
Integrated Meta-Omics Analysis Unveils the Pathways Modulating Tumorigenesis and Proliferation in High-Grade Meningioma
 , , , , and
, , , , and
Abstract
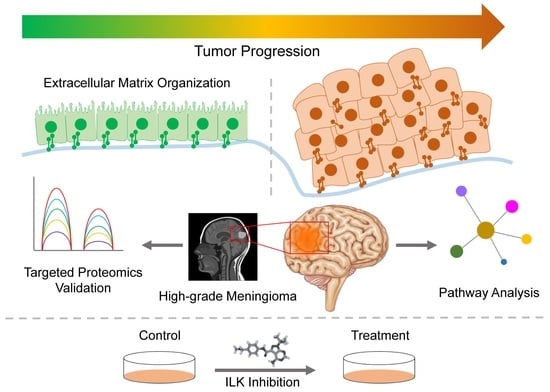
:
1. Introduction
2. Materials and Methods
2.1. Omic Data Mining, Literature Search, and Data Analysis
2.2. Pre-Processing, Normalization, and Statistical Data Analysis of Proteomic Datasets
2.3. Data Quality Check and Statistical Analysis of Transcriptomic Datasets
2.4. Protein–Protein Interaction Network (PPIN) and Pathway Enrichment Analysis
2.5. Sample Preparation for Targeted Proteomics Analysis
2.6. Transition List Preparation and Data Acquisition for Targeted Proteomics Analysis
2.7. Cell Culture, Stocks, and Doses of Inhibitor
2.8. Cell Proliferation Assay
2.9. Apoptosis Assay
2.10. Protein Extraction and Mass Spectrometry Data Acquisition
2.11. Label-Free Quantification and Biological Analysis of Cell Line Proteome Data
2.12. In Silico Drug Docking of Compounds Similar to Cpd22 as Well as FDA-Approved Drugs
3. Results
3.1. Integrated Omics Analysis of Meningioma Datasets
3.2. Pathway Mapping and Gene-Set Enrichment Analysis to Understand the Proteomic and Transcriptomic Alterations in High-Grade Meningioma
3.3. Validation of the Potential Common Markers Using MRM-based Targeted Proteomics
3.4. In Vitro Inhibition Using Cpd22 Reveals the Anti-Tumor Potential of the Pathway
3.5. Proteomic Alterations and Biological Pathway Perturbations Post-Treatment with an ILK Inhibitor in the Meningioma Cell Line
3.6. Screening of Compounds Using in Silico Docking for ILK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamashima, T. Human Meninges: Anatomy and Its Role in Meningioma Pathogenesis. In Meningiomas; Lee, J.H., Ed.; Springer: London, UK, 2009; pp. 15–24. ISBN 978-1-84882-910-7. [Google Scholar]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Paldor, I.; Awad, M.; Sufaro, Y.Z.; Kaye, A.H.; Shoshan, Y. Review of Controversies in Management of Non-Benign Meningioma. J. Clin. Neurosci. 2016, 31, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Negroni, C.; Hilton, D.A.; Ercolano, E.; Adams, C.L.; Kurian, K.M.; Baiz, D.; Hanemann, C.O. GATA-4, a Potential Novel Therapeutic Target for High-Grade Meningioma, Regulates miR-497, a Potential Novel Circulating Biomarker for High-Grade Meningioma. eBioMedicine 2020, 59, 102941. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Chuang, C.-C.; Wei, K.-C.; Chang, C.-N.; Lee, S.-T.; Wu, C.-T.; Hsu, Y.-H.; Lin, T.-K.; Hsu, P.-W.; Huang, Y.-C.; et al. Long Term Surgical Outcome and Prognostic Factors of Atypical and Malignant Meningiomas. Sci. Rep. 2016, 6, 35743. [Google Scholar] [CrossRef]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef]
- Sun, S.Q.; Hawasli, A.H.; Huang, J.; Chicoine, M.R.; Kim, A.H. An Evidence-Based Treatment Algorithm for the Management of WHO Grade II and III Meningiomas. FOC 2015, 38, E3. [Google Scholar] [CrossRef]
- Mukherjee, S.; Biswas, D.; Epari, S.; Shetty, P.; Moiyadi, A.; Ball, G.R.; Srivastava, S. Comprehensive Proteomic Analysis Reveals Distinct Functional Modules Associated with Skull Base and Supratentorial Meningiomas and Perturbations in Collagen Pathway Components. J. Proteom. 2021, 246, 104303. [Google Scholar] [CrossRef]
- Suppiah, S.; Nassiri, F.; Bi, W.L.; Dunn, I.F.; Hanemann, C.O.; Horbinski, C.M.; Hashizume, R.; James, C.D.; Mawrin, C.; Noushmehr, H.; et al. Molecular and Translational Advances in Meningiomas. Neuro-Oncology 2019, 21, i4–i17. [Google Scholar] [CrossRef]
- Karsy, M.; Hoang, N.; Barth, T.; Burt, L.; Dunson, W.; Gillespie, D.L.; Jensen, R.L. Combined Hydroxyurea and Verapamil in the Clinical Treatment of Refractory Meningioma: Human and Orthotopic Xenograft Studies. World Neurosurg. 2016, 86, 210–219. [Google Scholar] [CrossRef]
- Nassiri, F.; Tabatabai, G.; Aldape, K.; Zadeh, G. Challenges and Opportunities in Meningiomas: Recommendations from the International Consortium on Meningiomas. Neuro-Oncology 2019, 21, i2–i3. [Google Scholar] [CrossRef]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An Overview of Meningiomas. Future Oncol. 2018, 14, 2161–2177. [Google Scholar] [CrossRef]
- Burnett, B.A.; Womeldorff, M.R.; Jensen, R. Meningioma: Signaling Pathways and Tumor Growth. Handb. Clin. Neurol. 2020, 169, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Zhang, M.; Wu, W.W.; Mei, Y.; Dunn, I.F. Meningioma Genomics: Diagnostic, Prognostic, and Therapeutic Applications. Front. Surg. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 Inactivation in Tumor Biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coşkun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated Genomic Analyses of de Novo Pathways Underlying Atypical Meningiomas. Nat. Commun. 2017, 8, 14433. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Gill, C.M.; Loewenstern, J.; Rutland, J.W.; Arib, H.; Pain, M.; Umphlett, M.; Kinoshita, Y.; McBride, R.B.; Bederson, J.; Donovan, M.; et al. SWI/SNF Chromatin Remodeling Complex Alterations in Meningioma. J. Cancer Res. Clin. Oncol. 2021, 147, 3431–3440. [Google Scholar] [CrossRef]
- Dunn, J.; Ferluga, S.; Sharma, V.; Futschik, M.; Hilton, D.A.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Proteomic Analysis Discovers the Differential Expression of Novel Proteins and Phosphoproteins in Meningioma Including NEK9, HK2 and SET and Deregulation of RNA Metabolism. eBioMedicine 2019, 40, 77–91. [Google Scholar] [CrossRef]
- Papaioannou, M.-D.; Djuric, U.; Kao, J.; Karimi, S.; Zadeh, G.; Aldape, K.; Diamandis, P. Proteomic Analysis of Meningiomas Reveals Clinically Distinct Molecular Patterns. Neuro-Oncology 2019, 21, 1028–1038. [Google Scholar] [CrossRef]
- Mukherjee, S.; Biswas, D.; Gadre, R.; Jain, P.; Syed, N.; Stylianou, J.; Zeng, Q.; Mahadevan, A.; Epari, S.; Shetty, P.; et al. Comprehending Meningioma Signaling Cascades Using Multipronged Proteomics Approaches & Targeted Validation of Potential Markers. Front. Oncol. 2020, 10, 1600. [Google Scholar] [CrossRef]
- Kane, A.J.; Sughrue, M.E.; Rutkowski, M.J.; Shangari, G.; Fang, S.; McDermott, M.W.; Berger, M.S.; Parsa, A.T. Anatomic Location Is a Risk Factor for Atypical and Malignant Meningiomas. Cancer 2011, 117, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Mawrin, C.; Chung, C.; Preusser, M. Biology and Clinical Management Challenges in Meningioma. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e106–e115. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange Consortium in 2020: Enabling ‘Big Data’ Approaches in Proteomics. Nucleic Acids Res. 2019, 48, gkz984. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, M.; da Veiga Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M.; et al. Discovering and Linking Public Omics Data Sets Using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1418, pp. 93–110. ISBN 978-1-4939-3576-5. [Google Scholar]
- Parkinson, H.; Kapushesky, M.; Shojatalab, M.; Abeygunawardena, N.; Coulson, R.; Farne, A.; Holloway, E.; Kolesnykov, N.; Lilja, P.; Lukk, M.; et al. ArrayExpress—A Public Database of Microarray Experiments and Gene Expression Profiles. Nucleic Acids Res. 2007, 35, D747–D750. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Lenis, V.P.; Hilton, D.A.; Warta, R.; Herold-Mende, C.; Hanemann, C.O.; Futschik, M.E. Integration and Comparison of Transcriptomic and Proteomic Data for Meningioma. Cancers 2020, 12, 3270. [Google Scholar] [CrossRef]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data mining toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards More Transparent and Integrative Metabolomics Analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Millet, P. Overall MORPHEUS Toolset Flow. In Dynamic System Reconfiguration in Heterogeneous Platforms; Voros, N.S., Rosti, A., Hübner, M., Eds.; Lecture Notes in Electrical Engineering; Springer: Dordrecht, The Netherlands, 2009; Volume 40, pp. 109–117. ISBN 978-90-481-2426-8. [Google Scholar]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics—A Bioconductor Package for Quality Assessment of Microarray Data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Statistics for Biology and Health; Springer-Verlag: New York, NY, USA, 2005; pp. 397–420. ISBN 978-0-387-25146-2. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Lo Surdo, P.; Calderone, A.; Iannuccelli, M.; Licata, L.; Peluso, D.; Castagnoli, L.; Cesareni, G.; Perfetto, L. DISNOR: A Disease Network Open Resource. Nucleic Acids Res. 2018, 46, D527–D534. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2019, gkz1021. [Google Scholar] [CrossRef]
- Banerjee, A.; Biswas, D.; Barpanda, A.; Halder, A.; Sibal, S.; Kattimani, R.; Shah, A.; Mahadevan, A.; Goel, A.; Srivastava, S. The First Pituitary Proteome Landscape from Matched Anterior and Posterior Lobes for a Better Understanding of the Pituitary Gland. Mol. Cell. Proteom. MCP 2023, 22, 100478. [Google Scholar] [CrossRef]
- Ghantasala, S.; Pai, M.G.J.; Biswas, D.; Gahoi, N.; Mukherjee, S.; KP, M.; Nissa, M.U.; Srivastava, A.; Epari, S.; Shetty, P.; et al. Multiple Reaction Monitoring-Based Targeted Assays for the Validation of Protein Biomarkers in Brain Tumors. Front. Oncol. 2021, 11, 548243. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ghosh, S.; Biswas, D.; Rao, A.; Shetty, P.; Epari, S.; Moiyadi, A.; Srivastava, S. Clinical Proteomics for Meningioma: An Integrated Workflow for Quantitative Proteomics and Biomarker Validation in Formalin-Fixed Paraffin-Embedded Tissue Samples. OMICS J. Integr. Biol. 2022, 26, 512–520. [Google Scholar] [CrossRef]
- Halder, A.; Biswas, D.; Chauhan, A.; Saha, A.; Auromahima, S.; Yadav, D.; Nissa, M.U.; Iyer, G.; Parihari, S.; Sharma, G.; et al. A Large-Scale Targeted Proteomics of Serum and Tissue Shows the Utility of Classifying High Grade and Low Grade Meningioma Tumors. Clin. Proteom. 2023, 20, 41. [Google Scholar] [CrossRef]
- Barpanda, A.; Biswas, D.; Verma, A.; Parihari, S.; Singh, A.; Kapoor, S.; Kantharia, C.; Srivastava, S. Integrative Proteomic and Pharmacological Analysis of Colon Cancer Reveals the Classical Lipogenic Pathway with Prognostic and Therapeutic Opportunities. J. Proteome Res. 2023, 22, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Fukuda, K.; Gupta, S.; Chen, K.; Wu, C.; Qin, J. The Pseudoactive Site of ILK Is Essential for Its Binding to α-Parvin and Localization to Focal Adhesions. Mol. Cell 2009, 36, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A Web Application for Molecular Visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Batut, B.; Hiltemann, S.; Bagnacani, A.; Baker, D.; Bhardwaj, V.; Blank, C.; Bretaudeau, A.; Brillet-Guéguen, L.; Čech, M.; Chilton, J.; et al. Community-Driven Data Analysis Training for Biology. Cell Syst. 2018, 6, 752–758.e1. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Ahmed, N. Management of High-Grade Meningioma: Present, Past and Promising Future. In Central Nervous System Tumors—Primary and Secondary; Birol Sarica, F., Ed.; IntechOpen: London, UK, 2023; ISBN 978-1-80356-752-5. [Google Scholar]
- Bailey, D.D.; Montgomery, E.Y.; Garzon-Muvdi, T. Metastatic High-Grade Meningioma: A Case Report and Review of Risk Factors for Metastasis. Neuro-Oncol. Adv. 2023, 5, vdad014. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic Landscape of High-Grade Meningiomas. npj Genom. Med. 2017, 2, 15. [Google Scholar] [CrossRef]
- Shrivastava, R.K.; Sen, C.; Costantino, P.D.; Della Rocca, R. Sphenoorbital Meningiomas: Surgical Limitations and Lessons Learned in Their Long-Term Management. J. Neurosurg. 2005, 103, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.; Dixit, K.; Raizer, J. Recent Advances in Managing/Understanding Meningioma. F1000Res 2018, 7, 490. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.; Galanopoulos, T.; Tessa Hedley-Whyte, E.; Black, P.M.; Antoniades, H.N. Human Meningiomas Co-Express Platelet-Derived Growth Factor (Pdgf) and Pdgf-Receptor Genes and Their Protein Products. Int. J. Cancer 1990, 46, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Weisman, A.S.; Raguet, S.S.; Kelly, P.A. Characterization of the Epidermal Growth Factor Receptor in Human Meningioma. Cancer Res. 1987, 47, 2172–2176. [Google Scholar]
- Koul, D.; Shen, R.; Bergh, S.; Lu, Y.; De Groot, J.F.; Liu, T.J.; Mills, G.B.; Yung, W.K.A. Targeting Integrin-Linked Kinase Inhibits Akt Signaling Pathways and Decreases Tumor Progression of Human Glioblastoma. Mol. Cancer Ther. 2005, 4, 1681–1688. [Google Scholar] [CrossRef]
- Murphy, M.C.; Huston, J.; Glaser, K.J.; Manduca, A.; Meyer, F.B.; Lanzino, G.; Morris, J.M.; Felmlee, J.P.; Ehman, R.L. Preoperative Assessment of Meningioma Stiffness by Magnetic Resonance Elastography. J. Neurosurg. 2013, 118, 643–648. [Google Scholar] [CrossRef]
- Gatti, G.; Vilardo, L.; Musa, C.; Di Pietro, C.; Bonaventura, F.; Scavizzi, F.; Torcinaro, A.; Bucci, B.; Saporito, R.; Arisi, I.; et al. Role of Lamin A/C as Candidate Biomarker of Aggressiveness and Tumorigenicity in Glioblastoma Multiforme. Biomedicines 2021, 9, 1343. [Google Scholar] [CrossRef]
- Bordeleau, F.; Califano, J.P.; Negrón Abril, Y.L.; Mason, B.N.; LaValley, D.J.; Shin, S.J.; Weiss, R.S.; Reinhart-King, C.A. Tissue Stiffness Regulates Serine/Arginine-Rich Protein-Mediated Splicing of the Extra Domain B-Fibronectin Isoform in Tumors. Proc. Natl. Acad. Sci. USA 2015, 112, 8314–8319. [Google Scholar] [CrossRef]
- Kafka, A. Epithelial-to-Mesenchymal Transition Possible Role in Meningiomas. Front. Biosci. 2012, E4, 889–896. [Google Scholar] [CrossRef]
- Van Belle, K.; Herman, J.; Waer, M.; Sprangers, B.; Louat, T. OSU-T315 as an Interesting Lead Molecule for Novel B Cell-Specific Therapeutics. J. Immunol. Res. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Edwards, L.A.; Woo, J.; Huxham, L.A.; Verreault, M.; Dragowska, W.H.; Chiu, G.; Rajput, A.; Kyle, A.H.; Kalra, J.; Yapp, D.; et al. Suppression of VEGF Secretion and Changes in Glioblastoma Multiforme Microenvironment by Inhibition of Integrin-Linked Kinase (ILK). Mol. Cancer Ther. 2008, 7, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Zimu, Z.; Jia, Z.; Xian, F.; Rui, M.; Yuting, R.; Yuan, W.; Tianhong, W.; Mian, M.; Yinlong, L.; Enfang, S. Decreased Expression of PACSIN1 in Brain Glioma Samples Predicts Poor Prognosis. Front. Mol. Biosci. 2021, 8, 696072. [Google Scholar] [CrossRef]
- Biswas, D.; Shenoy, S.V.; Chauhan, A.; Halder, A.; Ghosh, B.; Padhye, A.; Auromahima, S.; Yadav, D.; Sasmal, S.; Dutta, S.; et al. BrainProt(TM) 3.0: Understanding Human Brain Diseases Using Comprehensively Curated & Integrated OMICS Datasets. bioRxiv 2023. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Pemigatinib: Hot Topics behind the First Approval of a Targeted Therapy in Cholangiocarcinoma. Cancer Treat. Res. Commun. 2021, 27, 100337. [Google Scholar] [CrossRef]
- Salhotra, A.; Sandhu, K.; O’Hearn, J.; Ali, H.; Nakamura, R.; Modi, B.G. A Critical Review of Belumosudil in Adult and Pediatric Patients with Chronic Graft-versus-Host Disease. Expert. Rev. Clin. Immunol. 2023, 19, 241–251. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | High-Grade | Low-Grade | Control |
|---|---|---|---|
| Age | |||
| N (N miss) | 13 (0) | 10 (0) | 9 (0) |
| Mean ± SD | 45.3 ± 13.2 | 48.2 ± 10.7 | 49.7 ± 29.7 |
| Min–Max | 23–67 | 31–74 | 0.5–88 |
| Median (IQR) | 46.5 (45–53) | 47 (40.5–57.5) | 45 (28–85) |
| Gender (%) | |||
| Female | 6 (46.2) | 7 (70) | 1 (12) |
| Male | 7 (53.8) | 3 (30) | 8 (88) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, D.; Halder, A.; Barpanda, A.; Ghosh, S.; Chauhan, A.; Bhat, L.; Epari, S.; Shetty, P.; Moiyadi, A.; Ball, G.R.; et al. Integrated Meta-Omics Analysis Unveils the Pathways Modulating Tumorigenesis and Proliferation in High-Grade Meningioma. Cells 2023, 12, 2483. https://doi.org/10.3390/cells12202483
Biswas D, Halder A, Barpanda A, Ghosh S, Chauhan A, Bhat L, Epari S, Shetty P, Moiyadi A, Ball GR, et al. Integrated Meta-Omics Analysis Unveils the Pathways Modulating Tumorigenesis and Proliferation in High-Grade Meningioma. Cells. 2023; 12(20):2483. https://doi.org/10.3390/cells12202483
Chicago/Turabian StyleBiswas, Deeptarup, Ankit Halder, Abhilash Barpanda, Susmita Ghosh, Aparna Chauhan, Lipika Bhat, Sridhar Epari, Prakash Shetty, Aliasgar Moiyadi, Graham Roy Ball, and et al. 2023. "Integrated Meta-Omics Analysis Unveils the Pathways Modulating Tumorigenesis and Proliferation in High-Grade Meningioma" Cells 12, no. 20: 2483. https://doi.org/10.3390/cells12202483