
Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations

1
Department of Pathology, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong
2
State Key Laboratory of Liver Research, The University of Hong Kong, Hong Kong
3
State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao
*
Author to whom correspondence should be addressed.
Cells 2023, 12(17), 2147; https://doi.org/10.3390/cells12172147
Submission received: 14 July 2023
/
Revised: 21 August 2023
/
Accepted: 23 August 2023
/
Published: 25 August 2023
(This article belongs to the Collection Exclusive Papers from Editorial Board Members and Invited Scholars in Cells)
Abstract
:In the emerging era of cancer immunotherapy, immune checkpoint blockades (ICBs) and adoptive cell transfer therapies (ACTs) have gained significant attention. However, their therapeutic efficacies are limited due to the presence of cold type tumors, immunosuppressive tumor microenvironment, and immune-related side effects. On the other hand, dendritic cell (DC)-based vaccines have been suggested as a new cancer immunotherapy regimen that can address the limitations encountered by ICBs and ACTs. Despite the success of the first generation of DC-based vaccines, represented by the first FDA-approved DC-based therapeutic cancer vaccine Provenge, several challenges remain unsolved. Therefore, new DC vaccine strategies have been actively investigated. This review addresses the limitations of the currently most adopted classical DC vaccine and evaluates new generations of DC vaccines in detail, including biomaterial-based, immunogenic cell death-inducing, mRNA-pulsed, DC small extracellular vesicle (sEV)-based, and tumor sEV-based DC vaccines. These innovative DC vaccines are envisioned to provide a significant breakthrough in cancer immunotherapy landscape and are expected to be supported by further preclinical and clinical studies.
1. Introduction
Immunotherapy has rapidly advanced for clinical cancer treatment, highlighting the importance of tumor–immune interactions in tumor regulation [1,2]. Despite recent significant progress in immune checkpoint blockades (ICBs) and adoptive cell therapies (ACTs) across a broad range of solid tumors, limited therapeutic efficacies and severe immune-related adverse events have rendered them from wide applications [3,4]. The highly immunosuppressive tumor microenvironment (TME), in which the numbers and functions of immunostimulatory cells such as effector T cells (TEFF) and antigen-presenting cells (APCs) are downregulated, remains one of the key determinant factors that subverts the therapeutic efficacies of existing cancer immunotherapies [5,6]. Although the cancer immunotherapy landscape is currently dominated by ICBs and ACTs, cancer vaccines that are supported by clear rationale and encouraging preclinical data for further development also appear promising [7,8].
Cancer vaccines carry distinct benefits that address the limitations of currently popular ICBs and ACTs. They can target an additional broader set of intracellular antigens, unlike ACTs that target distinguished tumor-specific surface antigens and require optimal target selection to circumvent antigen escape and “on-target off-tumor” toxicity [9,10]. They can also prime naïve tumor-reactive CD8+ T cells unlike ICBs, which are only responsive in patients with pre-existing antitumor immunity [11,12]. Cancer vaccines can also be implemented as a part of multimodal immunotherapeutic regimens.
Therapeutic cancer vaccines are designed to stimulate the patient’s antigen-specific adaptive immune response to eliminate cancer cells and generate durable patient responses [13]. Their purpose to eradicate cancer cells via antigen-specific cellular immunity is what makes them different from traditional preventative vaccines [14,15]. Cancer vaccine activity is mostly dependent on activated antigen-specific CD8+ T cells that differentiate into CD8+ TEFF or cytotoxic T lymphocytes (CTLs) to reject cancer cells [15]. Therefore, vaccine-elicited CD8+ T cells should ideally be of high T cell avidity or antigen-triggering sensitivity to recognize immunogenic peptide-major histocompatibility complex class I (pMHC I) complexes and effectively eliminate cancer cells [16,17]. In addition, vaccination should generate long-lived memory CD8+ T cells to prevent tumor relapse [18]. To achieve the ideal vaccine-elicited immune responses, the protective role of immune system components, including antigen presentation by appropriate APCs, induction of CD4+ TEFF or T helper (Th) cells, and suppression of immunosuppressive regulatory T cells (Treg) and TME, are also required [18,19,20].
Dendritic cells (DCs) are the most potent group of specialized APCs with key roles in initiation and regulation of innate and adaptive immune responses [21,22]. DCs comprise heterogenous populations, which can be broadly classified into conventional DCs (cDCs), consisting of two subsets (cDC1, cDC2), plasmacytoid DCs (pDCs) and monocyte-derived DCs (MoDCs), based on their ontogeny [23,24,25]. Recently, additional subsets and states of DCs such as DC3 are being revealed with advances in high-throughput single-cell analysis, which would facilitate a more accurate understanding of the functions and development of DCs [26,27,28,29,30,31,32,33,34]. As DCs initiate and modulate antigen-specific immunity and tolerance, the exploitation of their antigen-presenting capacities and heterogeneity provides great potential for improving antitumor immune response elicited by therapeutic vaccines [7,18,35].
DCs patrol their environment until they become activated upon encountering foreign pathogens or altered cells such as cancer cells as illustrated in Figure 1 [36]. Exogeneous danger signals, such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), activate and determine DC functions via pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) [37,38]. DCs then migrate to tumor-draining lymph nodes (TDLNs) and perform cross-presentation, which is a process where the acquired exogenous antigens are presented on MHC I molecules [39]. Activated, mature, antigen-loaded DCs cross-prime naïve T cells into antigen-specific TEFF cells, including CD4+ Th cells and CD8+ CTLs via T cell receptors (TCRs), and regulate immunogenic responses depending on the DC-derived cytokine environment [40,41,42,43]. Cross-presentation is essential to activate and cross-prime CTLs for defense against tumors [44]. However, the challenge to enhance cross-presentation abilities of DCs for effective cross-priming of CTLs often remains unmet in cancer immunotherapy [39,45]
2. Classical DC Vaccine
The two conventionally adapted methods for preparing DC-based vaccination are ex vivo differentiation of DCs from CD14+ monocyte precursors or CD34+ hematopoietic stem and progenitor cells (HSPCs) and direct targeting of antigens to DCs in vivo [20,46].
Reinfusion of ex vivo manipulated DCs is the most explored preparation method of DC-based vaccines, which is used in approximately 97% of clinical trials [47]. In this approach, CD14+ monocytes or CD34+ HSPCs are collected from patients by leukapheresis, differentiated into immature DCs in the presence of granulocyte macrophage-colony stimulating factor (GM-CSF) and interleukin-4 (IL-4), and simultaneously pulsed with tumor-associated antigens (TAAs) or tumor cell lysates while being stimulated in a maturation cocktail (Figure 2A) [46,48,49,50]. In most clinical studies, CD14+ monocytes are preferentially used to be differentiated into MoDCs because it is relatively easier to collect enough CD14+ monocytes as they represent 10% of peripheral blood mononuclear cells (PBMCs) [47]. Although CD34+-derived heterogenous APC populations were shown to stimulate CTLs more significantly than MoDCs (NCT00700167, NCT01456104), the limited numbers of CD34+ HSPCs (represent 0.1% of PBMCs) that can be isolated from apheresis products hinder them from clinical applications [51,52,53]. Due to the absence of standardized preparation of ex vivo manipulated DC vaccines, different studies follow various methods for DC sourcing, maturation, antigen loading, and administration [47]. So far, only sipuleucel-T (Provenge), the first FDA-approved therapeutic cancer vaccine, has demonstrated a satisfactory efficacy in phase III trials as an autologous ex vivo DC vaccine for metastatic castration-resistant prostate cancer (NCT00065442) [54,55,56]. The combination of Provenge with ICBs and a homeostatic cytokine IL-7 that enhances T and B cell development and proliferation has demonstrated encouraging clinical efficacies (Phase I, NCT01832870; Phase II, NCT01804465, NCT01881867) [57,58,59].
Another strategy that aims to directly target antigens to endogenous DCs in vivo has been explored to overcome the limitations of ex vivo manipulated DC vaccines [60]. This strategy involves antigen coupling to monoclonal antibodies (mAb) that are specific to DC surface molecules, including Clec9A, CD40, or DEC-205, to directly pulse DCs with antigens in vivo [61,62,63,64,65]. Despite effective TEFF-mediated antitumor responses and humoral immunity demonstrated in preclinical and clinical studies, clinical implementations have been discouraging [66]. Some of the drawbacks of this approach may be the requirement to ensure expressions of targeted receptor in the selected DC subpopulation and to co-administer DC maturation agents to prevent antigen tolerance in steady state [46,64,67].
3. Limitations of Classical DC Vaccine
Despite the early success of Provenge and generally accepted safety of conventional DC-based cancer vaccines, their clinical implications have generally been unsuccessful, with only 5–15% of patients benefiting from an objective immune response [68]. The limited efficacy of cancer vaccines may be mainly due to the presence of multiple immunosuppressive factors in the TME that act as immune rheostats or immunostats in short. In other words, the immunosuppressive factors modulate the antitumor T cell responses and act as a common rate-limiting step in clinical studies that altogether limit the clinical efficacies of cancer vaccines [69]. New generations of DC vaccines must therefore overcome multiple immunosuppressive mechanisms in the TME to improve TEFF-mediated antitumor responses.
Solid tumors produce soluble immunosuppressive mediators, such as indoleamine 2,3-dioxygenase (IDO), transforming growth factor beta (TGF-β), and vascular endothelial growth factor (VEGF), which not only suppress Th1 cells and CTL activity but also impair DC functions [70,71,72]. In addition, the TME attracts immunomodulatory populations such as Tregs and myeloid derived suppressor cells (MDSCs) and suppresses TAA expression to achieve immune evasion [73,74,75]. Tumor cells may also evade immune recognition by the overexpression of immune checkpoints such as cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death protein 1 (PD-1) [76]. Therefore, combination therapies incorporating ICBs and DC-based vaccines are being actively studied as potential improved therapeutic regimen (NCT01067287, NCT03035331, NCT04203901). Altogether, it is important to note that the effectiveness of DC vaccines may be altered post-administration of in vitro-modulated DCs to an in vivo immunosuppressive environment [21].
Despite MoDCs being the most adopted subset in clinical trials for DC vaccines, it remains unclear which subset is the most effective. Ex vivo generated MoDCs are suggested to be functionally dissimilar to the steady-state DC subsets present in the body, revealed by the disparities between their immune system-related transcripts at both transcriptional and phenotypical levels [20,77]. Moreover, the immunosuppressive TME limits the cross-presentation and T cell stimulation abilities of tumor-associated MoDCs [78]. In fact, studies suggest that endogenous DCs are required for T cell priming, as the capacity of ex vivo manipulated MoDCs as effective APCs in vivo is obscure [79]. In addition, MoDCs in the TME have limited capacity in migration to TDLNs [80,81]. The optimization of effective DC migration is thus another obstacle in the conventional DC vaccine regimen. To circumvent the complex migration cascade required for efficient homing of DCs to the TDLNs, in situ targeting of DCs has been proposed as an attractive alternative [82].
Therefore, a new generation of DC-based vaccines that incorporates heterogenous subsets of DCs to resemble the in vivo environment more closely may be beneficial [83]. For instance, both cDCs and pDCs have the potential to directly activate T cells in certain environmental cues [83]. Therefore, vaccines that can enhance the function of all DC subsets and cells that crosstalk with them may maximize tumor-specific T cell priming efficacy and vaccine longevity. In addition, ex vivo manipulation of MoDCs also requires extensive in vitro culture, which often disturbs their functionalities [68]. On top of this, despite the good safety profile of MoDC-based vaccines, their production according to good manufacturing procedure is highly expensive and laborious with inconsistent success [47]. The ultimate goal is to design new generations of immunotherapies that sustainably increase the magnitude of DC cross-presentation presentation and TEFF-derived antitumor immunity. Figure 2B illustrates the recent advances in DC-based vaccine regimen, which are discussed in detail below.
4. Biomaterial-Based DC Vaccines
The limited efficacies of conventional ex vivo differentiated DC vaccines were partially due to their inefficient migration to TDLNs and cross-presentation abilities when compared to endogenous DCs [51,52,81]. To overcome such limitations, several biomaterial-based strategies that directly target antigens to endogenous DCs have been developed [84]. These new strategies consist of implantable or injectable biomaterial-based scaffolds that allow in situ trafficking and the modulation of endogenous DCs [46]. The biomaterials are established to provide a spatiotemporally controlled delivery of chemoattractants, antigens, and adjuvants to recruit and activate desired endogenous DC populations [85]. The biomaterial-based scaffolds also regulate the kinetics of antigen exposure, which is critical in developing antigen-specific T cell response [86,87,88]. Moreover, unlike the traditional bolus vaccination strategy that delivers vaccine components in suspension over a short period of time, biomaterial-based scaffolds offer a new biomimetic immunogenic microenvironment in the body where DCs are activated and sustainably provided with antigens and stimulatory molecules over a period of at least two weeks [89,90].
Several implantable three-dimensional biomaterial-based scaffolds are based on a porous structure composed of poly(lactide-coglycolide) (PLG) [89]. PLG is FDA-approved for clinical use, prone to surface modification to enhance its unique scaffold characteristics, biocompatible, and biodegradable [91]. It has been shown that PLG scaffolds can encapsulate DC chemoattractants such as GM-CSF, and gradually release them over 15 days post-implantation [89,92]. In addition, PLG matrices are able to immobilize CpG-rich oligodeoxynucleotides (CpG-ODN), which are agonists of TLR9, as danger signals to activate DCs [9,93]. The spatiotemporal delivery of GM-CSF and CpG-ODN formulation via PLG matrices have successfully attracted and activated DCs in situ and induced prophylactic immunity against inoculations of murine B16-F10 melanoma cells [89].
In a subsequent study, a PLG scaffold that contained GM-CSF, CpG-ODN, and melanoma tumor lysates were assayed in B16-F10 murine melanoma models. Of note, the scaffold successfully recruited and activated heterogenous DC subsets, both pDCs and CD8+ cDCs [94]. Moreover, simultaneous trafficking and activation of pDCs and cDCs led to local accumulation of CTLs and superior antitumor responses than targeting a single DC subset [83]. Conventional ex vivo-developed cancer vaccines are unable to fully recapitulate the broad DC responses in vivo. Therefore, in situ generation of a heterogenous DC subsets by implantable immunostimulatory PLG matrices to activate robust CTL responses against established tumors is remarkable [94]. In addition, two-time vaccination with the PLG matrices led to a complete regression of established melanoma in ~47% of mice in preclinical studies, supporting the therapeutic efficacies of PLG scaffold vaccines [94]. The PLG matrices also increased the local production of IL-12, a Th1-promoting cytokine, which may be attributed to the CD8+ cDCs recruited that are adept at IL-12 production and CTL induction [95,96,97]. Vaccine efficacy highly correlated to the quantities of recruited CD8+ cDCs and pDCs, together with local GM-CSF and IL-12 concentrations [98]. Of note, the accumulation of CD8+ cDCs at the vaccine site is a marked feature of this strategy as CD8+ cDCs typically localize at secondary lymphoid structures [99]. It is suggested that the recruited pDCs supported the activation of CD8+ cDCs and priming of Th1 cells and CTLs, thereby inducing prophylactic immunity against tumor. In contrast to other vaccines that only incorporated GM-CSF as an adjuvant and showed a significant Treg increase at the vaccine site, the PLG matrix also incorporating CpG-ODN counteracted the immunosuppressive mechanisms as CD8+ CTLs outnumbered Tregs [100]. This acellular PLG scaffold in concert with GM-CSF, CpG-ODN, and TAAs may also be adapted for other types of solid cancers. A human version of this vaccine, named WDVAX, is currently in a phase I clinical trial for stage IV melanoma (NCT01753089) [90]. Furthermore, a combination of this PLG matrix with anti-CTLA-4 mAbs augmented vaccine-induced CTL activity and overall survival and led to tumor regression in B16-F10 melanoma mouse models [101]. This combination approach also obviated the need for multiple PLG vaccination, which may be attributed to the synergistic activation of multiple DC subsets and tumor-infiltrating CD8+ T cells [101].
The biomaterial-based vaccination system has expanded to injectable biomaterials such as hydrogels [46,102,103,104]. Alginate hydrogels are often obtained by cryogelatination, a technique which augments mechanical stability, pore connectivity, and shape memory to the injected hydrogels [105,106,107,108]. Injectable alginate gels are biocompatible and elicit a mild host response in vivo while providing sustained release of molecules [109,110,111]. In addition, these gels do not require surgical implantation, unlike PLG scaffold vaccines, to provide a niche for immunomodulation [112]. One of the challenges of ex vivo differentiated DC vaccine is the short half-life of pMHC on short-lived DC [113]. The alginate matrix may act as a local depot for antigens or pMHC to attract local DCs and sustain TEFF cell stimulation. Notably, the total number of local DCs recruited into the alginate gel was sustained [114]. Moreover, similar to the PLG scaffold that delivered GM-CSF and CpG-ODN, alginate hydrogel loaded with irradiated B16-F10 melanoma cells, GM-CSF, and CpG-ODN also elicited local infiltrations of cDCs and pDCs into the gel in a spatiotemporal manner in syngeneic mice [89,115]. The accumulation of a larger spectrum of DC subsets in situ elicited by these biomaterial scaffolds confer sustained DC functions and numbers, in contrast to conventional adoptive transfer ex vivo vaccines [79,93,116].
Combinatory strategies of loading other immunomodulatory agents in hydrogel-based platforms such as metabolism inhibitors or checkpoint blockades were found to augment antitumor immune response [117,118,119]. Metabolism inhibitors are envisioned as alternatives to more toxic cytokines that can counteract the immunosuppressive T cell activities. For instance, epacadostat, a small molecule inhibitor of IDO that suppresses TEFF, activated CD8+ T cells and suppressed the proliferation of Tregs [72,120,121]. Spatiotemporal controlled delivery of metabolism inhibitors via biomaterial scaffolds facilitated in improving poor pharmacokinetics and metabolic interventions [117,118,122]. Peritumoral injection of a hydrogel encapsulating GM-CSF and epacadostat in mice with subcutaneous (s.c.) 4T1 breast cancer cells recruited greater intratumoral (i.t.) cDCs with a 12-fold increase and upregulated expressions of a DC maturation marker CD86 compared to those injected with blank gels [123]. Moreover, the hydrogel successfully regulated the TME, as increased CTLs/Treg ratio and upregulated expression of T cell activation and proliferation marker CD69 on tumor-infiltrating CD8+ T cells was observed [123]. However, epacadostat does not completely inhibit the production of kynurenine (Kyn), a tryptophan catabolite by IDO, which promotes Treg and tumor-associated macrophage differentiation. Thus, epacadostat has failed to improve survival in Phase III clinical trials (NCT02752074) [124,125,126,127]. To address this limitation, one study reported a synergistic effect of alginate gels and a localized chemo-immunometabolic therapy in augmenting T cell activity and systemic anti-tumor immunity in poorly immunogenic 4T1 breast cancer and B16-F10 melanoma mouse models [128]. Enzyme-mediated Kyn elimination and chemotherapeutics were achieved by integrating kynureninase (KYNase) and a chemotherapeutic drug doxorubin (DOX) in alginate gels loaded with GM-CSF and CpG-ODN [128,129,130]. Previous studies have demonstrated increased in situ DC recruitment and systemic tumor-specific CD8+ T cells upon injection of alginate gels loaded with GM-CSF, CpG-ODN, and DOX conjugate [131]. The combinatory approach with chemo-immunometabolic therapy via alginate-based DC vaccine extended the systemic antitumor response, as numbers of tumor-specific CD8+ T cells increased in TDLNs, which could also induce abscopal effect in untreated tumors [128].
The spatiotemporal control of tumor antigens and different immunoadjuvants by the biomimetic biomaterial-based scaffolds successfully recruited endogenous DCs in situ and produced a more robust antitumor response. The scaffolds may incorporate different immunostimulatory molecules, metabolism inhibitors, and/or checkpoint blockades to maximize DC recruitment and activation, along with tumor-specific T cell responses, as well as modulating other immunosuppressive cells to modulate the TME. Although some scaffolds require surgical implantation and multiple doses, the recent development of injectable 3D structures and combination therapies alleviates such limitations and bypasses multiple immunizations [132,133]. Further studies are expected to provide more promising results of clinical implications of biomaterial-based DC vaccines.
5. Combinatory Immunogenic Cell Death-Inducing DC Vaccines
Conventional DC vaccines encounter unresponsiveness in cold tumors in which DC recruitment is defective, and thus T cell priming, activation, and infiltration are impaired [134,135,136]. Recently, increasing numbers of studies have shown that the induction of immunogenic cell death (ICD) is promising in converting cold tumors into hot tumors, thereby improving the immunogenic potential of DC vaccines in cold tumors [137,138,139]. ICD is a form of stress-induced regulated cell death response via different organellar and cellular stressors [140,141,142]. ICD is induced in cancer therapy by most common clinical treatments such as chemotherapy and radiotherapy, or emerging therapies such as photodynamic therapy (PDT) and photothermal therapy (PTT) [143,144,145,146,147]. ICD-inducing therapy is an attractive antitumor strategy for various solid tumors in view of its consequent antigenicity and adjuvanticity [144,148,149,150]. The antigenicity of ICD is determined by the level of tumor antigens from dying tumor cells [151,152,153]. The induction of ICD stimulates tumor cells to secrete, release, or surface-expose DAMPs and cytokines as adjuvants or danger signals [154,155,156]. Once emitted by dying cells, DAMPs recruit and activate innate and adaptive immune cells to elicit a specific antitumor immune response [155,157,158,159]. Antigenicity and adjuvanticity of anticancer treatment-induced ICD are critical for the development of long-term immunological memory against residual cancer cells and metastatic cells [160,161,162].
Canonical immunostimulatory DAMPs include, but are not limited to, calreticulin (CRT), heat shock proteins 70 (HSP70), high-mobility group box 1 (HMGB1), and ATP, and are associated with immunogenicity induced by DCs [140,161]. CRT confers immunogenicity by promoting phagocytosis of dying cells by DCs as an ‘eat-me’ signal [163,164]. Exogenous ATP, on the other hand, constitutes a ‘find-me’ signal that promotes DC recruitment and migration [165,166]. HMGB1 promotes activation, maturation, and cross-presentation abilities of DCs, while HSP70 increases the uptake of tumor cells and stimulates the migration and maturation of DCs [167,168,169,170]. Therefore, ICD activates tumor-specific immunity by tipping the cancer-immunity balance towards antitumor immunity while eradicating cancer cells and generating antigens in situ, which is in line with the objectives of cancer vaccines [69,139,171].
Radiotherapy not only induces ICD and abolishes highly proliferative cancer cells, but also promotes an abscopal effect, which is a rare phenomenon of tumor regression outside the field of local therapy [135,172,173]. However, tumor-specific immune response and abscopal effect induced by standalone radiotherapy are limited in immunosuppressive TME [137,174]. Moreover, radiotherapy can further exacerbate negative regulatory mechanisms by insufficient T cell priming and enhanced levels of Tregs and MDSCs [175,176]. To overcome the complex interaction between TME and radiotherapy, a combination of radiotherapy and immunotherapy is envisioned to generate an in situ cancer vaccine and enhance antitumor responses and abscopal effects [177]. Local administrations of immunostimulatory agents, along with radiotherapy, are increasingly being investigated to activate immature tolerogenic DCs into immunogenic DCs in established tumors. Many preclinical studies provided evidence that TLR agonists targeting TLR3, TLR9, and TLR4 together with local radiotherapy led to enhanced antitumor immunity [178]. The TLR3 agonist poly-ICLC was utilized to activate endogenous DCs together with Fms-like tyrosine kinase 3 ligand (Flt3L) as an adjuvant along with local radiotherapy as a treatment strategy for indolent non-Hodgkin’s lymphoma (iNHL) [179]. Such a combination functioned as an in situ vaccine, inducing greater i.t. infiltration of CD8+ T cells, DC activation, and cross-presentation [179]. Similarly, i.t. administration of Flt3L, local radiotherapy, and TLR3 stimulation followed by surgical resection demonstrated a potential multimodal therapy that can generate tumor-specific CTLs, achieve improved systemic tumor remission from 40% to 80%, and delay metastases in highly metastatic tumors [180]. Of note, both regimens observed increased expressions of PD-1 on CD8+ T cells, and thus synergized with PD-1/PD-L1 blockade to improve systemic tumor control and prolong survival [179,180].
These preclinical studies prompted a phase I/II trial of i.t. injection of Flt3L and poly-ICLC combined with radiotherapy in lymphoma patients (NCT01976585), which consistently demonstrated increased levels of PD-1+ CD8+ T cells and clinical efficacies. Furthermore, to confirm that ICB and radiotherapy synergistically facilitate antitumor immune response, a follow-up phase I/II clinical trial (NCT03789097) was performed. This demonstrated tolerability and early signs of efficacies in patients with iNHL, metastatic breast cancer, and head and neck squamous cell carcinoma [181]. Several clinical studies have also demonstrated that the combination of TLR9 agonist CpG-ODN and radiotherapy in various cancer models achieved systemic antitumor immunity and patient responses (phase I/II, NCT00185965; phase II, NCT00880581) [182,183]. These studies altogether support the combination of radiotherapy and immunotherapy in inducing synergistic effects that restrain not only local tumor growth but also distant metastases. The expansion of radiotherapy combined with immunotherapy may yield superior antitumor immunity compared to combination immunotherapies, which may result in increased toxicity [179]. While the combination of radiotherapy with other ICBs, such as CTLA-4, has also been robustly investigated (phase I/II, NCT00323882, NCT02221739; phase III, NCT00861614), further preclinical and clinical studies will demonstrate the promise of various combination strategies to build a robust in situ radiotherapy-based DC vaccine [184,185,186,187].
PDT-induced therapeutic ICD strategies have also been shown to be effective in various types of cancer and were approved by FDA [188]. PDT involves the use of a photosensitizer which is excited by a light of specific wavelength corresponding to the absorption spectrum of the photosensitizer in the presence of oxygen [151,189]. This results in the generation of singlet oxygen and other cytotoxic oxidants that trigger ICD in cancer cells [190]. Photosense (PS), a clinically approved photosensitizer, was found to successfully trigger ICD in several cancer cell types, including GL261 glioma cells [191]. ICD induced by PS-mediated PDT (PS-PDT) emitted CRT, HMGB1, and ATP, which successfully matured and activated DCs in vitro that produced IL-6 for T cell priming [191,192]. To demonstrate the efficacy of PS-PDT as a vaccine regimen, DCs were loaded ex vivo with GL261 glioma cells undergoing ICD post-PS-PDT (DC-GL261_PS-PDT) [160]. DC-GL261_PS-PDT not only protected mice against orthotopic challenge with GL261 cells prophylactically but also protected them therapeutically [160]. Moreover, the vaccinated mice showed significantly lower tumor mass, later onset of symptoms, and a significantly increased survival compared to control mice which were vaccinated with either PBS or freeze-thawed (F/T) GL261 cells [160]. Importantly, the DC vaccine pulsed with PS-PDT-induced dying cancer cell is proposed as an attractive approach for producing whole-tumor derived tumor-specific antigens (TSA) that may provide superior efficacy compared to conventional F/T cell lysates which often lead to weak immunogenicity and unregulated cell death by accidental necrosis [160,163,193,194]. In contrast to non-mutated self-antigen TAAs, TSAs or neoantigens are recognized as non-self-antigens that are generated by cancer cells as a result of tumor-specific alterations, such as mutation, RNA splicing, and post-translational modification [195]. Several studies have shown that using F/T cells as vaccines failed to generate antigen-specific CD8+ T cell-mediated immune responses [163,196,197,198,199].
It is noteworthy that autologous tumour cells possess a full repertoire of personal mutant TSAs including those that could be crucial for tumour rejection, which are absent in allogeneic tumor cell lines [200]. To promote antitumour immunity in a patient-personalized manner, a study developed an in situ DC vaccine that is administered via a single low-dose intravenous (i.v.) injection of an artificial vesicle polymersomal combination along with PDT without the need of loading DCs ex vivo [143]. A chimeric polymersome (CCPS) co-encapsulated a low-dose of DOX and PS. Chemotherapeutic agent DOX and PS-PDT induced ICD by releasing HMBG1 and CRT and forming reactive oxygen species [143,201]. Moreover, CCPS embedded with amine groups served as adjuvants and enhanced DC maturation and cross-presentation abilities by 19-fold compared to negative control group [202]. MC38 colorectal cancer-bearing mice treated with a single i.v. injection of CCPS_DOX_PS-PDT showed the highest serum levels of proinflammatory cytokines, IL-6, IL-12, and TNF-α, and the prominent elimination of primary tumors [143,203,204]. Furthermore, levels of mature migratory DCs and tumor-infiltrating CTLs were elevated in CCPS_DOX_PS-PDT vaccinated mice compared to those vaccinated with CCPS encapsulating DOX or PS alone, suggesting the importance of co-encapsulation of DOX and PS in developing immunogenicity via PDT. In addition, a significant abscopal effect was observed in mice treated with CCPS_DOX_PS-PDT [143]. Such a combination in in situ DC vaccines incorporating synthetic delivery vehicles, chemotherapeutic drugs, and PDT may potentially enhance DC maturation and therapeutic vaccine efficacy.
PTT induces ICD for tumor ablation with the heat converted from near-infrared laser (NIR) absorbed by photothermal agents that are accumulated in the tumor [205]. Recently, combinations of PTT with other therapeutic modalities such as nanomaterials, immunotherapies, or even PDT and conventional chemo- and radiotherapy were demonstrated to provide additive or synergistic therapeutic efficacies [206,207,208,209]. In particular, multiple studies have demonstrated that PTT combined with immunotherapy augmented the immune response by ICD of local cancer cells and the release of TSAs and cytokines, and inhibited metastases [210,211,212]. One study established a combination of PTT with immune-adjuvant nanomaterial that co-encapsulated a photothermal agent, indocyanine green (ICG), and an immune adjuvant TLR7 agonist imiquimod (R837) in a PLG scaffold, forming a PLG-ICG-R837 nanoparticle [213]. Upon NIR-induced PTT on primary tumors injected with PLG-ICG-R837, enhanced levels of DC recruitment into the tumor site, DC maturation in TDLNs, and sera levels of proinflammatory cytokines were observed [213]. Furthermore, it was shown that this combination with ICB established abscopal effects after the ablation of primary tumors and long-term anti-tumor immunogenicity, as secondary tumor growth of 4T1 breast cancer and CT26 colorectal cancer were completely inhibited [213]. Systemic administration of this formulation followed by PTT also offered a strong antitumor response without observable cytokine-storm-like side effects, supporting its efficacy in tumors that can hardly be reached [213]. Synergistic interactions between PTT and other therapeutic modalities, particularly immunotherapy, appear promising to enhance antitumor vaccine efficacy and immunological memory besides minimizing metastases, encouraging further studies [214].
6. mRNA-Pulsed DC Vaccines
The pulsing of DCs with tumor antigens is a widely used vaccination strategy in which autologous DCs are loaded with antigenic information and matured under favorable conditions [215]. The resulting DCs are then administered back into the patient to initiate antitumor immune responses [215]. Antigens that are used to pulse DCs are commonly obtained as whole-cell antigens from repeated F/T cancer samples and synthetic cancer antigenic peptides [215,216,217]. Cancer antigenic peptides are used in many clinical trials, but they involve substantial weaknesses [218]. For example, the selected peptides to be synthesized require prior identification of human leukocyte antigen (HLA) types and have HLA restriction of patients [219]. In addition, peptides may only serve to elicit either CD4+ or CD8+ T cell response, and the short half-life of HLA-antigenic peptide complex limits durable antigen presentation [21,113].
mRNA has emerged as an appealing candidate that offers both antigen delivery and innate immune activation under spatiotemporal control that circumvents the limitations of common tumor antigen sources [220]. mRNA pulsing of DCs is widely recognized due to its multiple advantages: the introduction of exogenous mRNA activates innate immune cells by stimulating various TLRs such as TLR3, TLR7, and TLR8, presenting strong intrinsic adjuvanticity; does not integrate into the genome, avoiding any insertional mutagenesis; can be readily produced in large amounts in vitro; can be engineered to increase immunogenicity and reduce inhibition of its translation; not subject to splicing, eliminating any uncertainty in protein products due to alternative splicing [215,221,222,223,224].
Since the feasibility of ex vivo mRNA-pulsed DC vaccine was first demonstrated 27 years ago, numerous preclinical and clinical studies have explored loading DCs with mRNAs as a new vaccination strategy [218,225]. Multiple studies have confirmed the ability of mRNA-pulsed DCs to induce potent tumor antigen-specific T cell responses in vivo [226,227,228,229]. Among several strategies developed to introduce mRNA directly into DC cytosol, electroporation has been demonstrated to be the most efficient method [230,231]. By means of weak electric pulse, mRNA is rapidly integrated into the cytosol, which prevents mRNA from degradation by ubiquitous extracellular ribonucleases [218]. Electroporation also alleviates the potential alteration of DC immunophenotype, maturation potential, migration capacity, and T cell-stimulatory potential [232,233].
DCs may be pulsed with mRNA from either source: whole tumor-derived mRNA or in vitro transcribed mRNA [218,234]. Tumor-derived mRNAs allow the delivery of complete repertoire of epitopes expressed in tumor, which circumvents immune escape due to antigen loss or downregulation [218]. The safety of autologous tumor mRNA-pulsed DC vaccines was demonstrated in clinical trials of various cancer types, including renal cell carcinoma (Phase I, NCT00006431) and advanced malignant melanoma (Phase I/II, NCT01278940) [235,236]. The latter study demonstrated vaccine-elicited T cell responses in about 51% of the patients and significantly enhanced survival in immune respondents [237]. However, relatively large amounts of autologous tumor cells and adequate accessibility of the tumor site are required to prepare whole tumor-derived mRNA [220]. In addition, tumors express many other molecules besides TSAs, such as self-antigens and inhibitory molecules [234]. These irrelevant peptides may become more abundant after a multitude of steps involved in the in vitro amplification of tumor-derived mRNAs and promote the under-representation and loss of immunogenic antigens or epitopes [238].
Therefore, in vitro generated mRNAs are preferentially employed in preparations of DC vaccines [238]. Although the safety profiles of in vitro generated mRNAs have been demonstrated in numerous clinical studies (Phase I/II, NCT00243529; Phase II, NCT00965224, NCT01446731, NCT02285413) [219,239,240,241,242], general clinical efficacies have been low due to the weak induction of T cell responses [220]. Thus, modifications to enhance the expression of mRNA-encoded antigen and translational efficiency and to diminish mRNA extracellular degradation level were performed [243]. Moreover, it has become increasingly clear that CD4+ T cells, especially Th1 cells, are important contributors in the antitumor activity on top of CD8+ T cells, and that they are essential for the formation of memory CD8+ T cells [244,245,246]. However, antigenic peptides that are processed from mRNA-encoded antigenic proteins in the cytosol of DCs and presented onto MHC I are only capable of stimulating CD8+ T cells. Since the activation of CD4+ T cells depends on MHC II presentation pathway, antigens are targeted to lysosomes by means of fusion to lysosomal sorting signals such as lysosome-associated membrane protein-1 (LAMP-1) or MHC I trafficking signal (MITD) [247,248,249]. Such LAMP-1- or MITD-fused chimeric proteins are processed and presented onto both MHC I and II to expand both CD4+ and CD8+ T cell responses and improve effector functions [219,250]. Clinical studies investigating the effect of LAMP-1 mRNA-loaded DC vaccines in glioblastoma have observed exceptional tumor-specific CD4+ and CD8+ T cell response and extended overall patient survival (Phase I, NCT00626483, NCT00639639; Phase II, NCT02366728) [251,252].
To further ameliorate DC vaccine efficacy, strategies that co-transfect DCs with mRNAs encoding immunostimulatory ligands and receptors to enhance DC maturation and T cell co-stimulation have been developed [220]. In preclinical studies, co-transfection with mRNAs encoding CD83, OX40 ligand (OX40L; CD134), 4-1BB ligand (4-1BBL; CD137L), CD40 ligand (CD40L), and glucocorticoid-induced tumor necrosis factor receptor ligand (GITRL) have shown enhanced DC co-stimulation and T cell priming [253,254,255,256,257]. Co-administration of antigenic and 4-1BBL mRNA significantly increased tumor-specific CTL response compared to only antigenic mRNA administration [256]. Another study demonstrated the synergistic effect of 4-1BBL and CD40L mRNA co-stimulation on enhancing both CD4+ and CD8+ T cell responses and alleviating Treg-suppressed CD8+ T cell proliferation [258]. Rocapuldencel-T is currently the most clinically advanced mRNA-pulsed DC vaccine, which integrates the co-transfection of autologous tumor-mRNA and CD40L mRNA [259]. However, it has failed to improve the overall survival of metastatic renal cell carcinoma patients in combination with standard treatment despite its tolerability and capability to increase T effector memory cells (TEM) [260]. Therefore, it led to withdrawal or termination of the clinical studies (Phase II, NCT02662634; Phase III, NCT01582672) [259,260]. Of note, TriMix formulation, a cocktail of mRNA-encoded adjuvants (CD70, CD40L, and constitutively active TLR4) that can be electroporated together with antigenic mRNA(s) was developed as an alternative approach [261,262,263]. Importantly, the electroporation of mature DCs with TriMix demonstrated its ability to reprogram Tregs to Th1-like cells with enhanced IL-12 secretion, therefore alleviating Treg inhibition of CD8+ T cells in vitro and in vivo [263,264].
The combination of mRNA-pulsed DCs and ICBs has also been investigated to maximize vaccine efficacies. In a single-arm phase II clinical study (NCT01302496), patients with stage III/IV malignant melanoma were treated with DCs, electroporated with TriMix, and mixed with multiple LAMP-1-fused melanoma-associated tumor mRNAs (TriMixDC-MEL), along with ipilimumab (IPI; CTLA-4 inhibitor) mAb [265]. The combination of TriMixDC-MEL and IPI was well tolerated and resulted in durable tumor reduction in patients with recurrent or refractory melanoma [266]. These promising preclinical data of co-transfection with antigenic tumor mRNA and IPI mAb may also encourage ICBs of CTLA-4 and/or PD-1 pathways [218]. Synergistic effects were observed in the co-transfection with mRNAs encoding anti-CTLA-4 and anti-PD-1 mAbs, which were associated with higher overall response rates and greater changes in tumor burden compared to single agent treatment, conferring it as the first FDA-approved treatment for advanced-staged melanoma [267,268]. However, the association of anti-CTLA-4 mAb with severe adverse effects and the superior efficacy and safety of anti-PD-1 mAb over IPI are encouraging more studies to focus on the combination of TriMixDC-MEL with anti-PD-1 treatment [269,270,271,272].
Co-transfection of DCs with mRNAs encoding stimulatory cytokines was also employed to enhance DC maturation and T-cell priming in autologous DC vaccines loaded with antigenic mRNA. Several stimulatory cytokines were explored, including GM-CSF, IL-12, and IL-15 [273,274,275,276,277]. IL-12 is a potent cytokine that mediates the differentiation of Th1 cells and stimulates the cytotoxic abilities of natural killer (NK) cells and CTLs [278,279]. However, electroporation of IL-12 mRNA alone into DCs did not affect cell survival or maturation status [280]. Bcl-2 is a critical pro-survival factor that exerts anti-apoptotic functions [281,282]. One study demonstrated the synergistic effects of electroporation of DCs with IL-12 and Bcl-2 mRNAs on priming CD8+ T cells and DC viability to assist vaccine potency and clinical efficacy [280]. In addition, an ongoing phase I clinical trial, in which IL-12 mRNA is administered intratumorally in combination with anti-PD-L1 mAb durvalumab in patients with advanced solid tumors, has shown the safety and tolerability of such combinational therapy (NCT03946800) [283,284].
In addition to ex vivo electroporation, several in vivo delivery methods to pulse DCs using “naked” mRNA have been developed because the administration route and delivery format considerably determines immune response [285]. For example, i.t. delivery of TriMix induced the maturation and migration of tumor infiltrating DCs toward TDLNs, stimulating antitumor responses against spontaneously acquired mRNAs [262,286,287]. The curative potential of i.t. delivery of TriMix is suggested to exploit autologous tumor antigenic repertoire without the need of tumor antigen co-delivery, and to stimulate CTLs against various tumor antigens in situ [220,287,288]. A phase I study of i.t. delivery of TriMix in patients with early breast cancer lesions is ongoing (NCT03788083) [287]. Intranodal (i.n.) injection delivers mRNA directly into the secondary lymphoid structure, which offers the advantage of targeting antigen delivery at the site of T cell activation, obviating the need for DC migration, and selective uptake by DCs to elicit prophylactic or therapeutic antitumor responses [285,289]. Simultaneous i.n. co-delivery of TriMix and antigenic mRNA recruited antigen-specific CD4+ and CD8+ T cells, and CTLs against various TAAs [264]. Clinical trials have also demonstrated the safety and tolerability of i.n. administration of TriMix combined with mRNAs, encoding melanoma-specific TAAs in patients with resected melanoma (Phase I, NCT03394937) [290,291]. Intradermal (i.d.) administration offers an ideal site of delivery, as various APCs reside in the skin [285,292], and the efficacies of i.d. delivery of self-adjuvanted mRNA-encoded TAAs have been established in various mouse cancer models and clinical studies [293,294,295]. On the other hand, i.v. administration of mRNA vaccines was concerned with rapid degradation of mRNAs by ribonucleases and poor target expression in secondary lymphoid structures [285,288]. An mRNA–liposome complex (mRNA-lipoplex) platform was generated to protect mRNA integrity and deliver mRNAs to target DCs in lymph nodes based on the net charge of the platform particles upon i.v. injection [13,296,297]. Since the safety of mRNA-lipoplex was demonstrated in animal models, several clinical trials were initiated (Phase I, NCT02410733, NCT02316457) [298,299]. Although numerous studies have shown efficacies of different mRNA-pulsed DC vaccine administration strategies, it has yet to come to a consensus as to which elicits a superior prophylactic or therapeutic response [218]. Recently, combining multiple routes has been proposed to induce a more systemic immune response in clinical trials of i.d. and i.v. administration of TriMix-MEL as a single agent (Phase Ib, NCT01066390; Phase II, NCT01676779) and in combination with IPI (Phase II, NCT01302496) [265,300,301,302].
7. DC-Derived Small Extracellular Vesicle (sEV) Vaccines
The major limitations of using conventional MoDCs to prepare DC vaccines are the immunosuppression in the TME, off-target toxicities, and the need for autologous cells [303]. To address such challenges encountered by conventional DC-based immunotherapies, cell-free DC-based vaccines incorporating DC-derived small extracellular vesicles (DCsEV) which are more resistant to immunosuppression are proposed [304]. Extracellular vesicles (EVs) are lipid-bound vesicles released by all cellular organisms and play key roles in intercellular crosstalk via the delivery of EV cargo, cell surface modifications, and target-cell modulation [305]. Small EVs (sEVs) are one of the heterogenous subsets of EVs, characterized by their size, and are of 30–150 nm or less than 200 nm in diameter [306]. EVs contain diverse cargo, including DNA, mRNA, non-coding RNAs, proteins, and lipids [307]. DCs process exogenous antigens in endosomal compartments that can fuse with the plasma membrane to release inert sEVs. DCsEVs play a significant role in DC-to-cell communications via transferring cargos that contain proteins, metabolites, and nucleic acids [304,308]. They carry functional surface antigenic pMHC I and pMHC II, as well as co-stimulatory molecules (CD80/86), and are thus capable of priming antigen-specific CD8+ T cells [309]. Of note, DCsEVs present more pMHC complexes, especially 10 to 100 folds greater pMHC II than DCs [304,310]. Additionally, they are highly biocompatible [309]. While maintaining the essential immunostimulatory properties of DCs, DcsEVs allow for frozen storage and are more amenable to strict vaccine manufacturing processes, and lack the risks associated with viable cellular or viral therapies such as the risk of in vivo replication [304,311].
Despite the strong safety profiles of DCsEVs as cancer immunotherapy, previous clinical trials of first-generation peptide-loaded DCsEVs from autologous MoDCs showed limited efficacies (Phase II, NCT01159288) [312,313,314]. Such a limited immunizing capacity may be due to the insufficient antigen presentation, elevation of Treg level, unknown efficiency of in vivo DCsEV trafficking, and surface expression of immunoregulatory molecules such as PD-L1 [312,313,314]. The second generation of DCsEVs were derived from IFN-ɣ-matured MoDCs [312,315]. Notably, the maturation status of donor DCs influenced the function of DCsEVs and shaped opposing immune responses [304,316]. DCsEVs derived from mature, stimulated MoDCs promoted the exchange of functional pMHC complexes with DCs and enhanced antigen-specific T cell responses, while DCsEVs constitutively released from immature DCs advanced T cell tolerance, Treg proliferation, and the suppression of CD4+ T-helper 17 cells and their production of immunostimulatory cytokine IL-17 [317,318]. IFN-ɣ-matured MoDCs generated DCsEVs that expressed elevated levels of pMHC II, costimulatory molecules, and intercellular adhesion molecules (ICAMs; important for DC homing to LNs), which thereby enhanced the immunostimulatory properties of DCsEVs [304,317]. However, the antigen-specific T cell immune response of the second generation of DCsEVs was still impeded, likely due to insufficient antigen presentation ability and poor in vivo distribution [304]. Several clinical trials on the basis of autologous DCsEVs are in the developing stages, as the safety of DCsEVs have been proved in clinical trials and their prospects in cancer therapy remain promising.
Many studies focus on peptide- or protein-loaded DCsEVs from MoDCs. Other types of DCsEVs that are capable of priming CD8+ T cell responses in vivo are needed to expand the studies of DCsEVs. Despite the presumed tolerogenic role of pDCs in tumors, several clinical trials have shown that vaccination with pDCs possibly leads to the induction of antitumor CD8+ T cell immunity [319,320]. Recently, a new subset of DCsEVs was discovered as the cross-presenting pDCs required bystander cDCs to cross-prime CD8+ T cells in vitro and in vivo by transferring antigens via pDC-derived small EVs (pDCsEVs) [321]. Therefore, pDCsEVs are suggested as a mean to integrate both cDCs and pDCs in cancer vaccines to achieve better anti-tumor efficacy [319]. Although further studies are needed to investigate whether pDCsEVs generated with pDC-targeted antigens exhibit enhanced cross-presentation capacity similar to DCs targeted with antigens and are capable of priming allogeneic CD8+ T cell response similar to protein-loaded DCsEVs, a new subset of DCsEVs suggests a significant mechanism to enhance T cell cross-priming and induce antitumor efficacy. While DCsEVs can potentially overcome the limitations of MoDC-based vaccine, further investigations are needed to elucidate the underlying mechanisms between DCsEVs and CD8+ T cell priming in vivo to advance their clinical application [68].
Recently, a third generation of DCsEV-based cancer vaccines has been developed using nanotechnology and molecular engineering [322]. DCsEVs are engineered to amplify certain immunostimulatory characteristics [304]. A novel DCsEV engineering approach has formed a “designer” vaccine as a universal immunotherapeutic strategy for hepatocellular carcinoma (HCC) [309]. These engineered DCsEVs present an HCC-targeting peptide P47, an antigenic epitope AFP212-A2, and a functional domain of high mobility group nucleosome-binding domain (HMGN1; N1ND) as an immunoadjuvant to recruit and activate DCs [309,323]. These engineered DCsEVs were i.v. injected and successfully enabled tumor-targeted N1ND-mediated recruitment and activation of endogenous cross-presenting DCs in the tumor of orthotopic HCC mice [309]. Moreover, antigen-specific induction of tumor-specific T cell responses and tumor suppression was achieved, supported by the elevated levels of IFN-ɣ-expressing CD8+ T cell and decreased levels of immunosuppressive cytokines such as IL-10 and TGF-β [324]. This study highlighted the capacity of engineered DCsEVs to induce tumor-specific immune response and to be presented as a generalizable approach as a personalized immunotherapy without having to identify patient-specific TSAs [309].
In addition to engineering immunostimulatory activities of DCsEVs, the incorporation of ICBs has been developed. A combination of DCsEVs and ICBs (anti-CTLA-4 therapy or PD-1/PD-L1 blockade) may further enhance the cross-priming of CD8+ T cells and alleviate the suppression of tumor-infiltrating CTLs [304,325]. For instance, anti-CTLA-4 mAb were anchored to the surface of DCsEVs derived from ovalbumin (OVA) antigen-pulsed, activated, and TLR-3 agonist poly(I:C)-matured immature DCs. Poly(I:C) is also a TLR-3 agonist from which its derivative poly-ICLC is generated [326]. This approach generated a bifunctional CTLA-4-mAb-modified DCsEVs (DCsEV-OVA-mAb) [327,328]. Poly(I:C) was demonstrated to effectively activate DCs, augment cross-presentation to CD4+ and CD8+ T cells, and enhance antitumor response in cervical cancer and melanoma immunotherapies. These poly(I:C)-matured DCs secreted DCsEVs with similar enhanced antitumor functions [329]. These functionalized DCsEV-OVA-mAb were enriched in MHC I and II and co-stimulatory molecule CD80. Compared to DCsEVs pulsed with OVA without mAb engineering, DCsEV-OVA-mAb induced a stronger T cell activation and proliferation in vivo, highlighting the importance of CTLA-4 mAb in the vaccine efficacy [327]. They also quickly migrated to TDLNs upon s.c. injection, increased i.t. CD4+ and CD8+ T cell migration, and significantly increased CTLs/Treg ratio in tumor [327]. Importantly, vaccination with DCsEV-OVA-mAb generated the most TEM cells, in line with previous studies that showed enhanced generation and function of CD8+ TEM cells post-CTLA-4 blockade therapy [330,331,332]. Altogether, surface modification of DCsEV with the incorporation of anti-CTLA-4 mAb provides a promising strategy to induce potent antitumor efficacy of DCsEV vaccination against cancer [327].
Moreover, a platform of genetically engineered DCsEVs incorporated with anti-PD-1 antibody on the surface which allows a direct presentation of tumor antigens to CD8+ T cells and stimulate strong CTL responses has been developed [333]. An immature DC2.4 cell line was engineered to display anti-PD-1-single-chain antibody fragment on their membrane surface. These α-PD-1-DCs were then activated and matured by recombinant adenovirus transduction of melanoma TSAs. The matured DCs were upregulated in the expressions of pMHC I, anti-PD-1 antibody, costimulatory molecules (CD80/86), and ICAM-I. The resultant DCsEVs collected and purified from these modified DCs were named ASPIRE [333]. Importantly, ASPIRE expressed the same immunostimulatory molecules as the engineered DCs. ASPIRE stimulated antitumoral CTL response in mice via direct transfer of pMHC I complex to endogenous DCs instead of intermediate DCsEV uptake by DCs, contrary to the mechanisms proposed by earlier studies [304,334,335]. When ASPIRE was inoculated into B16-F10 melanoma tumor-bearing mice, complete tumor rejection was achieved in all mice by successful migration of ASPIRE to TDLNs, where it contacted and activated CTLs. In addition, ASPIRE resulted in less PD-1+ dysfunctional CTLs in tumor-bearing mice than a combinatory treatment of unmodified antigen-presenting DCsEVs along with a free PD-1 mAb. The potential clinical translation of ASPIRE needs further investigation as it is currently unknown whether ASPIRE preparation can also be implemented in standard source of human autologous MoDCs for clinical use and whether it will establish similar immunostimulatory properties as the murine cell line preparation [336]. Nonetheless, ASPIRE presents a novel therapeutic vaccine platform to stimulate enhanced CTL immune response and overcome the immunosuppressive TME by the incorporation of anti-PD-1 ICB in DCsEV vaccines.
To develop novel cancer vaccines using DCsEVs, which is a highly heterogenous population that is often extremely difficult to obtain from a specific immune cell population without contamination with sEVs from other cell types, detailed characterization and careful collection of the sEVs are critical [306]. Recent progress in molecular engineering and the delivery of immune modulatory adjuvants have indeed promoted the antitumor response of DCsEV-based vaccines. Further understanding of sEVs, especially in their biogenesis from immune cells and interactions with tumor and other cell types as well as in vivo biodistribution tracking of them, are deemed as providing a significant breakthrough in sEV-based therapeutics.
8. Tumor-Derived sEV Vaccines
In addition to DCsEVs, tumor-derived sEVs (Tu-sEV) are proposed to be utilized as a source of tumor antigens to develop cancer vaccines [337]. Tu-sEVs share several similar functional characteristics with DCsEVs, but they shuttle information between tumor cells and the TME and are highly enriched in parental tumor antigens [338,339]. They also express distinct sets of proteins that facilitate their binding to and uptake by DCs, such as MHC, LAMP-1, CD9, and CD54 [340,341,342,343]. They can therefore stimulate a broad range of tumor-specific CTL responses against multiple antigenic epitopes [339,344]. Moreover, Tu-sEVs are easily isolated and purified by non- or minimally-invasive methods from patient’s plasma, ascites, and pleural effusions [338]. Tu-sEVs also have significant advantages such as antigen sources, attributed to their high immunogenicity from stressed tumor cells, more than irradiated tumor cell lysates [345,346]. Successful uptake and processing of Tu-sEVs by DCs can enhance the expression levels of co-stimulatory molecules, MHC, and CD11c, and lead to phenotypically and functionally mature DCs [347,348,349]. These features altogether support Tu-sEV as an attractive cell-free cancer vaccine candidate in a patient-personalized manner [350,351,352,353].
However, it is important to note that Tu-sEVs regulate immune responses through both immunosuppressive and immunostimulatory functions, promoting either immune escape or tumor regression [354,355,356]. Specifically, Tu-sEVs can induce the production of inhibitory cytokines, decrease the expression of co-stimulatory molecules, increase STAT3 expression, modulate DC differentiation, and inhibit maturation and the T cell stimulatory capacity of DCs [354,357,358]. Tu-sEVs impair DC maturation and antigen-specific responses via the downregulation of MHC II expression as well as the expansion of Tregs [357,359,360,361]. Earlier studies have, however, proven the feasibility and efficacy of Tu-sEVs as prophylactic and therapeutic cancer vaccines that not only elicit CTL responses but also prevent metastases in mouse models [339,345,351,362,363]. Meanwhile, possible Tu-sEV-derived immunosuppression has redirected further investigations of standalone Tu-sEV treatments to the development of Tu-sEV-engineering and DC loading to advance the immunogenicity of Tu-sEV-based cancer vaccines [337].
Cancer cells or Tu-sEVs themselves may be manipulated to increase the expression levels of tumor antigens or immunostimulatory molecules that are expressed on Tu-sEVs [337]. This may be achieved by transducing tumor cells with viral vectors transfected with genes encoding immunogenic antigens, such as mucin 1 (MUC1), which is a transmembrane glycoprotein that is overexpressed in many cancers, including prostate, breast, and ovarian cancer [364,365,366,367]. MUC1-transduced CT26 colon and TA3HA breast murine cancer cell lines secreted Tu-sEVs that successfully expressed target antigen MUC1 (MUC1-Tu-sEVs) [368]. Both autologous and allogenic MUC1-Tu-sEVs stimulated immune responses and inhibited tumor growth 2-fold greater than control Tu-sEVs, independent of their MHC types in vivo [368]. Furthermore, MUC1-Tu-sEVs activated splenocytes via DCs in a MUC1-dependent manner, inducing the secretion of Th1-type IFN-ɣ in vitro [368]. Recently, another study has directly anchored IFN-ɣ fusion protein onto the surface of RM-1 prostate cancer cell-derived Tu-sEVs (IFN-ɣ-Tu-sEVs) [369]. Vaccination with these IFN-ɣ-Tu-sEVs resulted in the highest levels of CD4+, CD8+, and IFN-ɣ+ CD8+ T cells [369]. Importantly, they decreased the levels of Tregs and PD-L1, and IDO expressions in the TME [369]. IFN-ɣ-Tu-sEVs also significantly inhibited tumor growth and prolonged the survival time of tumor-bearing mice [369]. Furthermore, another study incorporated interferon regulatory factor 1 (IRF-1), which is a tumor suppressor gene that regulates the expression of target genes such as MHC I, IL-15, and IFN-α [370]. Of note, IRF-1-Tu-sEVs displayed an enhanced antitumor response compared to IFN-ɣ-Tu-sEVs [370]. This functional response was mediated through elevated expressions of MHC I and IL-15Rα, resulting in increased tumor infiltrating CD4+ and CD8+ T cells [370]. These studies altogether support cancer cells or Tu-sEVs may be genetically engineered to enhance antitumor responses of Tu-sEV-based vaccines via elevated levels of immunostimulatory antigen and molecule expressions as well as both CD4+ and CD8+ T cells.
In addition to immunogenic tumor antigens, MHC II is another important molecule that could increase the immunogenicity of Tu-sEV-based vaccines [371]. Many cancer immunotherapies have focused on inducing MHCI I-restricted tumor-specific CTL responses because most tumor cells constitutively express MHC I, but not MHC II [372,373]. However, to optimally induce both humoral and cellular effector mechanisms, MHC II-restricted CD4+ T cells that support the maturation, proliferation, and functionality of CD8+ CTLs are also required [374]. Hence, a study transduced B16-F1 murine melanoma cells with MHC class II transcription activator (CIITA) gene to generate Tu-sEVs enriched in MHC II and tumor antigen TRP2 (CIITA-Tu-sEV) [375]. Compared to parental control Tu-sEVs, CIITA-Tu-sEVs exhibited greater DC maturation ability, which was reflected by higher MHC II and CD86 expression, and higher mRNA levels of inflammatory cytokine, TNF-α, maturation marker, CCR-7, and Th1-polarizing cytokine, IL-12 in vitro [375]. CIITA-Tu-sEVs also improved the preventative and therapeutic antitumor immune response and increased Th1 type antibody and IFN-ɣ levels in vivo in a dose-dependent manner [375]. Moreover, CIITA-Tu-sEVs exerted greater tumor rejection, increased survival rate, and overall survival by 20% at 60 days post-treatment [375]. Taken together, MHC II-expressing Tu-sEVs may be another potential candidate to be utilized in cancer vaccines to augment both cellular and humoral antitumor immune responses.
Engineered Tu-sEVs and vaccine adjuvant co-delivery system is also proposed to exhibit potent antitumor activity and to form an effective in situ DC vaccine. Murine melanoma B16-BL6 cells were transfected with a plasmid vector that encoded a fusion streptavidin (SAV)-lactadherin (LA) protein, which yielded genetically engineered SAV-Tu-sEVs [376]. SAV is a protein that binds to biotin with high affinity, and LA is an sEV-tropic protein that binds to the EV membrane [377,378]. SAV-Tu-sEVs were then incubated with biotinylated CpG-ODN to prepare CpG-ODN-modified SAV-Tu-sEVs (CpG-SAV-Tu-sEVs) [376]. These modified CpG-SAV-Tu-sEVs successfully delivered CpG-ODN to DCs, and activated and enhanced tumor antigen presentation abilities of DCs in vitro. Significantly increased levels of cytokine, such as TNF-α, IL-6, and IL-12p4, were also observed from CpG-SAV-Tu-sEV-pulsed DCs [376]. Moreover, immunization with these CpG-SAV-Tu-sEVs resulted in potent cellular and humoral immunity along with upregulation of Th1-related antibody as well as protective and therapeutic antitumor immunity [376]. CpG-SAV-Tu-sEVs demonstrated stronger in vivo antitumor effects in B16-BL6 tumor-bearing mice than separate administrations of CpG-ODN and Tu-sEV, suggesting the co-delivery of tumor antigens and adjuvants by genetically engineered Tu-sEVs as a potential immunotherapy approach.
The notion of Tu-sEV-mediated adjuvant delivery was supported by another study that evaluated the immunogenicity of mouse breast cancer cell-derived Tu-sEVs which were loaded with two immunoadjuvants, CpG-ODN and poly(I:C) (CpG-p(I:C)-Tu-sEV) [379,380]. These engineered CpG-p(I:C)-Tu-sEVs exhibited augmented immunostimulatory properties by activating antigen-specific primary and memory T cell responses and promoting tumor regression in tumor-bearing mice [379]. Similar to the Tu-sEV-mediated antigen-adjuvant co-delivery system above, these also elicited Th1-biased immunity reflected by elevated levels of Th1-related antibody and IFN-ɣ [379]. Altogether, this study also showed that Tu-sEV-based therapeutic vaccine can promote strong cellular and humoral antitumor immunity that can provide a personalized tumor therapy strategy.
In addition to genetic engineering of Tu-sEVs, their surface may also be modified to present immunoadjuvant. One study painted the surface of Tu-sEVs with the functional domain of HMGN1 (N1ND-Tu-sEV) via a vesicular anchor peptide [381]. DCs pulsed with these N1ND-Tu-sEVs-pulsed were activated and boosted CD8+ T cell levels. N1ND-Tu-sEVs also significantly increased DC migratory capacity and the generation and amplification of TEM, which contributed to long-lasting antitumor immunity and tumor suppression in different syngeneic immunogenic mouse models with large tumor burdens, most notably in low immunogenic orthotopic HCC [382]. Importantly, N1ND-painted serum sEVs from cancer patients could also induce tumor-specific cytolysis in vitro and promote DC activation [376]. This study demonstrated the potency of surface-modified Tu-sEVs to augment DC immunogenicity and to inhibit large established tumors, thus providing a platform to load immunoadjuvants onto Tu-sEVs to amplify the antitumor immunity of DC vaccines.
Tu-sEVs may also co-deliver ICD inducers and adjuvant to generate an in situ vaccine. One study loaded an ICD inducer, human neutrophil elastase (ELANE), and a TLR3 agonist Hiltonol onto breast cancer-derived Tu-sEVs to form an in situ vaccine (HELA-sEVs) [382]. These engineered HELA-sEVs were also enriched in breast-specific protein α-lactalbumin (α-LA), which granted them an enhanced targeting capability to specifically induce ICD in breast cancer cells [382]. HELA-sEV-induced ICD of breast cancer cells followed by TLR3 adjuvant-induced i.t. accumulation of cDC1s and cross-primed immunogenic CD8+ T cell responses. This significantly inhibited the tumor growth in a poorly immunogenic breast cancer mouse xenograft model and patient-derived tumor organoids [382]. Tu-sEV engineering strategies that allow the co-delivery of ICD inducer and adjuvant to trigger in situ ICD and cross-priming are promising for generating an effective in situ DC vaccine and may be extended to other types of solid cancers.
In the meantime, it is critical to carefully investigate the possible adverse roles of Tu-sEVs in impairing differentiation, maturation, and functions of DCs to harness their immunostimulatory capacity while avoiding immune escape [350,383]. Several mechanisms underlying the immune inhibitory effects of Tu-sEVs have been proven and more are being postulated, such as the interaction between Tu-sEV, PD-L1, and DC PD-1, reduced antigen sensing and costimulatory molecule expressions in DCs, and the production of VEGF and IL-10 to inhibit DC differentiation maturation [355,384,385,386,387,388,389]. Although numerous preclinical data support Tu-sEV as a potential source for cancer vaccine, and therapeutic interventions to harness Tu-sEV to stimulate antitumor response are being investigated, significant challenges remain to reject the opposing notion of Tu-sEVs as potential immune suppressors [383].
9. Conclusions and Perspectives
Cancer vaccines have emerged as an important breakthrough in solid cancer immunotherapy, supported by their safety and promising clinical potential. Despite being employed in most clinical trials, a MoDC-based conventional DC vaccine regimen has shown limited efficacies. In this review, several novel vaccine approaches that are capable of addressing the limitations of conventional DC vaccine have been discussed. The biomaterial-based and ICD-inducing DC vaccines have unique advantages to recruit and activate endogenous DCs, which may be combined to achieve synergistic effects to spatiotemporally provide immunoadjuvants and antigens in situ. In addition, in vitro generated adjuvant and antigen mRNAs are loaded onto DCs to enhance DC immunogenicity. On top of cell-based cancer vaccines, DC- and tumor-derived sEVs are also found to have comparable, if not greater, immunogenicity and induce promising antitumor responses, although further understanding of sEVs is needed. In addition to the discussed novel DC-based therapeutic approaches, combinations with other immunotherapies, including ICBs, recapitulation of heterogenous DC populations, DC maturation with various adjuvants, and different modes of vaccine delivery should also be considered. Despite significant advantages of the selected novel DC vaccines over the conventional DC vaccine, their clinical implications may require further improvements. Multimodal therapies are being robustly investigated to address some of the limitations of monotherapies in Table 1. For instance, limitations of ICD-inducing DC vaccines have been overcome by combinatory approaches with other ICD-inducing and biomaterial-based DC vaccines, as well as ICBs. In addition, biomimetic nanoparticles that can reverse the local immunosuppressive TME have been reported to exert synergistic antitumor responses with the new generations of DC vaccines [390,391,392]. Furthermore, recent advances in next generation sequencing-based patient-specific mutanome mapping have allowed for the identification of the entirety of somatic cancer mutations [393]. Such technology development and further understanding of DC immunogenicity are envisioned to further alleviate immune escape and induce effective antitumor immune responses, which are being investigated in several clinical trials with high frequency of T cell responses reported (Phase I, NCT02035956, NCT03289962, NCT02316457) [394,395,396]. Although much work remains to be done to achieve optimal universal or personalized DC-based immunotherapy, recent significant advances in novel DC vaccine regimen and upcoming clinical trials are expected to encourage therapeutic implementations of DC-based vaccines in the future.
Author Contributions
Conceptualization, writing—original draft preparation, K.-W.L. and X.M.; writing—review and editing, X.M. and J.W.P.Y.; K.-W.L. created the figure with BioRender.com (accessed on 27 June 2023). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by RGC Theme-based Research Scheme [Grant no. T12-716/22-R].
Acknowledgments
This project was supported by the Young Researcher Support Scheme of State Key Laboratory of Liver Research (The University of Hong Kong) [Project no. SKLLR/YRSS/2022-01].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic Immunity in Cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving Cancer Immunotherapy Using Nanomedicines: Progress, Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Phuengkham, H.; Ren, L.; Shin, I.W.; Lim, Y.T. Nanoengineered Immune Niches for Reprogramming the Immunosuppressive Tumor Microenvironment and Enhancing Cancer Immunotherapy. Adv. Mater. 2019, 31, 1803322. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic Cell Subsets in Cancer Immunity and Tumor Antigen Sensing. Cell Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer Vaccines: The next Immunotherapy Frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Prakash, M.D.; Cox, M.; Wilson, K.; Boer, J.C.; Cauchi, J.A.; Plebanski, M. Therapeutic Cancer Vaccines—T Cell Responses and Epigenetic Modulation. Front. Immunol. 2019, 9, 3109. [Google Scholar] [CrossRef]
- Appleton, E.; Hassan, J.; Chan Wah Hak, C.; Sivamanoharan, N.; Wilkins, A.; Samson, A.; Ono, M.; Harrington, K.J.; Melcher, A.; Wennerberg, E. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations with Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 754436. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, E.; Planès, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef]
- Kumbhari, A.; Kim, P.S.; Lee, P.P. Optimisation of Anti-Cancer Peptide Vaccines to Preferentially Elicit High-Avidity T Cells. J. Theor. Biol. 2020, 486, 110067. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer Immunotherapy via Dendritic Cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic Cell-Based Immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Balint, K.; Boudousquie, C.; Gannon, P.O.; Kandalaft, L.E. Personalized Dendritic Cell Vaccines-Recent Breakthroughs and Encouraging Clinical Results. Front. Immunol. 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic Cell Subsets in T Cell Programming: Location Dictates Function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)Fining the Dendritic Cell Lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M.; Merad, M. Expanding Dendritic Cell Nomenclature in the Single-Cell Era. Nat. Rev. Immunol. 2022, 22, 67–68. [Google Scholar] [CrossRef]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallée, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef]
- Marmonti, E.; Oliva-Ramirez, J.; Haymaker, C. Dendritic Cells: The Long and Evolving Road towards Successful Targetability in Cancer. Cells 2022, 11, 3028. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A Conserved Dendritic-Cell Regulatory Program Limits Antitumour Immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Decisions About Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.M.; Gallucci, S. The Dendritic Cell Response to Classic, Emerging, and Homeostatic Danger Signals. Implications for Autoimmunity. Front. Immunol. 2013, 4, 138. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Oliveira, M.M.S.; D’Aulerio, R.; Yong, T.; He, M.; Baptista, M.A.P.; Nylén, S.; Westerberg, L.S. Increased Cross-Presentation by Dendritic Cells and Enhanced Anti-Tumour Therapy Using the Arp2/3 Inhibitor CK666. Br. J. Cancer 2023, 128, 982–991. [Google Scholar] [CrossRef]
- Kurts, C.; Robinson, B.W.S.; Knolle, P.A. Cross-Priming in Health and Disease. Nat. Rev. Immunol. 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis e Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and Memory T-Cell Differentiation: Implications for Vaccine Development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory Cytokines as a Third Signal for T Cell Activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-Presentation by Dendritic Cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.-S.; Jiang, A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines 2020, 8, 706. [Google Scholar] [CrossRef]
- Calmeiro, J.; Carrascal, M.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Biomaterial-Based Platforms for in Situ Dendritic Cell Programming and Their Use in Antitumor Immunotherapy. J. Immunother. Cancer 2019, 7, 238. [Google Scholar] [CrossRef]
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor Dendritic Cell–Based Vaccines: Lessons from 20 Years of Clinical Trials and Future Perspectives. Transl. Res. 2016, 168, 74–95. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells Is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Ghorbaninezhad, F.; Asadzadeh, Z.; Masoumi, J.; Mokhtarzadeh, A.; Kazemi, T.; Aghebati-Maleki, L.; Shotorbani, S.S.; Shadbad, M.A.; Baghbanzadeh, A.; Hemmat, N.; et al. Dendritic Cell-Based Cancer Immunotherapy in the Era of Immune Checkpoint Inhibitors: From Bench to Bedside. Life Sci. 2022, 297, 120466. [Google Scholar] [CrossRef]
- Massa, C.; Thomas, C.; Wang, E.; Marincola, F.; Seliger, B. Different Maturation Cocktails Provide Dendritic Cells with Different Chemoattractive Properties. J. Transl. Med. 2015, 13, 175. [Google Scholar] [CrossRef]
- Ratzinger, G.; Baggers, J.; de Cos, M.A.; Yuan, J.; Dao, T.; Reagan, J.L.; Münz, C.; Heller, G.; Young, J.W. Mature Human Langerhans Cells Derived from CD34+ Hematopoietic Progenitors Stimulate Greater Cytolytic T Lymphocyte Activity in the Absence of Bioactive IL-12p70, by Either Single Peptide Presentation or Cross-Priming, Than Do Dermal-Interstitial or Monocyte-Derived Dendritic Cells. J. Immunol. 2004, 173, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rossi, M.; Ratzinger, G.; De Cos, M.A.; Chung, D.J.; Panageas, K.S.; Wolchock, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-Loaded Langerhans Cells, despite Increased IL15 Secretion and T-Cell Activation in Vitro, Elicit Antitumor T-Cell Responses Comparable to Peptide-Loaded Monocyte-Derived Dendritic Cells In Vivo. Clin. Cancer Res. 2011, 17, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.J.; Carvajal, R.D.; Postow, M.A.; Sharma, S.; Pronschinske, K.B.; Shyer, J.A.; Singh-Kandah, S.; Dickson, M.A.; D’Angelo, S.P.; Wolchok, J.D.; et al. Langerhans-Type Dendritic Cells Electroporated with TRP-2 MRNA Stimulate Cellular Immunity against Melanoma: Results of a Phase I Vaccine Trial. Oncoimmunology 2018, 7, e1372081. [Google Scholar] [CrossRef] [PubMed]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated Data from 2 Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trials of Active Cellular Immunotherapy with Sipuleucel-T in Advanced Prostate Cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in Prostate Cancer: The First FDA-Approved Therapeutic Cancer Vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Scholz, M.; Yep, S.; Chancey, M.; Kelly, C.; Chau, K.; Turner, J.; Lam, R.; Drake, C. Phase I Clinical Trial of Sipuleucel-T Combined with Escalating Doses of Ipilimumab in Progressive Metastatic Castrate-Resistant Prostate Cancer. Immunotargets Ther. 2017, 6, 11–16. [Google Scholar] [CrossRef]
- Sinha, M.; Zhang, L.; Subudhi, S.; Chen, B.; Marquez, J.; Liu, E.V.; Allaire, K.; Cheung, A.; Ng, S.; Nguyen, C.; et al. Pre-Existing Immune Status Associated with Response to Combination of Sipuleucel-T and Ipilimumab in Patients with Metastatic Castration-Resistant Prostate Cancer. J. Immunother. Cancer 2021, 9, e002254. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Morishima, C.; Szmulewitz, R.; Harshman, L.; Appleman, L.; Monk, P.; Bitting, R.L.; Kucuk, O.; Millard, F.; Seigne, J.D.; et al. IL-7 Expands Lymphocyte Populations and Enhances Immune Responses to Sipuleucel-T in Patients with Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Immunother. Cancer 2021, 9, e002903. [Google Scholar] [CrossRef]
- Gilboa, E. DC-Based Cancer Vaccines. J. Clin. Investig. 2007, 117, 1195–1203. [Google Scholar] [CrossRef]
- Kastenmüller, W.; Kastenmüller, K.; Kurts, C.; Seder, R.A. Dendritic Cell-Targeted Vaccines—Hope or Hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Benitez-Ribas, D.; Hoosemans, S.; Cambi, A.; Adema, G.J.; Figdor, C.G.; Tacken, P.J.; de Vries, I.J.M. DEC-205 Mediates Antigen Uptake and Presentation by Both Resting and Activated Human Plasmacytoid Dendritic Cells. Eur. J. Immunol. 2011, 41, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.-N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting Antigen to Mouse Dendritic Cells via Clec9A Induces Potent CD4 T Cell Responses Biased toward a Follicular Helper Phenotype. J. Immunol. 2011, 187, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Maraskovsky, E.; Heath, W.R. Targeting Dendritic Cells in Vivo for Cancer Therapy. Front. Immunol. 2012, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient Targeting of Protein Antigen to the Dendritic Cell Receptor DEC-205 in the Steady State Leads to Antigen Presentation on Major Histocompatibility Complex Class I Products and Peripheral CD8+ T Cell Tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Fu, C.; Ma, T.; Zhou, L.; Mi, Q.S.; Jiang, A. Dendritic Cell-Based Vaccines Against Cancer: Challenges, Advances and Future Opportunities. Immunol. Investig. 2022, 51, 2133–2158. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting Edge: Autocrine TGF-β Sustains Default Tolerogenesis by IDO-Competent Dendritic Cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T Cells, Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ Dendritic Cells Derived In Vitro from CD34+ Progenitors Closely Resemble Blood Dendritic Cells, Including Their Adjuvant Responsiveness, Contrary to Monocyte-Derived Dendritic Cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The Tumour Microenvironment Harbours Ontogenically Distinct Dendritic Cell Populations with Opposing Effects on Tumour Immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- Kleindienst, P.; Brocker, T. Endogenous Dendritic Cells Are Required for Amplification of T Cell Responses Induced by Dendritic Cell Vaccines In Vivo. J. Immunol. 2003, 170, 2817–2823. [Google Scholar] [CrossRef]
- Martín-Fontecha, A.; Sebastiani, S.; Höpken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of Dendritic Cell Migration to the Draining Lymph Node: Impact on T Lymphocyte Traffic and Priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Verdijk, P.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Boullart, A.C.I.; Kok, E.; Van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.M.; et al. Limited Amounts of Dendritic Cells Migrate into Thet-Cell Area of Lymph Nodes but Have High Immune Activating Potential in Melanoma Patients. Clin. Cancer Res. 2009, 15, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Adema, G.J.; De Vries, I.J.M.; Punt, C.J.A.; Figdor, C.G. Migration of Dendritic Cell Based Cancer Vaccines: In Vivo Veritas? Curr. Opin. Immunol. 2005, 17, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond CDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking Dendritic Cells into Medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Cai, L.; Xu, J.; Yang, Z.; Tong, R.; Dong, Z.; Wang, C.; Leong, K.W. Engineered Biomaterials for Cancer Immunotherapy. MedComm 2020, 1, 35–46. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine Delivery: A Matter of Size, Geometry, Kinetics and Molecular Patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Johansen, P.; Storni, T.; Rettig, L.; Qiu, Z.; Der-Sarkissian, A.; Smith, K.A.; Manolova, V.; Lang, K.S.; Senti, G.; Müllhaupt, B.; et al. Antigen Kinetics Determines Immune Reactivity. Proc. Natl. Acad. Sci. USA 2008, 105, 5189–5194. [Google Scholar] [CrossRef]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions In Vivo. J. Exp. Med. 2001, 194, 769–780. [Google Scholar] [CrossRef]
- Ali, O.A.; Huebsch, N.; Cao, L.; Dranoff, G.; Mooney, D.J. Infection-Mimicking Materials to Program Dendritic Cells in Situ. Nat. Mater. 2009, 8, 151–158. [Google Scholar] [CrossRef]
- Gu, L.; Mooney, D.J. Biomaterials and Emerging Anticancer Therapeutics: Engineering the Microenvironment. Nat. Rev. Cancer 2016, 16, 56–66. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An Overview of Poly(Lactic-Co-Glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of Dendritic Cell Development by GM-CSF: Molecular Control and Implications for Immune Homeostasis and Therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; Doherty, E.; Mooney, D.J.; Emerich, D. Relationship of Vaccine Efficacy to the Kinetics of DC and T-Cell Responses Induced by PLG-Based Cancer Vaccines. Biomatter 2011, 1, 66–75. [Google Scholar] [CrossRef]
- Ali, O.A.; Emerich, D.; Dranoff, G.; Mooney, D.J. In Situ Regulation of DC Subsets and T Cells Mediates Tumor Regression in Mice. Sci. Transl. Med. 2009, 1, 8ra19. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Murphy, K.M. Dendritic Cell Regulation of TH1-TH2 Development. Nat. Immunol. 2000, 1, 199–205. [Google Scholar] [CrossRef]
- den Haan, J.M.M.; Lehar, S.M.; Bevan, M.J. CD8+ but Not CD8− Dendritic Cells Cross-Prime Cytotoxic T Cells in Vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef]
- Ali, O.A.; Verbeke, C.; Johnson, C.; Sands, R.W.; Lewin, S.A.; White, D.; Doherty, E.; Dranoff, G.; Mooney, D.J. Identification of Immune Factors Regulating Antitumor Immunity Using Polymeric Vaccines with Multiple Adjuvants. Cancer Res. 2014, 74, 1670–1681. [Google Scholar] [CrossRef]
- Shortman, K.; Heath, W.R. The CD8+ Dendritic Cell Subset. Immunol. Rev. 2010, 234, 18–31. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 Blockade and GM-CSF Combination Immunotherapy Alters the Intratumor Balance of Effector and Regulatory T Cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Ali, O.A.; Lewin, S.A.; Dranoff, G.; Mooney, D.J. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-Cell Activity and Tumor Eradication. Cancer Immunol. Res. 2016, 4, 95–100. [Google Scholar] [CrossRef]
- Bencherif, S.A.; Sands, R.W.; Bhatta, D.; Arany, P.; Verbeke, C.S.; Edwards, D.A.; Mooney, D.J. Injectable Preformed Scaffolds with Shape-Memory Properties. Proc. Natl. Acad. Sci. USA 2012, 109, 19590–19595. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Qin, H.; Fernandez, I.; Wei, J.; Lin, J.; Kwak, L.W.; Roy, K. An Injectable Synthetic Immune-Priming Center Mediates Efficient T-Cell Class Switching and T-Helper 1 Response against B Cell Lymphoma. J. Control. Release 2011, 155, 184–192. [Google Scholar] [CrossRef]
- Shah, N.J.; Najibi, A.J.; Shih, T.Y.; Mao, A.S.; Sharda, A.; Scadden, D.T.; Mooney, D.J. A Biomaterial-Based Vaccine Eliciting Durable Tumour-Specific Responses against Acute Myeloid Leukaemia. Nat. Biomed. Eng. 2020, 4, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.M.A.; Ladewig, K.; Haylock, D.N.; McLean, K.M.; O’Connor, A.J. Cryogels for Biomedical Applications. J. Mater. Chem. B 2013, 1, 2682. [Google Scholar] [CrossRef]
- Hwang, Y.; Zhang, C.; Varghese, S. Poly(Ethylene Glycol) Cryogels as Potential Cell Scaffolds: Effect of Polymerization Conditions on Cryogel Microstructure and Properties. J. Mater. Chem. 2010, 20, 345–351. [Google Scholar] [CrossRef]
- Koshy, S.T.; Ferrante, T.C.; Lewin, S.A.; Mooney, D.J. Injectable, Porous, and Cell-Responsive Gelatin Cryogels. Biomaterials 2014, 35, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.Y.; Blacklow, S.O.; Li, A.W.; Freedman, B.R.; Bencherif, S.; Koshy, S.T.; Darnell, M.C.; Mooney, D.J. Injectable, Tough Alginate Cryogels as Cancer Vaccines. Adv. Healthc. Mater. 2018, 7, e1701469. [Google Scholar] [CrossRef] [PubMed]
- Augst, A.D.; Kong, H.J.; Mooney, D.J. Alginate Hydrogels as Biomaterials. Macromol. Biosci. 2006, 6, 623–633. [Google Scholar] [CrossRef]
- Najibi, A.J.; Shih, T.-Y.; Mooney, D.J. Cryogel Vaccines Effectively Induce Immune Responses Independent of Proximity to the Draining Lymph Nodes. Biomaterials 2022, 281, 121329. [Google Scholar] [CrossRef]
- Dellacherie, M.O.; Seo, B.R.; Mooney, D.J. Macroscale Biomaterials Strategies for Local Immunomodulation. Nat. Rev. Mater. 2019, 4, 379–397. [Google Scholar] [CrossRef]
- Adu-Berchie, K.; Mooney, D.J. Biomaterials as Local Niches for Immunomodulation. Acc. Chem. Res. 2020, 53, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Kukutsch, N.A.; Roßner, S.; Austyn, J.M.; Schuler, G.; Lutz, M.B. Formation and Kinetics of MHC Class I-Ovalbumin Peptide Complexes on Immature and Mature Murine Dendritic Cells. J. Investig. Dermatol. 2000, 115, 449–453. [Google Scholar] [CrossRef]
- Hori, Y.; Winans, A.M.; Huang, C.C.; Horrigan, E.M.; Irvine, D.J. Injectable Dendritic Cell-Carrying Alginate Gels for Immunization and Immunotherapy. Biomaterials 2008, 29, 3671–3682. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, S.A.; Sands, R.W.; Ali, O.A.; Li, W.A.; Lewin, S.A.; Braschler, T.M.; Shih, T.Y.; Verbeke, C.S.; Bhatta, D.; Dranoff, G.; et al. Injectable Cryogel-Based Whole-Cell Cancer Vaccines. Nat. Commun. 2015, 6, 7556. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial Watch: Dendritic Cell-Based Interventions for Cancer Therapy. Oncoimmunology 2012, 1, 1111–1134. [Google Scholar] [CrossRef]
- Wang, H.; Mooney, D.J. Biomaterial-Assisted Targeted Modulation of Immune Cells in Cancer Treatment. Nat. Mater. 2018, 17, 761–772. [Google Scholar] [CrossRef]
- Wang, C.; Ye, Y.; Hu, Q.; Bellotti, A.; Gu, Z. Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv. Mater. 2017, 29, 1606036. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Archibong, E.; Chen, Q.; Ruan, H.; Ahn, S.; Dukhovlinova, E.; Kang, Y.; Wen, D.; Dotti, G.; et al. Inhibition of Post-Surgery Tumour Recurrence via a Hydrogel Releasing CAR-T Cells and Anti-PDL1-Conjugated Platelets. Nat. Biomed. Eng. 2021, 5, 1038–1047. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, Y.-Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic Reprogramming of Terminally Exhausted CD8+ T Cells by IL-10 Enhances Anti-Tumor Immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef]
- Weber, J.S.; Mulé, J.J. Cancer Immunotherapy Meets Biomaterials. Nat. Biotechnol. 2015, 33, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Bhatta, R.; Liu, Y.; Bo, Y.; Wang, H. In Situ Dendritic Cell Recruitment and T Cell Activation for Cancer Immunotherapy. Front. Pharmacol. 2022, 13, 954955. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Wang, B.; Chen, J.; Caserto, J.S.; Wang, X.; Ma, M. An In Situ Hydrogel-Mediated Chemo-Immunometabolic Cancer Therapy. Nat. Commun. 2022, 13, 3821. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, C.; Li, J.; Cui, D.; Jiang, Y.; Pu, K. Activatable Polymer Nanoenzymes for Photodynamic Immunometabolic Cancer Therapy. Adv. Mater. 2021, 33, 2007247. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.-C.; et al. Reversal of Indoleamine 2,3-Dioxygenase–Mediated Cancer Immune Suppression by Systemic Kynurenine Depletion with a Therapeutic Enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Wang, H.; Najibi, A.J.; Sobral, M.C.; Seo, B.R.; Lee, J.Y.; Wu, D.; Li, A.W.; Verbeke, C.S.; Mooney, D.J. Biomaterial-Based Scaffold for in Situ Chemo-Immunotherapy to Treat Poorly Immunogenic Tumors. Nat. Commun. 2020, 11, 5696. [Google Scholar] [CrossRef] [PubMed]
- Li, A.W.; Sobral, M.C.; Badrinath, S.; Choi, Y.; Graveline, A.; Stafford, A.G.; Weaver, J.C.; Dellacherie, M.O.; Shih, T.-Y.; Ali, O.A.; et al. A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat. Mater. 2018, 17, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Dellacherie, M.O.; Li, A.; Lu, B.Y.; Verbeke, C.S.; Gu, L.; Stafford, A.G.; Doherty, E.J.; Mooney, D.J. Single-Shot Mesoporous Silica Rods Scaffold for Induction of Humoral Responses Against Small Antigens. Adv. Funct. Mater. 2020, 30, 2002448. [Google Scholar] [CrossRef]
- Kang, B.H.; Lee, H.K. Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors. Int. J. Mol. Sci. 2022, 23, 7325. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Hett, E.; Kroemer, G.; Marincola, F.M. Immunogenic Cell Death in Cancer: Concept and Therapeutic Implications. J. Transl. Med. 2023, 21, 162. [Google Scholar] [CrossRef]
- Jin, M.Z.; Wang, X.P. Immunogenic Cell Death-Based Cancer Vaccines. Front. Immunol. 2021, 12, 697964. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhu, G.; Wang, S.; Yu, G.; Yang, Z.; Lin, L.; Zhou, Z.; Liu, Y.; Dai, Y.; Zhang, F.; et al. In Situ Dendritic Cell Vaccine for Effective Cancer Immunotherapy. ACS Nano 2019, 13, 3083–3094. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising Efficacy and Reducing Toxicity of Anticancer Radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Morimoto, Y.; Tsujimoto, H.; Arake, M.; Harada, M.; Saitoh, D.; Hara, I.; Ozeki, E.; Satoh, A.; Takayama, E.; et al. Highly Reliable, Targeted Photothermal Cancer Therapy Combined with Thermal Dosimetry Using a near-Infrared Absorbent. Sci. Rep. 2020, 10, 9765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Wang, X.; Guan, X.; Zhang, W.; Ma, J. Recent Advances in Selective Photothermal Therapy of Tumor. J. Nanobiotechnol. 2021, 19, 335. [Google Scholar] [CrossRef]
- Bloy, N.; Garcia, P.; Laumont, C.M.; Pitt, J.M.; Sistigu, A.; Stoll, G.; Yamazaki, T.; Bonneil, E.; Buqué, A.; Humeau, J.; et al. Immunogenic Stress and Death of Cancer Cells: Contribution of Antigenicity vs Adjuvanticity to Immunosurveillance. Immunol. Rev. 2017, 280, 165–174. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding Cell Death Signals in Inflammation and Immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting Immunogenic Cancer Cell Death by Photodynamic Therapy: Past, Present and Future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.-P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying Cells Actively Regulate Adaptive Immune Responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Moserova, I.; Urbanova, L.; Bezu, L.; Kepp, O.; Cremer, I.; Salek, C.; Strnad, P.; Kroemer, G.; Galluzzi, L.; et al. Prognostic and Predictive Value of DAMPs and DAMP-Associated Processes in Cancer. Front. Immunol. 2015, 6, 402. [Google Scholar] [CrossRef]
- Hayashi, K.; Nikolos, F.; Lee, Y.C.; Jain, A.; Tsouko, E.; Gao, H.; Kasabyan, A.; Leung, H.E.; Osipov, A.; Jung, S.Y.; et al. Tipping the Immunostimulatory and Inhibitory DAMP Balance to Harness Immunogenic Cell Death. Nat. Commun. 2020, 11, 6299. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Vedunova, M.; Turubanova, V.; Vershinina, O.; Savyuk, M.; Efimova, I.; Mishchenko, T.; Raedt, R.; Vral, A.; Vanhove, C.; Korsakova, D.; et al. DC Vaccines Loaded with Glioma Cells Killed by Photodynamic Therapy Induce Th17 Anti-Tumor Immunity and Provide a Four-Gene Signature for Glioma Prognosis. Cell Death Dis. 2022, 13, 1062. [Google Scholar] [CrossRef]
- Wang, X.; Ji, J.; Zhang, H.; Fan, Z.; Zhang, L.; Shi, L.; Zhou, F.; Chen, W.R.; Wang, H.; Wang, X. Stimulation of Dendritic Cells by DAMPs in ALA-PDT Treated SCC Tumor Cells. Oncotarget 2015, 6, 44688–44702. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic Cell Stress and Death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin Promotes Immunity and Type I Interferon-Dependent Survival in Mice with Acute Myeloid Leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.-L.; et al. Molecular Mechanisms of ATP Secretion during Immunogenic Cell Death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and Extracellular Functions of Heat Shock Proteins: Repercussions in Cancer Therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human Tumor Cells Killed by Anthracyclines Induce a Tumor-Specific Immune Response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Saenz, R.; Futalan, D.; Leutenez, L.; Eekhout, F.; Fecteau, J.F.; Sundelius, S.; Sundqvist, S.; Larsson, M.; Hayashi, T.; Minev, B.; et al. TLR4-Dependent Activation of Dendritic Cells by an HMGB1-Derived Peptide Adjuvant. J. Transl. Med. 2014, 12, 211. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and Tolerogenic Cell Death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The Abscopal Effect of Local Radiotherapy: Using Immunotherapy to Make a Rare Event Clinically Relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic Effects of Local Radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient Regulatory T Cell Ablation Deters Oncogene-Driven Breast Cancer and Enhances Radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef]
- Haroun, R.; Naasri, S.; Oweida, A.J. Toll-Like Receptors and the Response to Radiotherapy in Solid Tumors: Challenges and Opportunities. Vaccines 2023, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic Clinical Tumor Regressions and Potentiation of PD1 Blockade with in Situ Vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Kajihara, R.; Yokoi, T.; Repasky, E.A.; Ito, F. Neoadjuvant in Situ Immunomodulation Enhances Systemic Antitumor Immunity against Highly Metastatic Tumors. Cancer Res. 2021, 81, 6183–6195. [Google Scholar] [CrossRef]
- Marron, T.; Fasano, J.; Doroshow, D.; Ostrowski, D.; Sorich, J.; Van-Voorthuysen, M.; Coffey, J.; Moshier, E.; Salazar, A.; Yellin, M.; et al. Flt3L-Primed in Situ Vaccination and Pembrolizumab Induce Systemic Tumor Regressions of Bulky Tumors in Patients with Lymphomas and ER/PR+ Breast Cancer. In Regular and Young Investigator Award Abstracts; BMJ Publishing Group Ltd.: London, UK, 2022; Volume 25, p. A622. [Google Scholar]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.L.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Long, S.R.; Hoppe, R.T.; Janssen, R.; Candia, A.F.; et al. In Situ Vaccination with a TLR9 Agonist and Local Low-Dose Radiation Induces Systemic Responses in Untreated Indolent Lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In Situ Vaccination with a TLR9 Agonist Induces Systemic Lymphoma Regression: A Phase I/II Study. J. Clin. Oncol. 2010, 28, 4324–4332. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus Placebo after Radiotherapy in Patients with Metastatic Castration-Resistant Prostate Cancer That Had Progressed after Docetaxel Chemotherapy (CA184-043): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab Alone or in Combination with Radiotherapy in Metastatic Castration-Resistant Prostate Cancer: Results from an Open-Label, Multicenter Phase I/II Study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic Therapy for Cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Niedre, M.J.; Yu, C.S.; Patterson, M.S.; Wilson, B.C. Singlet Oxygen Luminescence as an in Vivo Photodynamic Therapy Dose Metric: Validation in Normal Mouse Skin with Topical Amino-Levulinic Acid. Br. J. Cancer 2005, 92, 298–304. [Google Scholar] [CrossRef]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic Cell Death Induced by a New Photodynamic Therapy Based on Photosens and Photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The Two Faces of IL-6 in the Tumor Microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising Targets for Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, S.R.; Nave, H.; Korangy, F.; Schlote, K.; Pabst, R.; Jaffee, E.M.; Manns, M.P.; Greten, T.F. Apoptotic, but Not Necrotic, Tumor Cell Vaccines Induce a Potent Immune Response In Vivo. Int. J. Cancer 2003, 103, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gamrekelashvili, J.; Krüger, C.; von Wasielewski, R.; Hoffmann, M.; Huster, K.M.; Busch, D.H.; Manns, M.P.; Korangy, F.; Greten, T.F. Necrotic Tumor Cell Death In Vivo Impairs Tumor-Specific Immune Responses. J. Immunol. 2007, 178, 1573–1580. [Google Scholar] [CrossRef]
- Bartholomae, W.C.; Rininsland, F.H.; Eisenberg, J.C.; Boehm, B.O.; Lehmann, P.V.; Tary-Lehmann, M. T Cell Immunity Induced by Live, Necrotic, and Apoptotic Tumor Cells 1. J. Immunol. 2004, 173, 1012–1022. [Google Scholar] [CrossRef]
- Gamrekelashvili, J.; Greten, T.F.; Korangy, F. Immunogenicity of Necrotic Cell Death. Cell. Mol. Life Sci. 2015, 72, 273–283. [Google Scholar] [CrossRef]
- Harari, A.; Graciotti, M.; Bassani-Sternberg, M.; Kandalaft, L.E. Antitumour Dendritic Cell Vaccination in a Priming and Boosting Approach. Nat. Rev. Drug Discov. 2020, 19, 635–652. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X.; et al. A STING-Activating Nanovaccine for Cancer Immunotherapy. Nat. Nanotechnol. 2017, 12, 648–654. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef]
- Zakharova, M.; Ziegler, H.K. Paradoxical Anti-Inflammatory Actions of TNF-α: Inhibition of IL-12 and IL-23 via TNF Receptor 1 in Macrophages and Dendritic Cells. J. Immunol. 2005, 175, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Choi, K.Y. Advances in Nanomaterial-mediated Photothermal Cancer Therapies: Toward Clinical Applications. Biomedicines 2021, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, C.; Cui, W.; Gong, H.; Liang, C.; Shi, X.; Li, Z.; Sun, B.; Liu, Z. Combined Photothermal and Photodynamic Therapy Delivered by PEGylated MoS 2 Nanosheets. Nanoscale 2014, 6, 11219–11225. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xiao, Y.; Li, W.; Yang, Q.; Tan, L.; Jia, Y.; Qu, Y.; Qian, Z. Photosensitizer Micelles Together with IDO Inhibitor Enhance Cancer Photothermal Therapy and Immunotherapy. Adv. Sci. 2018, 5, 1700891. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, S.; Zhang, H.; Han, Y.; Liu, H.; Ren, F.; Sun, Q.; Li, Z.; Gao, M. Boosting the Radiosensitizing and Photothermal Performance of Cu 2– x Se Nanocrystals for Synergetic Radiophotothermal Therapy of Orthotopic Breast Cancer. ACS Nano 2019, 13, 1342–1353. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Ochyl, L.J.; Kuai, R.; Schwendeman, A.; Moon, J.J. Chemo-Photothermal Therapy Combination Elicits Anti-Tumor Immunity against Advanced Metastatic Cancer. Nat. Commun. 2018, 9, 1074. [Google Scholar] [CrossRef]
- Ng, C.W.; Li, J.; Pu, K. Recent Progresses in Phototherapy-Synergized Cancer Immunotherapy. Adv. Funct. Mater. 2018, 28, 1804688. [Google Scholar] [CrossRef]
- Wang, C.; Xu, L.; Liang, C.; Xiang, J.; Peng, R.; Liu, Z. Immunological Responses Triggered by Photothermal Therapy with Carbon Nanotubes in Combination with Anti-CTLA-4 Therapy to Inhibit Cancer Metastasis. Adv. Mater. 2014, 26, 8154–8162. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, J.; Zhang, Y.; Liu, M.; Lang, M.L.; Li, M.; Chen, W.R. Local Phototherapy Synergizes with Immunoadjuvant for Treatment of Pancreatic Cancer through Induced Immunogenic Tumor Vaccine. Clin. Cancer Res. 2018, 24, 5335–5346. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal Therapy with Immune-Adjuvant Nanoparticles Together with Checkpoint Blockade for Effective Cancer Immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical Development and Potential of Photothermal and Photodynamic Therapies for Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex Vivo Pulsed Dendritic Cell Vaccination against Cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The Clinical Application of Cancer Immunotherapy Based on Naturally Circulating Dendritic Cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Erdmann, M.; Haendle, I.; Voland, S.; Berger, T.; Schultz, E.; Strasser, E.; Dankerl, P.; Janka, R.; Schliep, S.; et al. Twelve-Year Survival and Immune Correlates in Dendritic Cell–Vaccinated Melanoma Patients. JCI Insight 2017, 2, e91438. [Google Scholar] [CrossRef]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. MRNA-Based Dendritic Cell Vaccines. Expert. Rev. Vaccines 2014, 14, 161–176. [Google Scholar] [CrossRef]
- Aarntzen, E.H.J.G.; Schreibelt, G.; Bol, K.; Lesterhuis, W.J.; Croockewit, A.J.; De Wilt, J.H.W.; Van Rossum, M.M.; Blokx, W.A.M.; Jacobs, J.F.M.; Duiveman-De Boer, T.; et al. Vaccination with MRNA-Electroporated Dendritic Cells Induces Robust Tumor Antigen-Specific CD4+ and CD8+ T Cells Responses in Stage III and IV Melanoma Patients. Clin. Cancer Res. 2012, 18, 5460–5470. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. MRNA Therapeutics in Cancer Immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Van Lint, S.; Heirman, C.; Thielemans, K.; Breckpot, K. mRNA: From a Chemical Blueprint for Protein Production to an Off-the-Shelf Therapeutic. Hum. Vaccines Immunother. 2013, 9, 265–274. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-ΚB by Toll-like Receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA Toolbox for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic Cells Pulsed with RNA Are Potent Antigen-Presenting Cells in Vitro and in Vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef]
- Müller, M.R.; Tsakou, G.; Grünebach, F.; Schmidt, S.M.; Brossart, P. Induction of Chronic Lymphocytic Leukemia (CLL)-Specific CD4- and CD8-Mediated T-Cell Responses Using RNA-Transfected Dendritic Cells. Blood 2004, 103, 1763–1769. [Google Scholar] [CrossRef]
- Milazzo, C.; Reichardt, V.L.; Müller, M.R.; Grünebach, F.; Brossart, P. Induction of Myeloma-Specific Cytotoxic T Cells Using Dendritic Cells Transfected with Tumor-Derived RNA. Blood 2003, 101, 977–982. [Google Scholar] [CrossRef]
- Nencioni, A.; Müller, M.R.; Grünebach, F.; Garuti, A.; Mingari, M.C.; Patrone, F.; Ballestrero, A.; Brossart, P. Dendritic Cells Transfected with Tumor RNA for the Induction of Antitumor CTL in Colorectal Cancer. Cancer Gene Ther. 2003, 10, 209–214. [Google Scholar] [CrossRef]
- Nair, S.K.; Morse, M.; Boczkowski, D.; Ian Cumming, R.; Vasovic, L.; Gilboa, E.; Kim Lyerly, H. Induction of Tumor-Specific Cytotoxic T Lymphocytes in Cancer Patients by Autologous Tumor RNA-Transfected Dendritic Cells. Ann. Surg. 2002, 235, 540–549. [Google Scholar] [CrossRef]
- Gilboa, E.; Vieweg, J. Cancer Immunotherapy with MRNA-Transfected Dendritic Cells. Immunol. Rev. 2004, 199, 251–263. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.I.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly Efficient Gene Delivery by MRNA Electroporation in Human Hematopoietic Cells: Superiority to Lipofection and Passive Pulsing of MRNA and to Electroporation of Plasmid CDNA for Tumor Antigen Loading of Dendritic Cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Van Nuffel, A.M.; Corthals, J.; Neyns, B.; Heirman, C.; Thielemans, K.; Bonehill, A. Immunotherapy of Cancer with Dendritic Cells Loaded with Tumor Antigens and Activated through MRNA Electroporation. Methods Mol. Biol. 2010, 629, 405–452. [Google Scholar] [CrossRef] [PubMed]
- Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. Electroporation of Mrna as Universal Technology Platform to Transfect a Variety of Primary Cells with Antigens and Functional Proteins. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1499, pp. 165–178. [Google Scholar]
- Markovic, S.N.; Dietz, A.B.; Greiner, C.W.; Maas, M.L.; Butler, G.W.; Padley, D.J.; Bulur, P.A.; Allred, J.B.; Creagan, E.T.; Ingle, J.N.; et al. Preparing Clinical-Grade Myeloid Dendritic Cells by Electroporation-Mediated Transfection of in Vitro Amplified Tumor-Derived MRNA and Safety Testing in Stage IV Malignant Melanoma. J. Transl. Med. 2006, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II Trial of Melanoma Therapy with Dendritic Cells Transfected with Autologous Tumor-MRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dannull, J.; Heiser, A.; Yancey, D.; Pruitt, S.; Madden, J.; Coleman, D.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Immunological and Clinical Responses in Metastatic Renal Cancer Patients Vaccinated with Tumor RNA-Transfected Dendritic Cells. Cancer Res. 2003, 63, 2127–2133. [Google Scholar]
- Kyte, J.A.; Aamdal, S.; Dueland, S.; Sæbøe-Larsen, S.; Inderberg, E.M.; Madsbu, U.E.; Skovlund, E.; Gaudernack, G.; Kvalheim, G. Immune Response and Long-Term Clinical Outcome in Advanced Melanoma Patients Vaccinated with Tumor-MRNA-Transfected Dendritic Cells. Oncoimmunology 2016, 5, e1232237. [Google Scholar] [CrossRef] [PubMed]
- Javorovic, M.; Pohla, H.; Frankenberger, B.; Wölfel, T.; Schendel, D.J. RNA Transfer by Electroporation into Mature Dendritic Cells Leading to Reactivation of Effector-Memory Cytotoxic T Lymphocytes: A Quantitative Analysis. Mol. Ther. 2005, 12, 734–743. [Google Scholar] [CrossRef]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic Cell Vaccination as Postremission Treatment to Prevent or Delay Relapse in Acute Myeloid Leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Kongsted, P.; Borch, T.H.; Ellebaek, E.; Iversen, T.Z.; Andersen, R.; Met, Ö.; Hansen, M.; Lindberg, H.; Sengeløv, L.; Svane, I.M. Dendritic Cell Vaccination in Combination with Docetaxel for Patients with Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase II Study. Cytotherapy 2017, 19, 500–513. [Google Scholar] [CrossRef]
- Bol, K.F.; Aarntzen, E.H.J.G.; in ’t Hout, F.E.M.; Schreibelt, G.; Creemers, J.H.A.; Lesterhuis, W.J.; Gerritsen, W.R.; Grunhagen, D.J.; Verhoef, C.; Punt, C.J.A.; et al. Favorable Overall Survival in Stage III Melanoma Patients after Adjuvant Dendritic Cell Vaccination. Oncoimmunology 2016, 5, e1057673. [Google Scholar] [CrossRef]
- Boudewijns, S.; Bloemendal, M.; de Haas, N.; Westdorp, H.; Bol, K.F.; Schreibelt, G.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Gorris, M.A.J.; Croockewit, A.; et al. Autologous Monocyte-Derived DC Vaccination Combined with Cisplatin in Stage III and IV Melanoma Patients: A Prospective, Randomized Phase 2 Trial. Cancer Immunol. Immunother. 2020, 69, 477–488. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, Ö.; Sahin, U. Modification of Antigen-Encoding RNA Increases Stability, Translational Efficacy, and T-Cell Stimulatory Capacity of Dendritic Cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Galaine, J.; Borg, C.; Godet, Y.; Adotévi, O. Interest of Tumor-Specific CD4 T Helper 1 Cells for Therapeutic Anticancer Vaccine. Vaccines 2015, 3, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The Multifaceted Role of CD4+ T Cells in CD8+ T Cell Memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ahrends, T.; Busselaar, J.; Severson, T.M.; Bąbała, N.; de Vries, E.; Bovens, A.; Wessels, L.; van Leeuwen, F.; Borst, J. CD4+ T Cell Help Creates Memory CD8+ T Cells with Innate and Help-Independent Recall Capacities. Nat. Commun. 2019, 10, 5531. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Vieweg, J.; Weizer, A.Z.; Dahm, P.; Yancey, D.; Turaga, V.; Higgins, J.; Boczkowski, D.; Gilboa, E.; Dannull, J. Enhanced Induction of Telomerase-Specific CD4+ T Cells Using Dendritic Cells Transfected with RNA Encoding a Chimeric Gene Product. Cancer Res. 2002, 62, 5041–5048. [Google Scholar]
- Benteyn, D.; Anguille, S.; Van Lint, S.; Heirman, C.; Van Nuffel, A.M.T.; Corthals, J.; Ochsenreither, S.; Waelput, W.; Van Beneden, K.; Breckpot, K.; et al. Design of an Optimized Wilms’ Tumor 1 (WT1) MRNA Construct for Enhanced WT1 Expression and Improved Immunogenicity in Vitro and in Vivo. Mol. Ther. Nucleic Acids 2013, 2, e134. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, Ö.; Sahin, U. Increased Antigen Presentation Efficiency by Coupling Antigens to MHC Class I Trafficking Signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; van der Bruggen, P.; Thielemans, K. Messenger RNA-Electroporated Dendritic Cells Presenting MAGE-A3 Simultaneously in HLA Class I and Class II Molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. MRNA Vaccine for Cancer Immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Dannull, J.; Nair, S.; Su, Z.; Boczkowski, D.; DeBeck, C.; Yang, B.; Gilboa, E.; Vieweg, J. Enhancing the Immunostimulatory Function of Dendritic Cells by Transfection with MRNA Encoding OX40 Ligand. Blood 2005, 105, 3206–3213. [Google Scholar] [CrossRef]
- Tcherepanova, I.Y.; Adams, M.D.; Feng, X.; Hinohara, A.; Horvatinovich, J.; Calderhead, D.; Healey, D.; Nicolette, C.A. Ectopic Expression of a Truncated CD40L Protein from Synthetic Post-Transcriptionally Capped RNA in Dendritic Cells Induces High Levels of IL-12 Secretion. BMC Mol. Biol. 2008, 9, 90. [Google Scholar] [CrossRef]
- Tuyaerts, S.; Van Meirvenne, S.; Bonehill, A.; Heirman, C.; Corthals, J.; Waldmann, H.; Breckpot, K.; Thielemans, K.; Aerts, J.L. Expression of Human GITRL on Myeloid Dendritic Cells Enhances Their Immunostimulatory Function but Does Not Abrogate the Suppressive Effect of CD4+CD25+ Regulatory T Cells. J. Leukoc. Biol. 2007, 82, 93–105. [Google Scholar] [CrossRef]
- Grünebach, F.; Kayser, K.; Weck, M.M.; Müller, M.R.; Appel, S.; Brossart, P. Cotransfection of Dendritic Cells with RNA Coding for HER-2/Neu and 4-1BBL Increases the Induction of Tumor Antigen Specific Cytotoxic T Lymphocytes. Cancer Gene Ther. 2005, 12, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Aerts-Toegaert, C.; Heirman, C.; Tuyaerts, S.; Corthals, J.; Aerts, J.L.; Bonehill, A.; Thielemans, K.; Breckpot, K.; Breckpot, K. CD83 Expression on Dendritic Cells and T Cells: Correlation with Effective Immune Responses. Eur. J. Immunol. 2007, 37, 686–695. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Heirman, C.; Corthals, J.; Empsen, C.; van Grunsven, L.A.; Allard, S.D.; Pen, J.; Lacor, P.; Thielemans, K.; Aerts, J.L. The Combination of 4-1BBL and CD40L Strongly Enhances the Capacity of Dendritic Cells to Stimulate HIV-Specific T Cell Responses. J. Leukoc. Biol. 2011, 89, 989–999. [Google Scholar] [CrossRef]
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H.; Karsh, L.I.; et al. Survival with AGS-003, an Autologous Dendritic Cell-Based Immunotherapy, in Combination with Sunitinib in Unfavorable Risk Patients with Advanced Renal Cell Carcinoma (RCC): Phase 2 Study Results. J. Immunother. Cancer 2015, 3, 14. [Google Scholar] [CrossRef]
- Figlin, R.A.; Tannir, N.M.; Uzzo, R.G.; Tykodi, S.S.; Chen, D.Y.T.; Master, V.; Kapoor, A.; Vaena, D.; Lowrance, W.; Bratslavsky, G.; et al. Results of the ADAPT Phase 3 Study of Rocapuldencel-T in Combination with Sunitinib as First-Line Therapy in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.M.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-Cell Stimulatory Capacity of Human Dendritic Cells by Co-Electroporation with CD40L, CD70 and Constitutively Active TLR4 Encoding MRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Bonehill, A.; Van Nuffel, A.M.T.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; Van Der Bruggen, P.; Neyns, B.; Thielemans, K. Single-Step Antigen Loading and Activation of Dendritic Cells by MRNA Electroporation for the Purpose of Therapeutic Vaccination in Melanoma Patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef]
- Pen, J.J.; De Keersmaecker, B.; Maenhout, S.K.; Van Nuffel, A.M.T.; Heirman, C.; Corthals, J.; Escors, D.; Bonehill, A.; Thielemans, K.; Breckpot, K.; et al. Modulation of Regulatory T Cell Function by Monocyte-Derived Dendritic Cells Matured through Electroporation with MRNA Encoding CD40 Ligand, Constitutively Active TLR4, and CD70. J. Immunol. 2013, 191, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Wilgenhof, S.; Heirman, C.; Corthals, J.; Breckpot, K.; Bonehill, A.; Neyns, B.; Thielemans, K. Optimized Dendritic Cell-Based Immunotherapy for Melanoma: The TriMix-Formula. Cancer Immunol. Immunother. 2014, 63, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived MRNA Electroporated Dendritic Cells (TriMixDC-MEL) plus Ipilimumab in Patientswith Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and Tumor Antigen MRNA Electroporated Dendritic Cell Vaccination plus Ipilimumab: Link between T-Cell Activation and Clinical Responses in Advanced Melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus Ipilimumab or Nivolumab Alone versus Ipilimumab Alone in Advanced Melanoma (CheckMate 067): 4-Year Outcomes of a Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Van den Bergh, J.; Willemen, Y.; Lion, E.; Van Acker, H.; De Reu, H.; Anguille, S.; Goossens, H.; Berneman, Z.; Van Tendeloo, V.; Smits, E. Transpresentation of Interleukin-15 by IL-15/IL-15Rα MRNA-Engineered Human Dendritic Cells Boosts Antitumoral Natural Killer Cell Activity. Oncotarget 2015, 6, 44123–44133. [Google Scholar] [CrossRef]
- Minkis, K.; Kavanagh, D.G.; Alter, G.; Bogunovic, D.; O’Neill, D.; Adams, S.; Pavlick, A.; Walker, B.D.; Brockman, M.A.; Gandhi, R.T.; et al. Type 2 Bias of T Cells Expanded from the Blood of Melanoma Patients Switched to Type 1 by IL-12p70 MRNA-Transfected Dendritic Cells. Cancer Res. 2008, 68, 9441–9450. [Google Scholar] [CrossRef]
- Bontkes, H.J.; Kramer, D.; Ruizendaal, J.J.; Kueter, E.W.M.; van Tendeloo, V.F.I.; Meijer, C.J.L.M.; Hooijberg, E. Dendritic Cells Transfected with Interleukin-12 and Tumor-Associated Antigen Messenger RNA Induce High Avidity Cytotoxic T Cells. Gene Ther. 2007, 14, 366–375. [Google Scholar] [CrossRef]
- Naka, T.; Iwahashi, M.; Nakamura, M.; Ojima, T.; Nakamori, M.; Ueda, K.; Katsuda, M.; Miyazawa, M.; Ishida, K.; Yamaue, H. Tumor Vaccine Therapy against Recrudescent Tumor Using Dendritic Cells Simultaneously Transfected with Tumor RNA and Granulocyte Macrophage Colony-Stimulating Factor RNA. Cancer Sci. 2008, 99, 407–413. [Google Scholar] [CrossRef]
- de Mey, W.; Locy, H.; De Ridder, K.; De Schrijver, P.; Autaers, D.; Lakdimi, A.; Esprit, A.; Franceschini, L.; Thielemans, K.; Verdonck, M.; et al. An MRNA Mix Redirects Dendritic Cells towards an Antiviral Program, Inducing Anticancer Cytotoxic Stem Cell and Central Memory CD8+ T Cells. Front. Immunol. 2023, 14, 1111523. [Google Scholar] [CrossRef] [PubMed]
- Heufler, C.; Koch, F.; Stanzl, U.; Topar, G.; Wysocka, M.; Trinchieri, G.; Enk, A.; Steinman, R.M.; Romani, N.; Schuler, G. Interleukin-12 Is Produced by Dendritic Cells and Mediates T Helper 1 Development as Well as Interferon-γ Production by T Helper 1 Cells. Eur. J. Immunol. 1996, 26, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The Interleukin-12/ Interleukin-12-Receptor System: Role in Normal and Pathologic Immune Responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Wang, Q.T.; Sun, S.N.; Li, S.Y.; Shang, H.; He, Y.W. Enhanced Human T Lymphocyte Antigen Priming by Cytokine-Matured Dendritic Cells Overexpressing Bcl-2 and IL-12. Front. Cell Dev. Biol. 2020, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Nopora, A.; Brocker, T. Bcl-2 Controls Dendritic Cell Longevity In Vivo. J. Immunol. 2002, 169, 3006–3014. [Google Scholar] [CrossRef]
- Granucci, F.; Vizzardelli, C.; Pavelka, N.; Feau, S.; Persico, M.; Virzi, E.; Rescigno, M.; Moro, G.; Ricciardi-Castagnoli, P. Inducible IL-2 Production by Dendritic Cells Revealed by Global Gene Expression Analysis. Nat. Immunol. 2001, 2, 882–888. [Google Scholar] [CrossRef]
- Castañón, E.; Zamarin, D.; Carneiro, B.A.; Marron, T.; Patel, S.P.; Subbiah, V.; Mehmi, I.; Oberoi, H.K.; El-Khoueiry, A.; Ridgway, B.; et al. Abstract CT004: Intratumoral (IT) MEDI1191 + Durvalumab (D): Update on the First-in-Human Study in Advanced Solid Tumors. Cancer Res. 2023, 83, CT004. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Zamarin, D.; Marron, T.; Mehmi, I.; Patel, S.P.; Subbiah, V.; El-Khoueiry, A.; Grand, D.; Garcia-Reyes, K.; Goel, S.; et al. Abstract CT183: First-in-Human Study of MEDI1191 (MRNA Encoding IL-12) plus Durvalumab in Patients (Pts) with Advanced Solid Tumors. Cancer Res. 2022, 82, CT183. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines-a New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen MRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van Der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix MRNA Results in T-Cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol Res. 2016, 4, 146–156. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Dewitte, H.; Lentacker, I.; Heirman, C.; Breckpot, K.; Thielemans, K. The ReNAissanCe of MRNA-Based Cancer Therapy. Expert. Rev. Vaccines 2014, 14, 235–251. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-Based Pharmacotherapy for Tumors: From Bench to Clinic and Back. Biomed. Pharmacother. 2020, 125, 109997. [Google Scholar] [CrossRef]
- Arance Fernandez, A.M.; Baurain, J.-F.; Vulsteke, C.; Rutten, A.; Soria, A.; Carrasco, J.; Neyns, B.; De Keersmaecker, B.; Van Assche, T.; Lindmark, B. A Phase I Study (E011-MEL) of a TriMix-Based MRNA Immunotherapy (ECI-006) in Resected Melanoma Patients: Analysis of Safety and Immunogenicity. J. Clin. Oncol. 2019, 37, 2641. [Google Scholar] [CrossRef]
- Clausen, B.E.; Stoitzner, P. Functional Specialization of Skin Dendritic Cell Subsets in Regulating T Cell Responses. Front. Immunol. 2015, 6, 534. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.-G.; Garbe, C. Direct Injection of Protamine-Protected MRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-Adjuvanted MRNA Vaccination in Advanced Prostate Cancer Patients: A First-in-Man Phase I/IIa Study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-Based Vaccines with Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA Vaccine Drives Immunity in Checkpoint-Inhibitor-Treated Melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Schmidt, M.; Vogler, I.; Derhovanessian, E.; Omokoko, T.; Godehardt, E.; Attig, S.; Cortini, A.; Newrzela, S.; Grützner, J.; Bolte, S.; et al. 88MO T-Cell Responses Induced by an Individualized Neoantigen Specific Immune Therapy in Post (Neo)Adjuvant Patients with Triple Negative Breast Cancer. Ann. Oncol. 2020, 31, S276. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Neyns, B.; Thielemans, K. Clinical Trials with MRNA Electroporated Dendritic Cells for Stage III/IV Melanoma Patients. J. Immunother. Cancer 2015, 3, P211. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A Phase IB Study on Intravenous Synthetic MRNA Electroporated Dendritic Cell Immunotherapy in Pretreated Advanced Melanoma Patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; van Dam, P.J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A Randomized Controlled Phase II Clinical Trial on MRNA Electroporated Autologous Monocyte-Derived Dendritic Cells (TriMixDC-MEL) as Adjuvant Treatment for Stage III/IV Melanoma Patients Who Are Disease-Free Following the Resection of Macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef]
- Kalluri, R.; McAndrews, K.M. The Role of Extracellular Vesicles in Cancer. Cell 2023, 186, 1610–1626. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic Cell-Derived Exosomes for Cancer Therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A Brief History of Nearly Everything—The Rise and Rise of Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular Vesicles in Cancer—Implications for Future Improvements in Cancer Care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zuo, B.; Zhang, Y.; Zhao, K.; Wu, L.; Qi, H.; Yang, R.; Gao, X.; Geng, M.; Wu, Y.; Jing, R.; et al. Universal Immunotherapeutic Strategy for Hepatocellular Carcinoma with Exosome Vaccines That Engage Adaptive and Innate Immune Responses. J. Hematol. Oncol. 2022, 15, 46. [Google Scholar] [CrossRef]
- Chen, X.; Feng, J.; Chen, W.; Shao, S.; Chen, L.; Wan, H. Small Extracellular Vesicles: From Promoting Pre-Metastatic Niche Formation to Therapeutic Strategies in Breast Cancer. Cell Commun. Signal. 2022, 20, 141. [Google Scholar] [CrossRef]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic Potential of Extracellular Vesicles. Front. Immunol. 2014, 5, 518. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic Cell-Derived Exosomes as Maintenance Immunotherapy after First Line Chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of Metastatic Melanoma Patients with Autologous Dendritic Cell (DC) Derived-Exosomes: Results of the First Phase 1 Clinical Trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A Phase I Study of Dexosome Immunotherapy in Patients with Advanced Non-Small Cell Lung Cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Ploix, S.; Lapierre, V.; Théry, C.; Commere, P.-H.; Tramalloni, D.; Gorrichon, K.; Virault-Rocroy, P.; Tursz, T.; Lantz, O.; et al. Updated Technology to Produce Highly Immunogenic Dendritic Cell-Derived Exosomes of Clinical Grade. J. Immunother. 2011, 34, 65–75. [Google Scholar] [CrossRef]
- Qian, K.; Fu, W.; Li, T.; Zhao, J.; Lei, C.; Hu, S. The Roles of Small Extracellular Vesicles in Cancer and Immune Regulation and Translational Potential in Cancer Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 286. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Nicco, C.; Lombard, B.; Véron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Théry, C. ICAM-1 on Exosomes from Mature Dendritic Cells Is Critical for Efficient Naive T-Cell Priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef]
- Pang, X.-L.; Wang, Z.-G.; Liu, L.; Feng, Y.-H.; Wang, J.-X.; Xie, H.-C.; Yang, X.-L.; Li, J.-F.; Feng, G.-W. Immature Dendritic Cells Derived Exosomes Promotes Immune Tolerance by Regulating T Cell Differentiation in Renal Transplantation. Aging 2019, 11, 8911–8924. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.-S.; Jiang, A. Plasmacytoid Dendritic Cells and Cancer Immunotherapy. Cells 2022, 11, 222. [Google Scholar] [CrossRef]
- Tel, J.; Erik, H.J.G.A.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Wim, J.G.O.; Van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid Dendritic Cells Cross-Prime Naive CD8 T Cells by Transferring Antigen to Conventional Dendritic Cells through Exosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23730–23741. [Google Scholar] [CrossRef]
- Greening, D.W.; Xu, R.; Ale, A.; Hagemeyer, C.E.; Chen, W. Extracellular Vesicles as next Generation Immunotherapeutics. Semin. Cancer Biol. 2023, 90, 73–100. [Google Scholar] [CrossRef]
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H.F. Dendritic Cell-Derived Exosomes Elicit Tumor Regression in Autochthonous Hepatocellular Carcinoma Mouse Models. J. Hepatol. 2017, 67, 739–748. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of Antitumor Immunity by IL-10 and TGF-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4 and CD8 Regulatory T Cells 1. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Miao, Y.; Wang, X.; Huang, X.; Dai, J. Recent Progress of Dendritic Cell-Derived Exosomes (Dex) as an Anti-Cancer Nanovaccine. Biomed. Pharmacother. 2022, 152, 113250. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.; Verhezen, T.; van der Heijden, S.; Berneman, Z.N.; Peeters, M.; Lardon, F.; Wouters, A.; Smits, E.L.J.M. A Systematic Review on Poly(I:C) and Poly-ICLC in Glioblastoma: Adjuvants Coordinating the Unlocking of Immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 213. [Google Scholar] [CrossRef] [PubMed]
- Phung, C.D.; Pham, T.T.; Nguyen, H.T.; Nguyen, T.T.; Ou, W.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; Kim, J.O. Anti-CTLA-4 Antibody-Functionalized Dendritic Cell-Derived Exosomes Targeting Tumor-Draining Lymph Nodes for Effective Induction of Antitumor T-Cell Responses. Acta Biomater. 2020, 115, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.K.; Redecke, V.; Prilliman, K.R.; Takabayashi, K.; Corr, M.; Tallant, T.; DiDonato, J.; Dziarski, R.; Akira, S.; Schoenberger, S.P.; et al. A Subset of Toll-Like Receptor Ligands Induces Cross-Presentation by Bone Marrow-Derived Dendritic Cells. J. Immunol. 2003, 170, 4102–4110. [Google Scholar] [CrossRef]
- Tran, T.H.; Tran, T.T.P.; Truong, D.H.; Nguyen, H.T.; Pham, T.T.; Yong, C.S.; Kim, J.O. Toll-like Receptor-Targeted Particles: A Paradigm to Manipulate the Tumor Microenvironment for Cancer Immunotherapy. Acta Biomater. 2019, 94, 82–96. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Pedicord, V.A.; Montalvo, W.; Leiner, I.M.; Allison, J.P. Single Dose of Anti-CTLA-4 Enhances CD8+ T-Cell Memory Formation, Function, and Maintenance. Proc. Natl. Acad. Sci. USA 2011, 108, 266–271. [Google Scholar] [CrossRef]
- Liu, C.; Liu, X.; Xiang, X.; Pang, X.; Chen, S.; Zhang, Y.; Ren, E.; Zhang, L.; Liu, X.; Lv, P.; et al. A Nanovaccine for Antigen Self-Presentation and Immunosuppression Reversal as a Personalized Cancer Immunotherapy Strategy. Nat. Nanotechnol. 2022, 17, 531–540. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering Dendritic Cell Vaccines to Improve Cancer Immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Cianciaruso, C.; Hansen, S.K.; De Palma, M. EVIR: Chimeric Receptors That Enhance Dendritic Cell Cross-Dressing with Tumor Antigens. Nat. Methods 2018, 15, 183–186. [Google Scholar] [CrossRef]
- Mohammadzadeh, Y.; De Palma, M. Boosting Dendritic Cell Nanovaccines. Nat. Nanotechnol. 2022, 17, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Naseri, M.; Bozorgmehr, M.; Zöller, M.; Ranaei Pirmardan, E.; Madjd, Z. Tumor-Derived Exosomes: The next Generation of Promising Cell-Free Vaccines in Cancer Immunotherapy. Oncoimmunology 2020, 9, 1779991. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant Effusions and Immunogenic Tumour-Derived Exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-Derived Exosomes Are a Source of Shared Tumor Rejection Antigens for CTL Cross-Priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef]
- Hao, S.; Bai, O.; Li, F.; Yuan, J.; Laferte, S.; Xiang, J. Mature Dendritic Cells Pulsed with Exosomes Stimulate Efficient Cytotoxic T-Lymphocyte Responses and Antitumour Immunity. Immunology 2007, 120, 90–102. [Google Scholar] [CrossRef]
- Rappa, G.; Santos, M.F.; Green, T.M.; Karbanová, J.; Hassler, J.; Bai, Y.; Barsky, S.H.; Corbeil, D.; Lorico, A. Nuclear Transport of Cancer Extracellular Vesicle-Derived Biomaterials through Nuclear Envelope Invagination-Associated Late Endosomes. Oncotarget 2017, 8, 14443–14461. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-Induced Exosomes Contribute to a More Aggressive and Chemoresistant Ovarian Cancer Phenotype: A Novel Mechanism Linking STAT3/Rab Proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved Vaccine Efficacy of Tumor Exosome Compared to Tumor Lysate Loaded Dendritic Cells in Mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, J.; Shao, C.; Liu, S.; Yu, Y.; Wang, Q.; Cao, X. Efficient Induction of Antitumor T Cell Immunity by Exosomes Derived from Heat-Shocked Lymphoma Cells. Eur. J. Immunol. 2006, 36, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Bu, N.; Wu, H.; Sun, B.; Zhang, G.; Zhan, S.; Zhang, R.; Zhou, L. Exosome-Loaded Dendritic Cells Elicit Tumor-Specific CD8+ Cytotoxic T Cells in Patients with Glioma. J. Neurooncol. 2011, 104, 659–667. [Google Scholar] [CrossRef]
- Li, W.; Mu, D.; Tian, F.; Hu, Y.; Jiang, T.; Han, Y.; Chen, J.; Han, G.; Li, X. Exosomes Derived from Rab27a-Overexpressing Tumor Cells Elicit Efficient Induction of Antitumor Immunity. Mol. Med. Rep. 2013, 8, 1876–1882. [Google Scholar] [CrossRef]
- Czernek, L.; Düchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef]
- André, F.; Schartz, N.E.C.; Chaput, N.; Flament, C.; Raposo, G.; Amigorena, S.; Angevin, E.; Zitvogel, L. Tumor-Derived Exosomes: A New Source of Tumor Rejection Antigens. Vaccine 2002, 20, A28–A31. [Google Scholar] [CrossRef]
- Bu, N.; Li, Q.-L.; Feng, Q.; Sun, B.-Z. Immune Protection Effect of Exosomes against Attack of L1210 Tumor Cells. Leuk. Lymphoma. 2006, 47, 913–918. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Peng, Y.; Yu, S.; Liu, J.; Wu, L.; Zhang, L.; Wu, Q.; Chang, X.; Yu, X.; et al. Dendritic Cells Loaded with Tumor Derived Exosomes for Cancer Immunotherapy. Oncotarget 2018, 9, 2887–2894. [Google Scholar] [CrossRef]
- Jafari, A.; Babajani, A.; Abdollahpour-Alitappeh, M.; Ahmadi, N.; Rezaei-Tavirani, M. Exosomes and Cancer: From Molecular Mechanisms to Clinical Applications. Med. Oncol. 2021, 38, 45. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor Exosomes Inhibit Differentiation of Bone Marrow Dendritic Cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor Exosomes Block Dendritic Cells Maturation to Decrease the T Cell Immune Response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Huang, S.; Li, Y.; Zhang, J.; Rong, J.; Ye, S. Epidermal Growth Factor Receptor-Containing Exosomes Induce Tumor-Specific Regulatory T Cells. Cancer Investig. 2013, 31, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Cao, X. The Exosomes in Tumor Immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef]
- Yang, C.; Kim, S.H.; Bianco, N.R.; Robbins, P.D. Tumor-Derived Exosomes Confer Antigen-Specific Immunosuppression in a Murine Delayed-Type Hypersensitivity Model. PLoS ONE 2011, 6, e22517. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-Derived Microvesicles Induce, Expand and Up-Regulate Biological Activities of Human Regulatory T Cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Park, K.-S.; Yoon, Y.J.; Lee, J.; Moon, H.-G.; Jang, S.C.; Choi, K.-H.; Kim, Y.-K.; Gho, Y.S. Therapeutic Effects of Autologous Tumor-Derived Nanovesicles on Melanoma Growth and Metastasis. PLoS ONE 2012, 7, e33330. [Google Scholar] [CrossRef]
- Ren, G.; Wang, Y.; Yuan, S.; Wang, B. Dendritic Cells Loaded with HeLa-Derived Exosomes Simulate an Antitumor Immune Response. Oncol. Lett. 2018, 15, 6636–6640. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; An, Y.; Jin, T.; Zhang, F.; He, P. Detection of MUC1 Protein on Tumor Cells and Their Derived Exosomes for Breast Cancer Surveillance with an Electrochemiluminescence Aptasensor. J. Electroanal. Chem. 2021, 882, 115011. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in Cancer: Function, Prognosis and Therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene Expression Profiling Identifies Clinically Relevant Subtypes of Prostate Cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Sekine, H.; Ohno, T.; Abe, M.; Keefe, K.; Kufe, D.W. Use of a Murine Monoclonal Antibody for Detection of Circulating Plasma DF3 Antigen Levels in Breast Cancer Patients. J. Clin. Investig. 1985, 75, 1671–1678. [Google Scholar] [CrossRef]
- Cho, J.A.; Yeo, D.J.; Son, H.Y.; Kim, H.W.; Jung, D.S.; Ko, J.K.; Jason, S.K.; Kim, Y.N.; Kim, C.W. Exosomes: A New Delivery System for Tumor Antigens in Cancer Immunotherapy. Int. J. Cancer 2005, 114, 613–622. [Google Scholar] [CrossRef]
- Shi, X.; Sun, J.; Li, H.; Lin, H.; Xie, W.; Li, J.; Tan, W. Antitumor Efficacy of Interferon-γ-Modified Exosomal Vaccine in Prostate Cancer. Prostate 2020, 80, 811–823. [Google Scholar] [CrossRef]
- Yang, M.Q.; Du, Q.; Varley, P.R.; Goswami, J.; Liang, Z.; Wang, R.; Li, H.; Stolz, D.B.; Geller, D.A. Interferon Regulatory Factor 1 Priming of Tumour-Derived Exosomes Enhances the Antitumour Immune Response. Br. J. Cancer 2018, 118, 62–71. [Google Scholar] [CrossRef]
- Accolla, R.S.; Ramia, E.; Tedeschi, A.; Forlani, G. CIITA-Driven MHC Class II Expressing Tumor Cells as Antigen Presenting Cell Performers: Toward the Construction of an Optimal Anti-Tumor Vaccine. Front. Immunol. 2019, 10, 1806. [Google Scholar] [CrossRef]
- Daar, A.S.; Fuggle, S.V.; Fabre, J.W.; Ting, A.; Morris, P.J. The Detailed Distribution of MHC Class II Antigens in Normal Human Organs. Transplantation 1984, 38, 293–298. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC Class I Antigens, Immune Surveillance, and Tumor Immune Escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Tosi, G. Optimal MHC-II-Restricted Tumor Antigen Presentation to CD4+ T Helper Cells: The Key Issue for Development of Anti-Tumor Vaccines. J. Transl. Med. 2012, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.H.; Cho, J.A.; Kim, C.W. Introduction of the CIITA Gene into Tumor Cells Produces Exosomes with Enhanced Anti-Tumor Effects. Exp. Mol. Med. 2011, 43, 281–290. [Google Scholar] [CrossRef]
- Morishita, M.; Takahashi, Y.; Matsumoto, A.; Nishikawa, M.; Takakura, Y. Exosome-Based Tumor Antigens–Adjuvant Co-Delivery Utilizing Genetically Engineered Tumor Cell-Derived Exosomes with Immunostimulatory CpG DNA. Biomaterials 2016, 111, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Takakura, Y. In Vivo Tracking of Extracellular Vesicles in Mice Using Fusion Protein Comprising Lactadherin and Gaussia Luciferase. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1660, pp. 245–254. [Google Scholar]
- Green, N.M. The Use of [14C] Biotin for Kinetic Studies and for Assay. Biochem. J. 1963, 89, 585–591. [Google Scholar] [CrossRef]
- Yildirim, M.; Yildirim, T.C.; Turay, N.; Bildik, T.; Ibibik, B.; Evcili, I.; Ersan, P.G.; Tokat, U.M.; Sahin, O.; Gursel, I. TLR Ligand Loaded Exosome Mediated Immunotherapy of Established Mammary Tumor in Mice. Immunol. Lett. 2021, 239, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Kadima, A.N.; Cole, D.J.; Gillanders, W.E. Defining the Antigen-Specific T-Cell Response to Vaccination and Poly(I:C)/TLR3 Signaling. J. Immunother. 2005, 28, 220–228. [Google Scholar] [CrossRef]
- Zuo, B.; Qi, H.; Lu, Z.; Chen, L.; Sun, B.; Yang, R.; Zhang, Y.; Liu, Z.; Gao, X.; You, A.; et al. Alarmin-Painted Exosomes Elicit Persistent Antitumor Immunity in Large Established Tumors in Mice. Nat. Commun. 2020, 11, 1790. [Google Scholar] [CrossRef]
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered Exosomes as an in Situ DC-Primed Vaccine to Boost Antitumor Immunity in Breast Cancer. Mol. Cancer 2022, 21, 45. [Google Scholar] [CrossRef]
- Hosseini, R.; Asef-Kabiri, L.; Yousefi, H.; Sarvnaz, H.; Salehi, M.; Akbari, M.E.; Eskandari, N. The Roles of Tumor-Derived Exosomes in Altered Differentiation, Maturation and Function of Dendritic Cells. Mol Cancer 2021, 20, 83. [Google Scholar] [CrossRef]
- Wang, Y.; Yi, J.; Chen, X.; Zhang, Y.; Xu, M.; Yang, Z. The Regulation of Cancer Cell Migration by Lung Cancer Cell-Derived Exosomes through TGF-β and IL-10. Oncol. Lett. 2016, 11, 1527–1530. [Google Scholar] [CrossRef]
- Lenart, M.; Rutkowska-Zapala, M.; Baj-Krzyworzeka, M.; Szatanek, R.; Węglarczyk, K.; Smallie, T.; Ziegler-Heitbrock, L.; Zembala, M.; Siedlar, M. Hyaluronan Carried by Tumor-Derived Microvesicles Induces IL-10 Production in Classical (CD14++ CD16−) Monocytes via PI3K/Akt/MTOR-Dependent Signalling Pathway. Immunobiology 2017, 222, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, L.; Li, D.; Yin, Y.; Zhang, C.-Y.; Li, J.; Zhang, Y. Microvesicle-Delivery MiR-150 Promotes Tumorigenesis by up-Regulating VEGF, and the Neutralization of MiR-150 Attenuate Tumor Development. Protein Cell 2013, 4, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.-P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-Associated Hsp72 from Tumor-Derived Exosomes Mediates STAT3-Dependent Immunosuppressive Function of Mouse and Human Myeloid-Derived Suppressor Cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-Derived Exosomes, Myeloid-Derived Suppressor Cells, and Tumor Microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune Modulation of T-Cell and NK (Natural Killer) Cell Activities by TEXs (Tumour-Derived Exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Ji, B.; Wei, M.; Yang, B. Recent Advances in Nanomedicines for Photodynamic Therapy (PDT)-Driven Cancer Immunotherapy. Theranostics 2022, 12, 434–458. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Moon, J.J. Photothermal Therapy Combined with Neoantigen Cancer Vaccination for Effective Immunotherapy against Large Established Tumors and Distant Metastasis. Adv. Ther. 2021, 4, 2100093. [Google Scholar] [CrossRef]
- Roth, G.A.; Picece, V.C.T.M.; Ou, B.S.; Luo, W.; Pulendran, B.; Appel, E.A. Designing Spatial and Temporal Control of Vaccine Responses. Nat. Rev. Mater. 2021, 7, 174–195. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards Personalized, Tumour-Specific, Therapeutic Vaccines for Cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Ibn-Salem, J.; Sorn, P.; Suchan, M.; Holtsträter, C.; Lahrmann, U.; Vogler, I.; Schmoldt, K.; Lang, F.; Schrörs, B.; et al. Accurate Detection of Tumor-Specific Gene Fusions Reveals Strongly Immunogenic Personal Neo-Antigens. Nat. Biotechnol. 2022, 40, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Song, H.; Qin, Y.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Engineering Dendritic-Cell-Based Vaccines and PD-1 Blockade in Self-Assembled Peptide Nanofibrous Hydrogel to Amplify Antitumor T-Cell Immunity. Nano Lett. 2018, 18, 4377–4385. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of tumor antigen cross-presentation by DCs to T cells in TDLNs. DAMPs from tumor cells activate DCs by binding to PRRs in the TME. Mature DCs capture tumor antigens and migrate to TDLNs to perform cross-presentation by presenting acquired tumor antigens on MHC I molecules. Activated, mature, antigen-loaded DCs then cross-prime naïve T cells into antigen-specific TEFF cells including CD4+ Th cells and CD8+ CTLs via TCRs. Cross-presentation allows the activation and cross-priming of CTLs to induce effective antitumor responses. DC: dendritic cell; TDLNs: tumor draining lymph nodes; DAMP: damage-associated molecular patterns; PRRs: pattern recognition receptors; TME: tumor microenvironment; MHC I: major histocompatibility complex I; MHC II: major histocompatibility complex II; TEFF: effector T cells; CD4+ Th cells: CD4+ T helper cells; CD8+ CTLs: CD8+ cytotoxic T lymphocytes; TCR: T cell receptor.
Figure 1.
Schematic diagram of tumor antigen cross-presentation by DCs to T cells in TDLNs. DAMPs from tumor cells activate DCs by binding to PRRs in the TME. Mature DCs capture tumor antigens and migrate to TDLNs to perform cross-presentation by presenting acquired tumor antigens on MHC I molecules. Activated, mature, antigen-loaded DCs then cross-prime naïve T cells into antigen-specific TEFF cells including CD4+ Th cells and CD8+ CTLs via TCRs. Cross-presentation allows the activation and cross-priming of CTLs to induce effective antitumor responses. DC: dendritic cell; TDLNs: tumor draining lymph nodes; DAMP: damage-associated molecular patterns; PRRs: pattern recognition receptors; TME: tumor microenvironment; MHC I: major histocompatibility complex I; MHC II: major histocompatibility complex II; TEFF: effector T cells; CD4+ Th cells: CD4+ T helper cells; CD8+ CTLs: CD8+ cytotoxic T lymphocytes; TCR: T cell receptor.

Figure 2.
Schematic diagram of conventional ex vivo MoDC-derived DC vaccine and new generations of DC vaccines. (A) Patient’s PBMCs are obtained by leukapheresis and CD14+ monocytes are selected by plastic adherence or immunomagnetic beads. Monocytes are differentiated into immature DCs in the presence of GM-CSF and IL-4 on day 6. Immature DCs are then matured in a maturation cocktail consisted of TNF-α, IL-1β, IL-6, and PGE2, while being simultaneously pulsed with autologous tumor cell lysates or antigens on day 7 for 48 hours. The mature, antigen-loaded DCs are reinfused to the patient. (B) Biomaterial-based DC vaccine uses PLG scaffold and hydrogel, which can encapsulate chemoattractant, TLR agonist, tumor cell lysates, and/or metabolism inhibitors or checkpoint inhibitors. PLG scaffold and hydrogel can be implanted or injected, respectively, into the patient to recruit, mature, and activate endogenous DCs in situ. ICD-based DC vaccines use radiotherapy, PDT, or PTT to generate an in situ DC vaccine, which may be facilitated by other adjuvants, immunotherapies, and/or chemotherapy, and combined with biomaterials to enhance the vaccine-induced immune response. mRNA-pulsed DC vaccines involve the electroporation of immature DCs with mRNAs that encode tumor antigens, adjuvants, immunostimulatory ligands, and/or cytokines. DCsEV-based vaccines also make use of immature DCs, which are matured and antigen-loaded with maturation cocktail and tumor peptides or proteins. sEVs secreted from matured, antigen-loaded DCs are collected and undergo surface modifications to enhance their immunogenicity and are injected to the patient. Tu-sEV-based DC vaccines manipulate cancer cells and/or Tu-sEVs to increase the levels of cross-presentation by Tu-sEVs. This may be achieved by transducing tumor cells with viral vectors transfected with genes encoding immunogenic antigens, performing surface modifications, and/or directly modifying Tu-sEV surface. DC: dendritic cell; MoDC: monocyte-derived DCs; PMBC: peripheral blood mononuclear cells; GM-CSF: granulocyte-macrophage colony-stimulating factor; IL-4: interleukin 4; Ag: antigen; TNF-α: tumor necrosis factor α; PGE2: prostaglandin E2; PLG: poly(lactide-coglycolide); TLR: toll-like receptor; CpG-ODN: CpG oligodeoxynucleotides; ICD: immunogenic cell death; PDT: photodynamic therapy; PTT: photothermal therapy; sEV: small extracellular vesicle; DCsEV: DC-derived sEV: Tu-sEV: tumor-derived sEV.
Figure 2.
Schematic diagram of conventional ex vivo MoDC-derived DC vaccine and new generations of DC vaccines. (A) Patient’s PBMCs are obtained by leukapheresis and CD14+ monocytes are selected by plastic adherence or immunomagnetic beads. Monocytes are differentiated into immature DCs in the presence of GM-CSF and IL-4 on day 6. Immature DCs are then matured in a maturation cocktail consisted of TNF-α, IL-1β, IL-6, and PGE2, while being simultaneously pulsed with autologous tumor cell lysates or antigens on day 7 for 48 hours. The mature, antigen-loaded DCs are reinfused to the patient. (B) Biomaterial-based DC vaccine uses PLG scaffold and hydrogel, which can encapsulate chemoattractant, TLR agonist, tumor cell lysates, and/or metabolism inhibitors or checkpoint inhibitors. PLG scaffold and hydrogel can be implanted or injected, respectively, into the patient to recruit, mature, and activate endogenous DCs in situ. ICD-based DC vaccines use radiotherapy, PDT, or PTT to generate an in situ DC vaccine, which may be facilitated by other adjuvants, immunotherapies, and/or chemotherapy, and combined with biomaterials to enhance the vaccine-induced immune response. mRNA-pulsed DC vaccines involve the electroporation of immature DCs with mRNAs that encode tumor antigens, adjuvants, immunostimulatory ligands, and/or cytokines. DCsEV-based vaccines also make use of immature DCs, which are matured and antigen-loaded with maturation cocktail and tumor peptides or proteins. sEVs secreted from matured, antigen-loaded DCs are collected and undergo surface modifications to enhance their immunogenicity and are injected to the patient. Tu-sEV-based DC vaccines manipulate cancer cells and/or Tu-sEVs to increase the levels of cross-presentation by Tu-sEVs. This may be achieved by transducing tumor cells with viral vectors transfected with genes encoding immunogenic antigens, performing surface modifications, and/or directly modifying Tu-sEV surface. DC: dendritic cell; MoDC: monocyte-derived DCs; PMBC: peripheral blood mononuclear cells; GM-CSF: granulocyte-macrophage colony-stimulating factor; IL-4: interleukin 4; Ag: antigen; TNF-α: tumor necrosis factor α; PGE2: prostaglandin E2; PLG: poly(lactide-coglycolide); TLR: toll-like receptor; CpG-ODN: CpG oligodeoxynucleotides; ICD: immunogenic cell death; PDT: photodynamic therapy; PTT: photothermal therapy; sEV: small extracellular vesicle; DCsEV: DC-derived sEV: Tu-sEV: tumor-derived sEV.


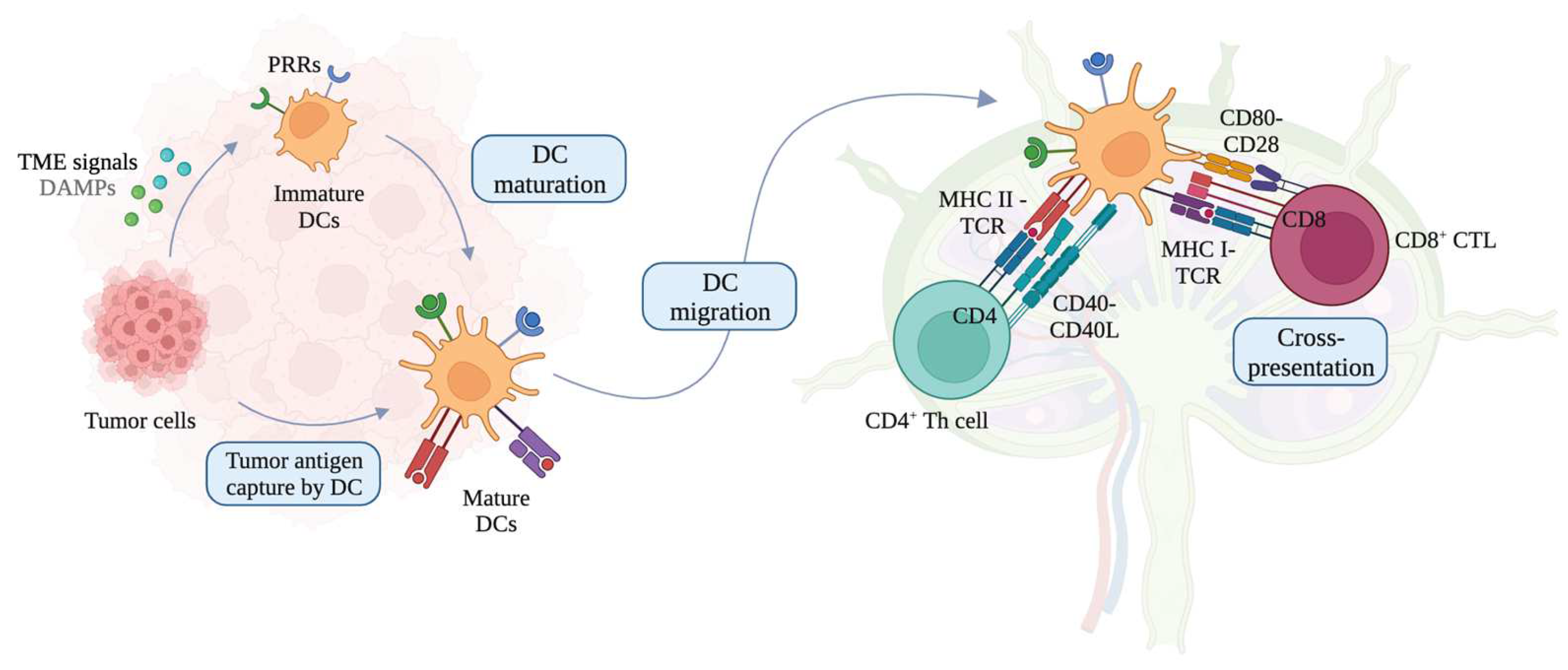{kind=link}
{kind=link}
Table 1.
Advantages and limitations of conventional MoDC-based vaccine and new generations of DC vaccines.
Table 1.
Advantages and limitations of conventional MoDC-based vaccine and new generations of DC vaccines.
| DC Vaccine Type | Advantages | Limitations | Reference |
|---|---|---|---|
| Conventional MoDC-based |
|
| NCT01832870 (Phase I); NCT01804465, NCT01881867 (Phase II); NCT00065442 (Phase III) [56,57,58,59] |
| Biomaterial-based |
|
| NCT01753089 (Phase I) [89,94,101,103,114,115,123,128,131,132,133,397] |
| Combinatory ICD-inducing |
|
| NCT01976585, NCT03789097, NCT00185965, NCT00323882, NCT02221739, (Phase I/II); NCT00880581 (Phase II); NCT00861614 (Phase III) [143,160,181,182,183,184,185,187,202,208,211,213] |
| mRNA-based |
|
| NCT00626483, NCT00639639, NCT03946800, NCT03788083, NCT03394937, NCT02410733, NCT02316457 (Phase I); NCT01066390 (Phase Ib); NCT02366728, NCT01302496, NCT01676779 (Phase II) [248,252,256,258,263,265,266,280,283,284,287,289,291,298,299,302] |
| DCsEV-based |
|
| NCT01159288 (Phase II) [309,312,313,314,315,318,321,327,333] |
| Tu-sEV-based |
|
| [368,369,370,376,379,381,382] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, K.-W.; Yam, J.W.P.; Mao, X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells 2023, 12, 2147. https://doi.org/10.3390/cells12172147
AMA Style
Lee K-W, Yam JWP, Mao X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells. 2023; 12(17):2147. https://doi.org/10.3390/cells12172147
Chicago/Turabian StyleLee, Kyu-Won, Judy Wai Ping Yam, and Xiaowen Mao. 2023. "Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations" Cells 12, no. 17: 2147. https://doi.org/10.3390/cells12172147
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.