
Loss of Lipid Carrier ApoE Exacerbates Brain Glial and Inflammatory Responses after Lysosomal GBA1 Inhibition
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Systemic Inhibition of Lysosomal GBA1 Enzyme in Mice
2.3. GBA1 Activity Assay
2.4. Immunofluorescence and Immunohistochemistry
2.5. Tissue Homogenization and Protein Extraction
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
3.1. Prolonged Glycolipid Elevation by GBA1 Inhibition Increases Brain ApoE Protein
3.2. Loss of ApoE Amplified Brain Regional Microgliosis in Response to GBA1 Inhibition
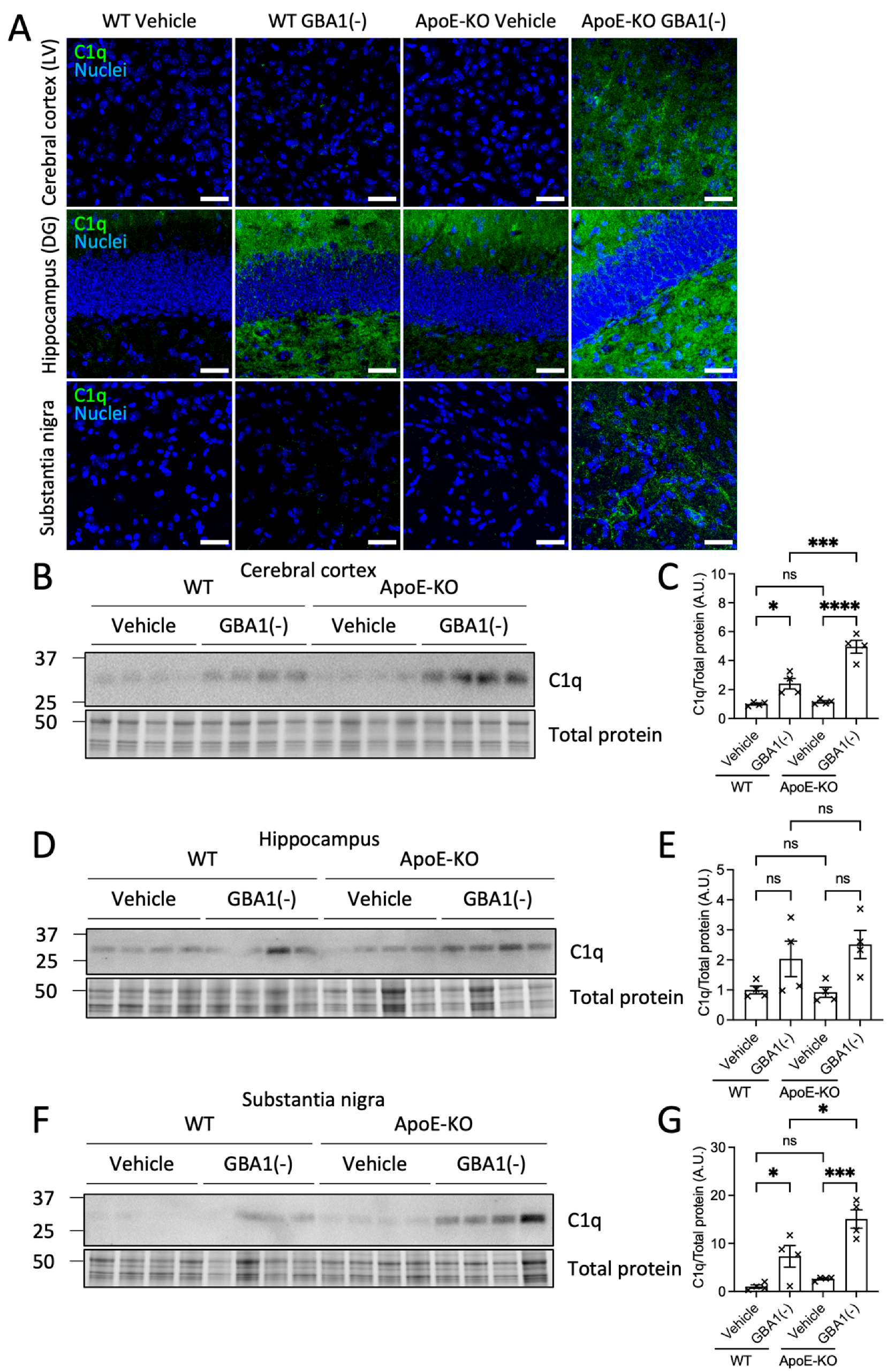
3.3. Loss of ApoE Elevated Brain Regional C1q Deposition after GBA1 Inhibition
3.4. Loss of ApoE Increased LAMP1 Lysosomal Labeling Associated with Microglia after GBA1 Inhibition
3.5. Increased Lysosome, but Not Autophagosome, Membrane Formation after GBA1 Inhibition Was Amplified by Loss of ApoE
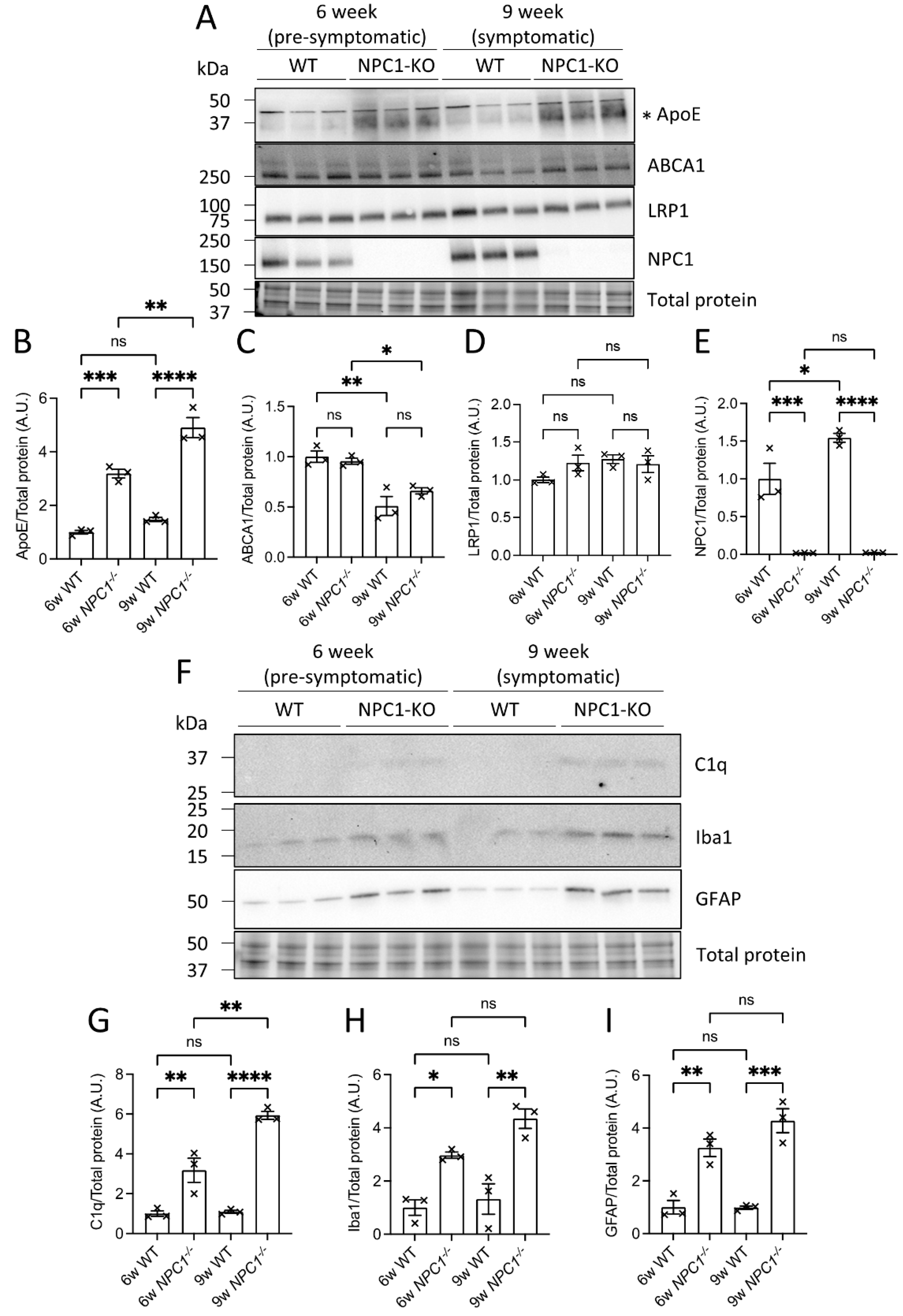
3.6. ABCA1 and LRP1 Protein Levels Were Reduced in the Absence of ApoE, Whereas NPC1 Protein Levels Increased in Response to GBA1 Inhibition
3.7. ApoE and Complement Factor C1q Protein Levels Were Elevated in the Brains of NPC1−/− Mice
4. Discussion
4.1. Summary of Experiments and Findings
4.2. Brain ApoE Lipid Transport Function Is Critical to Adapt to Lipid Perturbations
4.3. Do ApoE Responses to Glycolipid Stress Limit Complement Factor C1q Activation?
4.4. ApoE Function Is Critical following Astrocyte and Microglial Activation
4.5. Lipid Transport and Lysosomal Mechanisms Integrate Intracellular and Extracellular Lipid Adaptations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Szwedo, A.A.; Dalen, I.; Pedersen, K.F.; Camacho, M.; Bäckström, D.; Forsgren, L.; Tzoulis, C.; Winder-Rhodes, S.; Hudson, G.; Liu, G.; et al. GBA and APOE Impact Cognitive Decline in Parkinson’s Disease: A 10-Year Population-Based Study. Mov. Disord. 2022, 37, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Heckman, M.G.; Murray, M.E.; Soto, A.I.; Walton, R.L.; Diehl, N.N.; van Gerpen, J.A.; Uitti, R.J.; Wszolek, Z.K.; Ertekin-Taner, N.; et al. APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 2018, 91, e1182–e1195. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Sanchez, J.S.; Almeida, M.C.; Boassa, D.; Acosta-Uribe, J.; Vila-Castelar, C.; Ramirez-Gomez, L.; Baena, A.; Aguillon, D.; Villalba-Moreno, N.D.; et al. Distinct tau neuropathology and cellular profiles of an APOE3 Christchurch homozygote protected against autosomal dominant Alzheimer’s dementia. Acta Neuropathol. 2022, 144, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Isacson, O.; Brekk, O.R.; Hallett, P.J. Novel Results and Concepts Emerging From Lipid Cell Biology Relevant to Degenerative Brain Aging and Disease. Front. Neurol. 2019, 10, 1053. [Google Scholar] [CrossRef]
- de Leeuw, S.M.; Kirschner, A.W.T.; Lindner, K.; Rust, R.; Budny, V.; Wolski, W.E.; Gavin, A.-C.; Nitsch, R.M.; Tackenberg, C. APOE2, E3, and E4 differentially modulate cellular homeostasis, cholesterol metabolism, and inflammatory response in isogenic iPSC-derived astrocytes. Stem Cell Rep. 2021, 17, 110–126. [Google Scholar] [CrossRef]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e716. [Google Scholar] [CrossRef]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef]
- Cooper, O.; Hallett, P.; Isacson, O. Upstream lipid and metabolic systems are potential causes of Alzheimer’s disease, Parkinson’s disease and dementias. FEBS J. 2022. [Google Scholar] [CrossRef]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Lindner, K.; Beckenbauer, K.; van Ek, L.C.; Titeca, K.; de Leeuw, S.M.; Awwad, K.; Hanke, F.; Korepanova, A.V.; Rybin, V.; van der Kam, E.L.; et al. Isoform- and cell-state-specific lipidation of ApoE in astrocytes. Cell Rep. 2022, 38, 110435. [Google Scholar] [CrossRef] [PubMed]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef]
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell type-specific lipid storage changes in Parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.-R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Central Nervous System Lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains: Thematic Review Series: ApoE and Lipid Homeostasis in Alzheimer’s Disease. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E structure: Insights into function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Pinals, R.L.; Tsai, L.H. Building in vitro models of the brain to understand the role of APOE in Alzheimer’s disease. Life Sci. Alliance 2022, 5, e202201542. [Google Scholar] [CrossRef]
- Leduc, V.; Domenger, D.; De Beaumont, L.; Lalonde, D.; Bélanger-Jasmin, S.; Poirier, J. Function and comorbidities of apolipoprotein e in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011, 2011, 974361. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Smith, D.A.; Morris, L.; Fletcher, C.; Colaco, A.; Huebecker, M.; Tordo, J.; Palomar, N.; Massaro, G.; Henckaerts, E.; et al. AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann–Pick type C1 disease. Hum. Mol. Genet. 2018, 27, 3079–3098. [Google Scholar] [CrossRef]
- Kaya, E.; Smith, D.A.; Smith, C.; Morris, L.; Bremova-Ertl, T.; Cortina-Borja, M.; Fineran, P.; Morten, K.J.; Poulton, J.; Boland, B.; et al. Acetyl-leucine slows disease progression in lysosomal storage disorders. Brain Commun. 2020, 3, fcaa148. [Google Scholar] [CrossRef]
- Roney, J.C.; Li, S.; Farfel-Becker, T.; Huang, N.; Sun, T.; Xie, Y.; Cheng, X.-T.; Lin, M.-Y.; Platt, F.M.; Sheng, Z.-H. Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev. Cell 2021, 56, 1452–1468.e1458. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Zigdon, H.; Meshcheriakova, A.; Klein, A.D.; Yaacobi, C.; Eilam, R.; Kenwood, B.M.; Rahim, A.A.; Massaro, G.; Merrill, A.H., Jr.; et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol. 2016, 239, 496–509. [Google Scholar] [CrossRef]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Reboulet, R.; Quinn, B.; Huelsken, J.; Witte, D.; Grabowski, G.A. Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol. Genet. Metab. 2008, 94, 190–203. [Google Scholar] [CrossRef]
- Korecka, J.A.; Thomas, R.; Christensen, D.P.; Hinrich, A.J.; Ferrari, E.J.; Levy, S.A.; Hastings, M.L.; Hallett, P.J.; Isacson, O. Mitochondrial clearance and maturation of autophagosomes are compromised in LRRK2 G2019S familial Parkinson’s disease patient fibroblasts. Hum. Mol. Genet. 2019, 28, 3232–3243. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef]
- Hallett, P.J.; Engelender, S.; Isacson, O. Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson’s disease. J. Neuroinflammation 2019, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Dinkel, L.; Müller, S.A.; Sebastian Monasor, L.; Schifferer, M.; Cantuti-Castelvetri, L.; König, J.; Vidatic, L.; Bremova-Ertl, T.; Lieberman, A.P.; et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.E.; Klein, A.D.; Hong, J.; Dimbil, U.J.; Scott, M.P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 2012, 21, 2946–2960. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. npj Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Mus, L.; Siani, F.; Giuliano, C.; Ghezzi, C.; Cerri, S.; Blandini, F. Development and biochemical characterization of a mouse model of Parkinson’s disease bearing defective glucocerebrosidase activity. Neurobiol. Dis. 2019, 124, 289–296. [Google Scholar] [CrossRef]
- Chu, Y.; Jin, X.; Parada, I.; Pesic, A.; Stevens, B.; Barres, B.; Prince, D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7975–7980. [Google Scholar] [CrossRef]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.-H.; Huang, E.J.; Rowitch, D.H.; et al. A Dramatic Increase of C1q Protein in the CNS during Normal Aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef]
- Dejanovic, B.; Wu, T.; Tsai, M.-C.; Graykowski, D.; Gandham, V.D.; Rose, C.M.; Bakalarski, C.E.; Ngu, H.; Wang, Y.; Pandey, S.; et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2022, 2, 837–850. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Remolina Serrano, J.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Zhu, H.; Kono, M.; Hoachlander-Hobby, L.E.; Huso, V.L.; Proia, R.L. Genetic defects in the sphingolipid degradation pathway and their effects on microglia in neurodegenerative disease. Cell Signal 2021, 78, 109879. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Reinshagen, V.; Zhou, S.; Burgess, B.L.; Bernier, L.; McIsaac, S.A.; Chan, J.Y.; Tansley, G.H.; Cohn, J.S.; Hayden, M.R.; Wellington, C.L. Deficiency of ABCA1 Impairs Apolipoprotein E Metabolism in Brain*. J. Biol. Chem. 2004, 279, 41197–41207. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef]
- Sucunza, D.; Rico, A.J.; Roda, E.; Collantes, M.; González-Aseguinolaza, G.; Rodríguez-Pérez, A.I.; Peñuelas, I.; Vázquez, A.; Labandeira-García, J.L.; Broccoli, V.; et al. Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. Int. J. Mol. Sci. 2021, 22, 4825. [Google Scholar] [CrossRef]
- Brekk, O.R.; Korecka, J.A.; Crapart, C.C.; Huebecker, M.; MacBain, Z.K.; Rosenthal, S.A.; Sena-Esteves, M.; Priestman, D.A.; Platt, F.M.; Isacson, O.; et al. Upregulating β-hexosaminidase activity in rodents prevents α-synuclein lipid associations and protects dopaminergic neurons from α-synuclein-mediated neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Khalil, Y.A.; Rabès, J.-P.; Boileau, C.; Varret, M. APOE gene variants in primary dyslipidemia. Atherosclerosis 2021, 328, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Calle, R.; Konings, S.C.; Frontiñán-Rubio, J.; García-Revilla, J.; Camprubí-Ferrer, L.; Svensson, M.; Martinson, I.; Boza-Serrano, A.; Venero, J.L.; Nielsen, H.M.; et al. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Giannisis, A.; Al-Grety, A.; Carlsson, H.; Patra, K.; Twohig, D.; Sando, S.B.; Lauridsen, C.; Berge, G.; Grøntvedt, G.R.; Bråthen, G.; et al. Plasma apolipoprotein E levels in longitudinally followed patients with mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 115. [Google Scholar] [CrossRef]
- Giannisis, A.; Patra, K.; Edlund, A.K.; Nieto, L.A.; Benedicto-Gras, J.; Moussaud, S.; de la Rosa, A.; Twohig, D.; Bengtsson, T.; Fu, Y.; et al. Brain integrity is altered by hepatic APOE ε4 in humanized-liver mice. Mol. Psychiatry 2022, 27, 3533–3543. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connolly, K.J.; Margaria, J.; Di Biase, E.; Cooper, O.; Hallett, P.J.; Isacson, O. Loss of Lipid Carrier ApoE Exacerbates Brain Glial and Inflammatory Responses after Lysosomal GBA1 Inhibition. Cells 2023, 12, 2564. https://doi.org/10.3390/cells12212564
Connolly KJ, Margaria J, Di Biase E, Cooper O, Hallett PJ, Isacson O. Loss of Lipid Carrier ApoE Exacerbates Brain Glial and Inflammatory Responses after Lysosomal GBA1 Inhibition. Cells. 2023; 12(21):2564. https://doi.org/10.3390/cells12212564
Chicago/Turabian StyleConnolly, Kyle J., Juliette Margaria, Erika Di Biase, Oliver Cooper, Penelope J. Hallett, and Ole Isacson. 2023. "Loss of Lipid Carrier ApoE Exacerbates Brain Glial and Inflammatory Responses after Lysosomal GBA1 Inhibition" Cells 12, no. 21: 2564. https://doi.org/10.3390/cells12212564