.8. Clonogenic Assays
2.9. ChIP-Sequencing
2.10. RNA-Sequencing
2.11. Bioinformatics
2.12. Statistics
3. Results
3.1. GR Depletion Leads to Decreased LEDGF/p75 Protein Expression in Prostate Cancer Cells
3.2. GR and LEDGF/p75 Are Upregulated in Enzalutamide-Resistant LNCaP Cells
3.3. ChIP-Seq Analysis Reveals GR Binding Sites in the Promoter Region of LEDGF/p75
3.4. LEDGF/p75 and GR Interact in DTX-Resistant PCa Cells
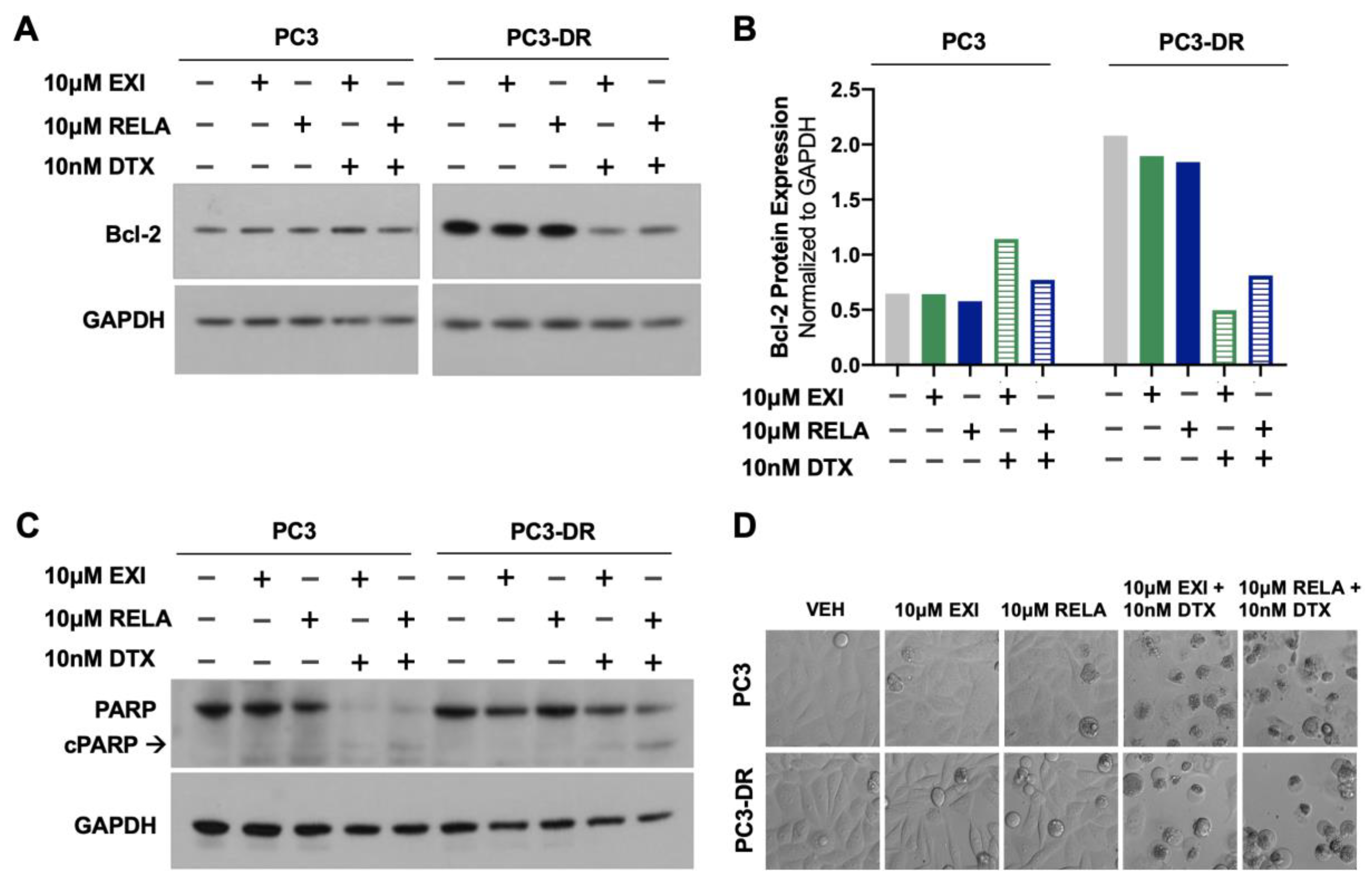
3.5. Selective GR Modulators Resensitize DTX-Resistant Cells to DTX
3.6. Targeting of GR and LEDGF/p75 Decreases Clonogenicity in PC3-DR Cells in the Presence of Docetaxel
3.7. RNA-Seq Analysis of Genes Differentially Regulated after GR or LEDGF/p75 Knockdown in DTX-Resistant PCa Cells Reveals Unique and Overlapping Transcriptomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Damodaran, S.; Kyriakopoulos, C.E.; Jarrard, D.F. Newly Diagnosed Metastatic Prostate Cancer: Has the Paradigm Changed? Urol. Clin. N. Am. 2017, 44, 611–621. [Google Scholar] [CrossRef]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar]
- Ehsani, M.; David, F.O.; Baniahmad, A. Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer. Cancers 2021, 13, 1534. [Google Scholar] [CrossRef]
- Chen, K.; O’Brien, J.; McVey, A.; Jenjitranant, P.; Kelly, B.D.; Kasivisvanathan, V.; Lawrentschuk, N.; Murphy, D.G.; Azad, A.A. Combination treatment in metastatic prostate cancer: Is the bar too high or have we fallen short? Nat. Rev. Urol. 2023, 20, 116–123. [Google Scholar]
- Chang, C.Y.; Walther, P.J.; McDonnell, D.P. Glucocorticoids manifest androgenic activity in a cell line derived from a metastatic prostate cancer. Cancer Res. 2001, 61, 8712–8717. [Google Scholar]
- Richards, J.; Lim, A.C.; Hay, C.W.; Taylor, A.E.; Wingate, A.; Nowakowska, K.; Pezaro, C.; Carreira, S.; Goodall, J.; Arlt, W.; et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: A rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012, 72, 2176–2182. [Google Scholar]
- Zhao, X.Y.; Malloy, P.J.; Krishnan, A.V.; Swami, S.; Navone, N.M.; Peehl, D.M.; Feldman, D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat. Med. 2000, 6, 703–706. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, J.; Liu, Y.; Shi, Z.; Xuan, Z.; Yang, K.; Xu, C.; Bai, Y.; Fu, M.; Xiao, Q.; et al. The Crucial Role of AR-V7 in Enzalutamide-Resistance of Castration-Resistant Prostate Cancer. Cancers 2022, 14, 4877. [Google Scholar] [CrossRef]
- Shim, M.; Kim, Y.; Park, Y.; Ahn, H. Taxane-based Chemotherapy Induced Androgen Receptor Splice Variant 7 in Patients with Castration-Resistant Prostate Cancer: A Tissue-based Analysis. Sci. Rep. 2019, 9, 16794. [Google Scholar]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [Green Version]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.E.; et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife 2017, 6, e20183. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Wang, P.; Wongvipat, J.; Karthaus, W.R.; Abida, W.; Armenia, J.; Rockowitz, S.; Drier, Y.; Bernstein, B.E.; Long, H.W.; et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife 2017, 6, e27861. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Gryder, B.E.; Nguyen, Y.T.M.; Alilin, A.N.; Grayson, A.R.; Bajwa, W.; Jansson, K.H.; Beshiri, M.L.; Agarwal, S.; Rodriguez-Nieves, J.A.; et al. Targeting the PI3K/AKT Pathway Overcomes Enzalutamide Resistance by Inhibiting Induction of the Glucocorticoid Receptor. Mol. Cancer Ther. 2020, 19, 1436–1447. [Google Scholar] [CrossRef]
- Smith, R.; Liu, M.; Liby, T.; Bayani, N.; Bucher, E.; Chiotti, K.; Derrick, D.; Chauchereau, A.; Heiser, L.; Alumkal, J.; et al. Enzalutamide response in a panel of prostate cancer cell lines reveals a role for glucocorticoid receptor in enzalutamide resistant disease. Sci. Rep. 2020, 10, 21750. [Google Scholar] [CrossRef]
- Puhr, M.; Eigentler, A.; Handle, F.; Hackl, H.; Ploner, C.; Heidegger, I.; Schaefer, G.; Brandt, M.P.; Hoefer, J.; Van der Pluijm, G.; et al. Targeting the glucocorticoid receptor signature gene Mono Amine Oxidase-A enhances the efficacy of chemo- and anti-androgen therapy in advanced prostate cancer. Oncogene 2021, 40, 3087–3100. [Google Scholar] [CrossRef]
- Moll, J.M.; Hofland, J.; Teubel, W.J.; de Ridder, C.M.A.; Taylor, A.E.; Graeser, R.; Arlt, W.; Jenster, G.W.; van Weerden, W.M. Abiraterone switches castration-resistant prostate cancer dependency from adrenal androgens towards androgen receptor variants and glucocorticoid receptor signalling. Prostate 2022, 82, 505–516. [Google Scholar] [CrossRef]
- Sakellakis, M.; Flores, L.J. Is the glucocorticoid receptor a key player in prostate cancer?: A literature review. Medicine 2022, 101, e29716. [Google Scholar]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Woods-Burnham, L.; Cajigas-Du Ross, C.K.; Love, A.; Basu, A.; Sanchez-Hernandez, E.S.; Martinez, S.R.; Ortiz-Hernández, G.L.; Stiel, L.; Durán, A.M.; Wilson, C.; et al. Glucocorticoids Induce Stress Oncoproteins Associated with Therapy-Resistance in African American and European American Prostate Cancer Cells. Sci. Rep. 2018, 8, 15063. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.R.; Elix, C.C.; Ochoa, P.T.; Sanchez-Hernandez, E.S.; Alkashgari, H.R.; Ortiz-Hernandez, G.L.; Zhang, L.; Casiano, C.A. Glucocorticoid Receptor and beta-Catenin Interact in Prostate Cancer Cells and Their Co-Inhibition Attenuates Tumorsphere Formation, Stemness, and Docetaxel Resistance. Int. J. Mol. Sci. 2023, 24, 7130. [Google Scholar] [CrossRef]
- Buck, S.A.J.; Koolen, S.L.W.; Mathijssen, R.H.J.; de Wit, R.; van Soest, R.J. Cross-resistance and drug sequence in prostate cancer. Drug Resist. Updat. 2021, 56, 100761. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B.; et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De Leon, M.; Casiano, C.A. Expression of the stress response oncoprotein LEDGF/p75 in human cancer: A study of 21 tumor types. PLoS ONE 2012, 7, e30132. [Google Scholar]
- Rios-Colon, L.; Cajigas-Du Ross, C.K.; Basu, A.; Elix, C.; Alicea-Polanco, I.; Sanchez, T.W.; Radhakrishnan, V.; Chen, C.S.; Casiano, C.A. Targeting the stress oncoprotein LEDGF/p75 to sensitize chemoresistant prostate cancer cells to taxanes. Oncotarget 2017, 8, 24915–24931. [Google Scholar] [CrossRef] [Green Version]
- Daugaard, M.; Kirkegaard-Sorensen, T.; Ostenfeld, M.S.; Aaboe, M.; Hoyer-Hansen, M.; Orntoft, T.F.; Rohde, M.; Jaattela, M. Lens epithelium-derived growth factor is an Hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, O.; Doskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Sapoznik, S.; Cohen, B.; Tzuman, Y.; Meir, G.; Ben-Dor, S.; Harmelin, A.; Neeman, M. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009, 69, 9306–9314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargavan, B.; Fatma, N.; Chhunchha, B.; Singh, V.; Kubo, E.; Singh, D.P. LEDGF gene silencing impairs the tumorigenicity of prostate cancer DU145 cells by abating the expression of Hsp27 and activation of the Akt/ERK signaling pathway. Cell Death Dis. 2012, 3, e316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammer, P.; Groth-Pedersen, L.; Kirkegaard, T.; Daugaard, M.; Rytter, A.; Szyniarowski, P.; Høyer-Hansen, M.; Povlsen, L.K.; Nylandsted, J.; Larsen, J.E.; et al. BAMLET activates a lysosomal cell death program in cancer cells. Mol Cancer Ther. 2010, 9, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Leitz, J.; Reuschenbach, M.; Lohrey, C.; Honegger, A.; Accardi, R.; Tommasino, M.; Llano, M.; von Knebel Doeberitz, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Oncogenic human papillomaviruses activate the tumor-associated lens epithelial-derived growth factor (LEDGF) gene. PLoS Pathog. 2014, 10, e1003957. [Google Scholar] [CrossRef]
- Liedtke, V.; Schroder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack, P.; Rodiger, S.; Schenk, L. LEDGF/p75 Is Required for an Efficient DNA Damage Response. Int. J. Mol. Sci. 2021, 22, 5866. [Google Scholar] [CrossRef]
- Canella, A.; Van Belle, S.; Brouns, T.; Nigita, G.; Carlon, M.S.; Christ, F.; Debyser, Z. LEDGF/p75-mediated chemoresistance of mixed-lineage leukemia involves cell survival pathways and super enhancer activators. Cancer Gene Ther. 2022, 29, 133–140. [Google Scholar]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Ochoa, P.T.; Elix, C.C.; Alkashgari, H.R.; McMullen, J.R.W.; Soto, U.; Martinez, S.R.; Diaz Osterman, C.J.; Mahler, M.; et al. The LEDGF/p75 Integrase Binding Domain Interactome Contributes to the Survival, Clonogenicity, and Tumorsphere Formation of Docetaxel-Resistant Prostate Cancer Cells. Cells 2021, 10, 2723. [Google Scholar] [CrossRef]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Ling Li, X.; Lin, Y.C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/p75 promotes tumorigenicity in breast cancer cells by promoting the transcription of cell cycle genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Singh, D.P.; Fatma, N.; Chylack, L.T., Jr.; Shinohara, T. Activation of LEDGF gene by thermal-and oxidative-stresses. Biochem. Biophys. Res. Commun. 2000, 276, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T., Jr.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar]
- Matsui, H.; Lin, L.R.; Singh, D.P.; Shinohara, T.; Reddy, V.N. Lens epithelium-derived growth factor: Increased survival and decreased DNA breakage of human RPE cells induced by oxidative stress. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2935–2941. [Google Scholar]
- Wu, X.; Daniels, T.; Molinaro, C.; Lilly, M.B.; Casiano, C.A. Caspase cleavage of the nuclear autoantigen LEDGF/p75 abrogates its pro-survival function: Implications for autoimmunity in atopic disorders. Cell Death Differ. 2002, 9, 915–925. [Google Scholar] [PubMed] [Green Version]
- Singh, D.P.; Fatma, N.; Kimura, A.; Chylack, L.T., Jr.; Shinohara, T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem. Biophys. Res. Commun. 2001, 283, 943–955. [Google Scholar] [CrossRef]
- Basu, A.; Drame, A.; Munoz, R.; Gijsbers, R.; Debyser, Z.; De Leon, M.; Casiano, C.A. Pathway specific gene expression profiling reveals oxidative stress genes potentially regulated by transcription co-activator LEDGF/p75 in prostate cancer cells. Prostate 2012, 72, 597–611. [Google Scholar] [PubMed] [Green Version]
- Basu, A.; Cajigas-Du Ross, C.K.; Rios-Colon, L.; Mediavilla-Varela, M.; Daniels-Wells, T.R.; Leoh, L.S.; Rojas, H.; Banerjee, H.; Martinez, S.R.; Acevedo-Martinez, S.; et al. LEDGF/p75 Overexpression Attenuates Oxidative Stress-Induced Necrosis and Upregulates the Oxidoreductase ERP57/PDIA3/GRP58 in Prostate Cancer. PLoS ONE 2016, 11, e0146549. [Google Scholar]
- Singh, D.P.; Ohguro, N.; Chylack, L.T., Jr.; Shinohara, T. Lens epithelium-derived growth factor: Increased resistance to thermal and oxidative stresses. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1444–1451. [Google Scholar]
- Van Belle, S.; El Ashkar, S.; Cermakova, K.; Matthijssens, F.; Goossens, S.; Canella, A.; Hodges, C.H.; Christ, F.; De Rijck, J.; Van Vlierberghe, P.; et al. Unlike Its Paralog LEDGF/p75, HRP-2 Is Dispensable for MLL-R Leukemogenesis but Important for Leukemic Cell Survival. Cells 2021, 10, 192. [Google Scholar]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.A.; Kara, H.O.; Yu, J.R.; Lee, C.H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein-protein and protein-chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Cermakova, K.; Horejsi, M.; Prochazkova, K.; Fabry, M.; Sharma, S.; Christ, F.; Demeulemeester, J.; Debyser, Z.; Rijck, J.; et al. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015, 6, 7968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar]
- Oki, S.; Ohta, T. ChIP Atlas. Available online: https://chip-atlas.org/peak_browser (accessed on 10 May 2023).
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [PubMed] [Green Version]
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.P.; Lundin, M.; Konsti, J.; et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011, 30, 3962–3976. [Google Scholar] [PubMed]
- Bergeron, B.P.; Diedrich, J.D.; Zhang, Y.; Barnett, K.R.; Dong, Q.; Ferguson, D.C.; Autry, R.J.; Yang, W.; Hansen, B.S.; Smith, C.; et al. Epigenomic profiling of glucocorticoid responses identifies cis-regulatory disruptions impacting steroid resistance in childhood acute lymphoblastic leukemia. Leukemia 2022, 36, 2374–2383. [Google Scholar]
- NIH SRA Toolkit. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud/ (accessed on 15 May 2023).
- Andrews, S. FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 May 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raney, B.J.; Dreszer, T.R.; Barber, G.P.; Clawson, H.; Fujita, P.A.; Wang, T.; Nguyen, N.; Paten, B.; Zweig, A.S.; Karolchik, D.; et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30, 1003–1005. [Google Scholar]
- Nassar, L.R.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [PubMed]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar]
- Cajigas-Du Ross, C.K.; Martinez, S.R.; Woods-Burnham, L.; Duran, A.M.; Roy, S.; Basu, A.; Ramirez, J.A.; Ortiz-Hernandez, G.L.; Rios-Colon, L.; Chirshev, E.; et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget 2018, 9, 30363–30384. [Google Scholar] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [PubMed] [Green Version]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar]
- Dietz, F.; Franken, S.; Yoshida, K.; Nakamura, H.; Kappler, J.; Gieselmann, V. The family of hepatoma-derived growth factor proteins: Characterization of a new member HRP-4 and classification of its subfamilies. Biochem. J. 2002, 366 Pt 2, 491–500. [Google Scholar]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [PubMed] [Green Version]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [PubMed] [Green Version]
- Grand, F.H.; Koduru, P.; Cross, N.C.; Allen, S.L. NUP98-LEDGF fusion and t(9;11) in transformed chronic myeloid leukemia. Leuk. Res. 2005, 29, 1469–1472. [Google Scholar]
- Gallego Hernanz, M.P.; Torregrosa Diaz, J.M.; Sorel, N.; Bobin, A.; Dindinaud, E.; Bouyer, S.; Desmier, D.; Brizard, F.; Leleu, X.; Maillard, N.; et al. Long-term molecular remission in a patient with acute myeloid leukemia harboring a new NUP98-LEDGF rearrangement. Cancer Med. 2019, 8, 1765–1770. [Google Scholar]
- Wang, H.; Farnung, L.; Dienemann, C.; Cramer, P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2020, 27, 8–13. [Google Scholar] [PubMed] [Green Version]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar]
- Claessens, F.; Joniau, S.; Helsen, C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell. Mol. Life Sci. 2017, 74, 2217–2228. [Google Scholar] [PubMed] [Green Version]
- Munster, P.N.; Greenstein, A.E.; Fleming, G.F.; Borazanci, E.; Sharma, M.R.; Custodio, J.M.; Tudor, I.C.; Pashova, H.I.; Shepherd, S.P.; Grauer, A.; et al. Overcoming Taxane Resistance: Preclinical and Phase 1 Studies of Relacorilant, a Selective Glucocorticoid Receptor Modulator, with Nab-Paclitaxel in Solid Tumors. Clin. Cancer Res. 2022, 28, 3214–3224. [Google Scholar] [PubMed]
- Greenstein, A.E.; Hunt, H.J. Glucocorticoid receptor antagonism promotes apoptosis in solid tumor cells. Oncotarget 2021, 12, 1243–1255. [Google Scholar] [PubMed]
- van Soest, R.J.; van Royen, M.E.; de Morree, E.S.; Moll, J.M.; Teubel, W.; Wiemer, E.A.; Mathijssen, R.H.; de Wit, R.; van Weerden, W.M. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2013, 49, 3821–3830. [Google Scholar]
- van Soest, R.J.; de Wit, R. Irrefutable evidence for the use of docetaxel in newly diagnosed metastatic prostate cancer: Results from the STAMPEDE and CHAARTED trials. BMC Med. 2015, 13, 304. [Google Scholar]
- Shiota, M.; Dejima, T.; Yamamoto, Y.; Takeuchi, A.; Imada, K.; Kashiwagi, E.; Inokuchi, J.; Tatsugami, K.; Kajioka, S.; Uchiumi, T.; et al. Collateral resistance to taxanes in enzalutamide-resistant prostate cancer through aberrant androgen receptor and its variants. Cancer Sci. 2018, 109, 3224–3234. [Google Scholar]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar]
- Onstenk, W.; Sieuwerts, A.M.; Kraan, J.; Van, M.; Nieuweboer, A.J.; Mathijssen, R.H.; Hamberg, P.; Meulenbeld, H.J.; De Laere, B.; Dirix, L.Y.; et al. Efficacy of Cabazitaxel in Castration-resistant Prostate Cancer Is Independent of the Presence of AR-V7 in Circulating Tumor Cells. Eur. Urol. 2015, 68, 939–945. [Google Scholar] [PubMed]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/beta-Catenin Pathway Overcomes Resistance to Enzalutamide in Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar]
- Yeh, Y.; Guo, Q.; Connelly, Z.; Cheng, S.; Yang, S.; Prieto-Dominguez, N.; Yu, X. Wnt/Beta-Catenin Signaling and Prostate Cancer Therapy Resistance. Adv. Exp. Med. Biol. 2019, 1210, 351–378. [Google Scholar]
- Bian, P.; Dou, Z.; Jia, Z.; Li, W.; Pan, D. Activated Wnt/beta-Catenin signaling contributes to E3 ubiquitin ligase EDD-conferred docetaxel resistance in prostate cancer. Life Sci. 2020, 254, 116816. [Google Scholar]
- Isaacsson Velho, P.; Fu, W.; Wang, H.; Mirkheshti, N.; Qazi, F.; Lima, F.A.S.; Shaukat, F.; Carducci, M.A.; Denmeade, S.R.; Paller, C.J.; et al. Wnt-pathway Activating Mutations Are Associated with Resistance to First-line Abiraterone and Enzalutamide in Castration-resistant Prostate Cancer. Eur. Urol. 2020, 77, 14–21. [Google Scholar] [PubMed]
- Wang, C.; Chen, Q.; Xu, H. Wnt/beta-catenin signal transduction pathway in prostate cancer and associated drug resistance. Discov. Oncol. 2021, 12, 40. [Google Scholar] [PubMed]
- Pudova, E.; Kobelyatskaya, A.; Katunina, I.; Snezhkina, A.; Nyushko, K.; Fedorova, M.; Pavlov, V.; Bulavkina, E.; Dalina, A.; Tkachev, S.; et al. Docetaxel Resistance in Castration-Resistant Prostate Cancer: Transcriptomic Determinants and the Effect of Inhibiting Wnt/beta-Catenin Signaling by XAV939. Int. J. Mol. Sci. 2022, 23, 12837. [Google Scholar]
- Palit, S.A.; Vis, D.; Stelloo, S.; Lieftink, C.; Prekovic, S.; Bekers, E.; Hofland, I.; Sustic, T.; Wolters, L.; Beijersbergen, R.; et al. TLE3 loss confers AR inhibitor resistance by facilitating GR-mediated human prostate cancer cell growth. Elife 2019, 8, e47430. [Google Scholar]
- Vandegraaff, N.; Devroe, E.; Turlure, F.; Silver, P.A.; Engelman, A. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology 2006, 346, 415–426. [Google Scholar]
- Abazid, A.; Martin, B.; Choinowski, A.; McNeill, R.V.; Brandenburg, L.O.; Ziegler, P.; Zimmermann, U.; Burchardt, M.; Erb, H.; Stope, M.B. The androgen receptor antagonist enzalutamide induces apoptosis, dysregulates the heat shock protein system, and diminishes the androgen receptor and estrogen receptor beta1 expression in prostate cancer cells. J. Cell. Biochem. 2019, 120, 16711–16722. [Google Scholar]
- Xie, Y.; Wang, L.; Khan, M.A.; Hamburger, A.W.; Guang, W.; Passaniti, A.; Munir, K.; Ross, D.D.; Dean, M.; Hussain, A. Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells. Cancers 2021, 13, 633. [Google Scholar] [PubMed]
- Chang, C.Y.; Chen, J.T.; Chen, T.H.; Chen, R.M. Enzalutamide Induces Apoptotic Insults to Human Drug-Resistant and -Sensitive Glioblastoma Cells via an Intrinsic Bax-Mitochondrion-Cytochrome C Caspase Cascade Activation Pathway. Molecules 2022, 27, 6666. [Google Scholar] [PubMed]
- Alsawalha, L.; Ahram, M.; Abdullah, M.S.; Dalmizrak, O. Enzalutamide Overcomes Dihydrotestosterone-Induced Chemoresistance in Triple- Negative Breast Cancer Cells via Apoptosis. Anticancer Agents Med. Chem. 2022, 22, 3038–3048. [Google Scholar]
- Xiang, Z.; Sun, Y.; You, B.; Zhang, M.; Huang, C.; Yu, J.; You, X.; Wu, D.; Chang, C. Suppressing BCL-XL increased the high dose androgens therapeutic effect to better induce the Enzalutamide-resistant prostate cancer autophagic cell death. Cell Death Dis. 2021, 12, 68. [Google Scholar]
- Pilling, A.B.; Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar]
- Pilling, A.B.; Hwang, O.; Boudreault, A.; Laurent, A.; Hwang, C. IAP Antagonists Enhance Apoptotic Response to Enzalutamide in Castration-Resistant Prostate Cancer Cells via Autocrine TNF-alpha Signaling. Prostate 2017, 77, 866–877. [Google Scholar] [PubMed]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sorensen, C.S.; Petersen, N.H.; Sorensen, P.H.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [PubMed]
- Shinohara, T.; Singh, D.P.; Fatma, N. LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Eye Res. 2002, 21, 341–358. [Google Scholar]
- Leoh, L.S.; van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar]
- Ortiz-Hernandez, G.L.; Sanchez-Hernandez, E.S.; Casiano, C.A. Twenty years of research on the DFS70/LEDGF autoantibody-autoantigen system: Many lessons learned but still many questions. Autoimmun. Highlights 2020, 11, 3. [Google Scholar]
- Debyser, Z.; Bruggemans, A.; Van Belle, S.; Janssens, J.; Christ, F. LEDGINs, Inhibitors of the Interaction Between HIV-1 Integrase and LEDGF/p75, Are Potent Antivirals with a Potential to Cure HIV Infection. Adv. Exp. Med. Biol. 2021, 1322, 97–114. [Google Scholar] [PubMed]
- Singh, P.K.; Li, W.; Bedwell, G.J.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors. Viruses 2022, 14, 1883. [Google Scholar] [CrossRef] [PubMed]
- Ribeirinho-Soares, S.; Padua, D.; Amaral, A.L.; Valentini, E.; Azevedo, D.; Marques, C.; Barros, R.; Macedo, F.; Mesquita, P.; Almeida, R. Prognostic significance of MUC2, CDX2 and SOX2 in stage II colorectal cancer patients. BMC Cancer 2021, 21, 359. [Google Scholar]
- Chou, C.L.; Chen, T.J.; Tian, Y.F.; Chan, T.C.; Yeh, C.F.; Li, W.S.; Tsai, H.H.; Li, C.F.; Lai, H.Y. Upregulated MUC2 Is an Unfavorable Prognostic Indicator for Rectal Cancer Patients Undergoing Preoperative CCRT. J. Clin. Med. 2021, 10, 3030. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Satoh, T.; Iwakawa, M.; Nakawatari, M.; Oki, A.; Matsumoto, K.; Okada, S.; Minaguchi, T.; Yoshikawa, H.; Imai, T. Villin1, a diagnostic marker for endometrial adenocarcinoma with high grade nuclear atypia. Cancer Biol. Ther. 2011, 12, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbazan, J.; Muinelo-Romay, L.; Vieito, M.; Candamio, S.; Diaz-Lopez, A.; Cano, A.; Gomez-Tato, A.; Casares de Cal Mde, L.; Abal, M.; Lopez-Lopez, R. A multimarker panel for circulating tumor cells detection predicts patient outcome and therapy response in metastatic colorectal cancer. Int. J. Cancer 2014, 135, 2633–2643. [Google Scholar] [PubMed]
- Wu, Z.; Lun, P.; Ji, T.; Niu, J.; Sun, X.; Liu, X.; Xu, J. LncRNA SNHG25 Promotes Glioma Progression Through Activating MAPK Signaling. Mol. Neurobiol. 2022, 59, 6993–7005. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, S.; Qi, Y.; Tian, J.; Xu, F. Long noncoding RNA SNHG25 promotes the malignancy of endometrial cancer by sponging microRNA-497-5p and increasing FASN expression. J. Ovarian Res. 2021, 14, 163. [Google Scholar] [CrossRef]
- Zhiyu, Z.; Qi, Z.; Zhen, S.; Jianglei, Z.; Jun, O. Small nucleolar RNA host gene 25 is a long non-coding RNA helps diagnose and predict outcomes in prostate cancer. Cancer Treat. Res. Commun. 2023, 35, 100. [Google Scholar]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Hernandez, E.S.; Ochoa, P.T.; Suzuki, T.; Ortiz-Hernandez, G.L.; Unternaehrer, J.J.; Alkashgari, H.R.; Diaz Osterman, C.J.; Martinez, S.R.; Chen, Z.; Kremsky, I.; et al. Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells. Cells 2023, 12, 2046. https://doi.org/10.3390/cells12162046
Sanchez-Hernandez ES, Ochoa PT, Suzuki T, Ortiz-Hernandez GL, Unternaehrer JJ, Alkashgari HR, Diaz Osterman CJ, Martinez SR, Chen Z, Kremsky I, et al. Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells. Cells. 2023; 12(16):2046. https://doi.org/10.3390/cells12162046
Chicago/Turabian StyleSanchez-Hernandez, Evelyn S., Pedro T. Ochoa, Tise Suzuki, Greisha L. Ortiz-Hernandez, Juli J. Unternaehrer, Hossam R. Alkashgari, Carlos J. Diaz Osterman, Shannalee R. Martinez, Zhong Chen, Isaac Kremsky, and et al. 2023. "Glucocorticoid Receptor Regulates and Interacts with LEDGF/p75 to Promote Docetaxel Resistance in Prostate Cancer Cells" Cells 12, no. 16: 2046. https://doi.org/10.3390/cells12162046