
Differential Expression of miRNAs, lncRNAs, and circRNAs between Ovaries and Testes in Common Carp (Cyprinus carpio)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and RNA Extraction
2.3. Library Preparation and Sequencing
2.4. Data Quality Control and Read Alignment
2.5. Bioinformatics Analysis of smaRNA Sequences
2.6. Processing of lncRNA Sequencing Data
2.7. Identification of circRNAs and Prediction of Coding Potential
2.8. Differential Expression Analysis
2.9. Enrichment Analysis of Differential Candidate Genes
2.10. Quantitative PCR
2.11. ceRNA Analysis
3. Results
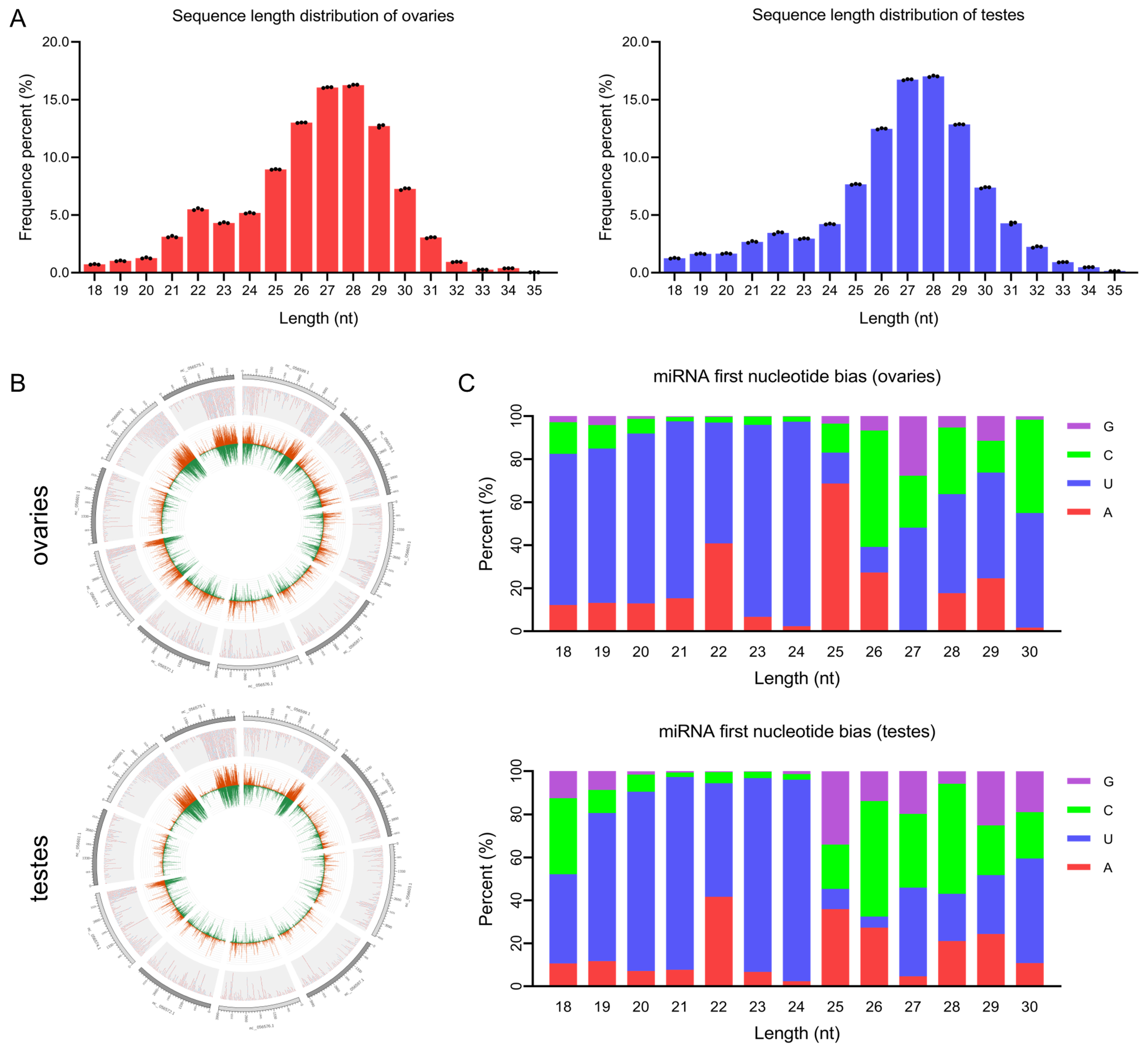
3.1. Overview of Gonadal smaRNA Sequencing of Common Carp
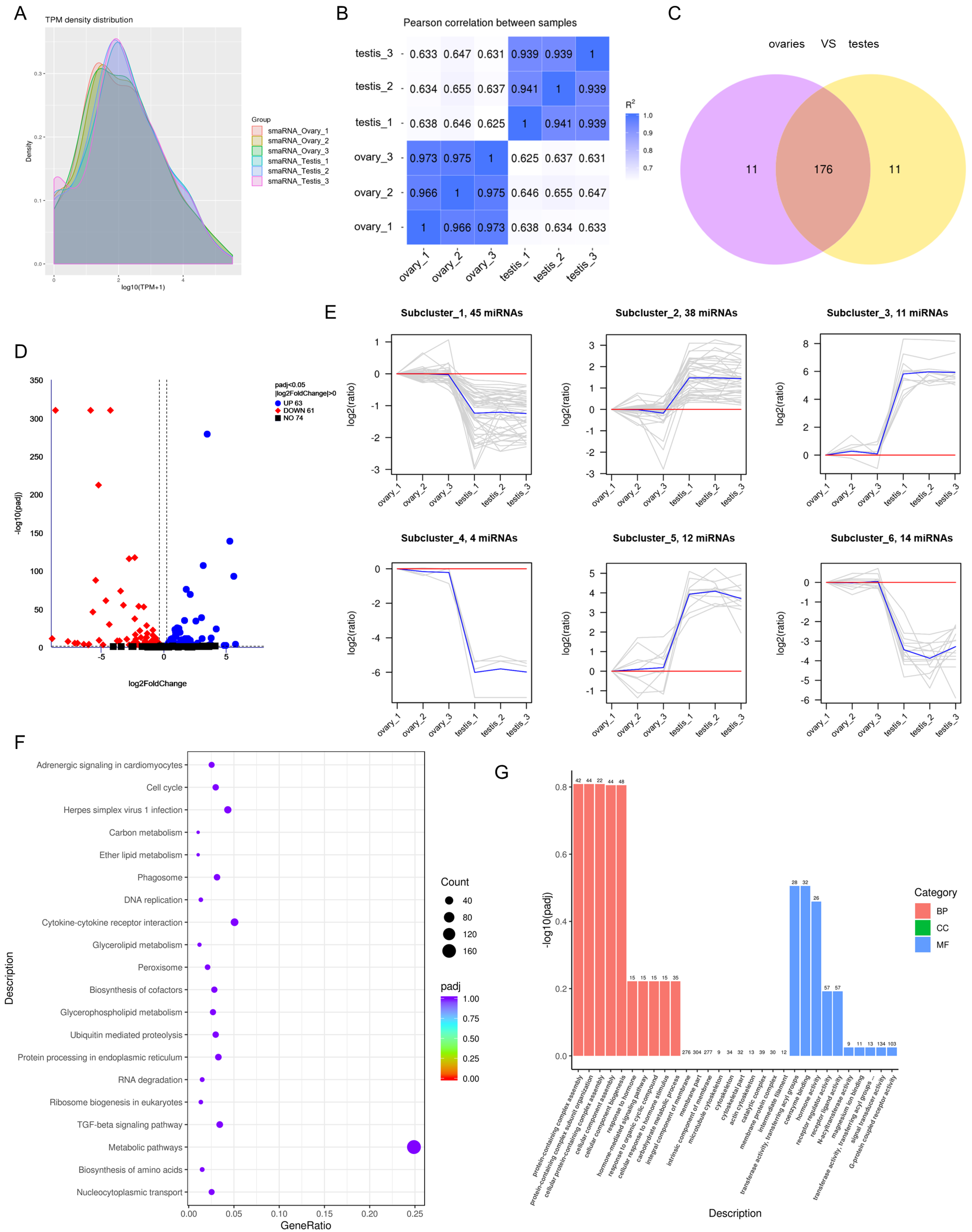
3.2. Analysis of miRNA in Common Carp Gonads
3.3. Differential Expression of miRNAs between Ovaries and Testes
3.4. Target Gene Prediction and Functional Enrichment of miRNAs
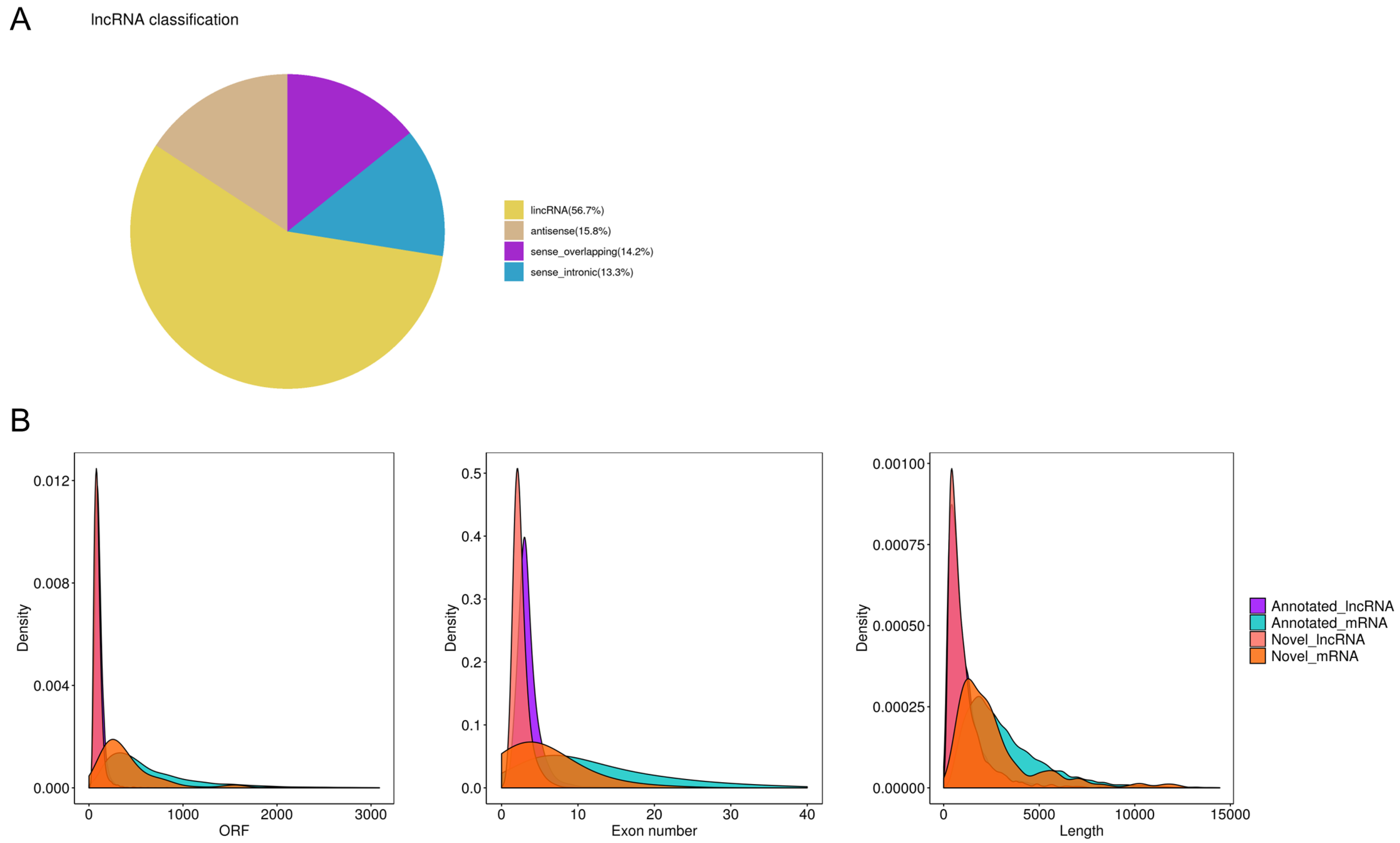
3.5. Overview of Gonadal lncRNA Sequencing
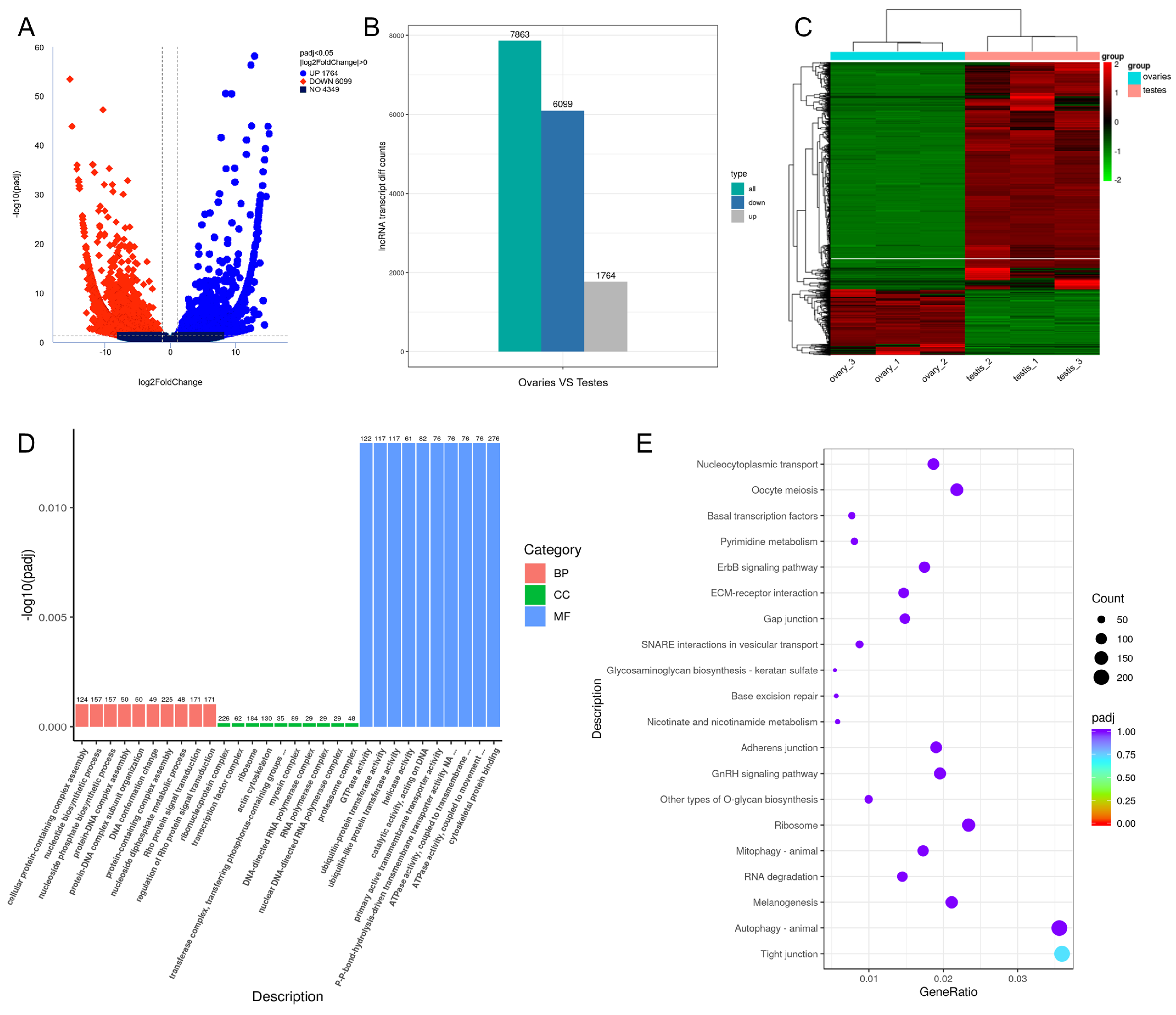
3.6. Differential Expression of lncRNAs
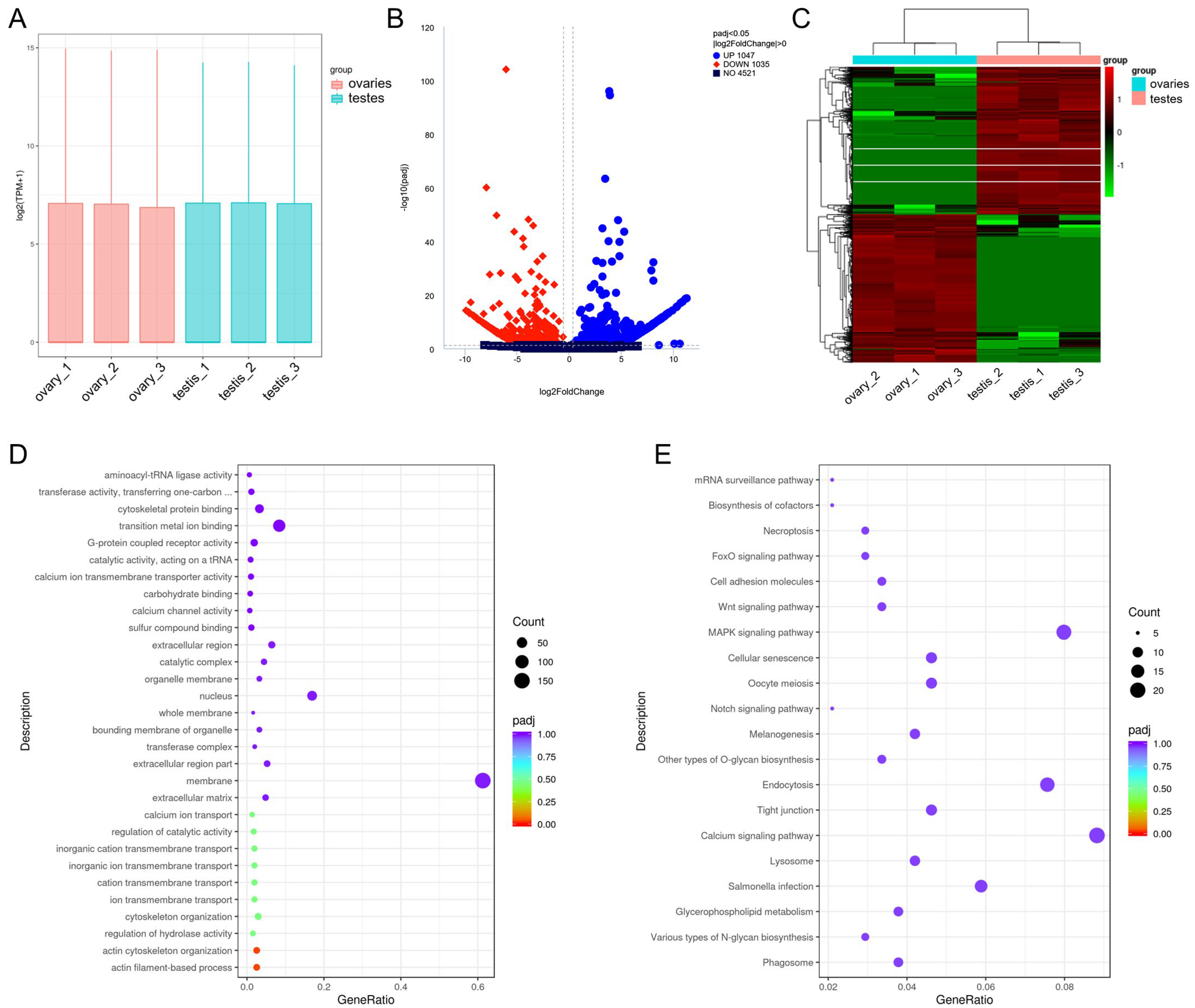
3.7. Differentially Expressed mRNAs
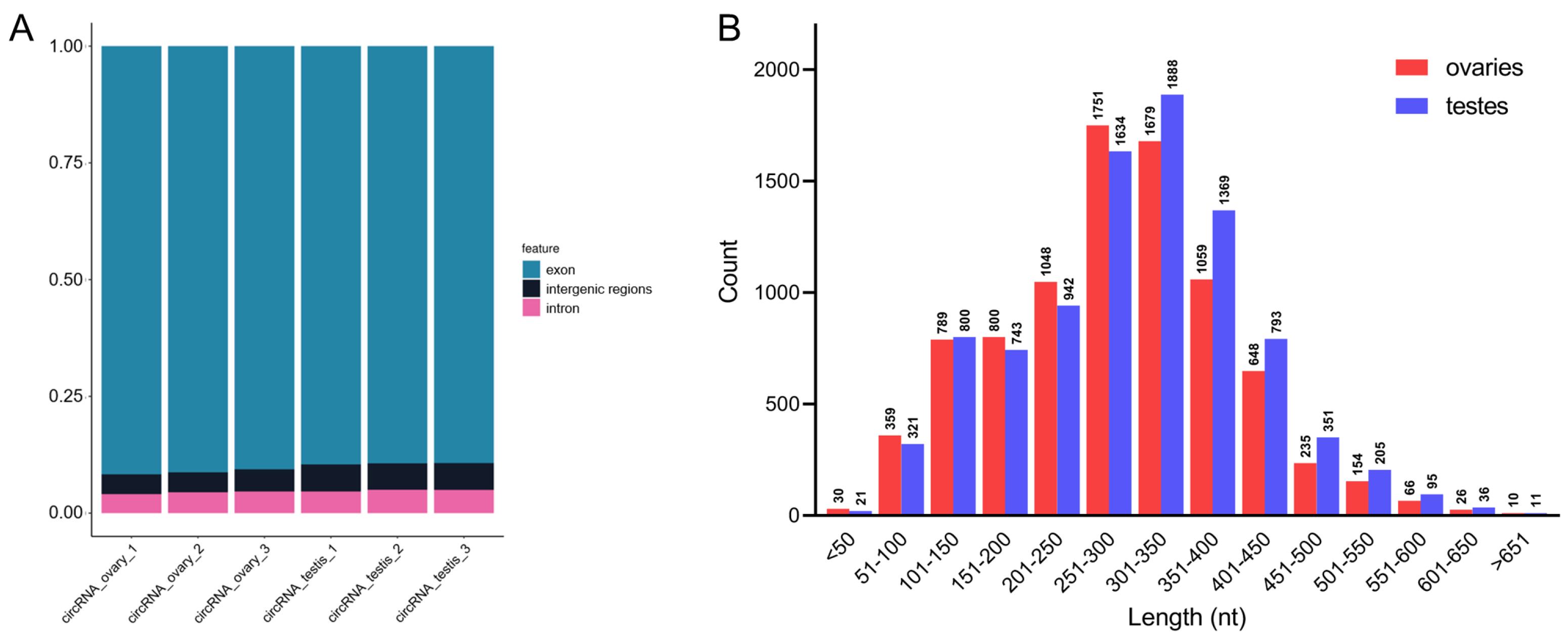
3.8. Identification of Gonadal circRNAs
3.9. Expression Profiles and Functional Analysis of circRNAs in Gonads
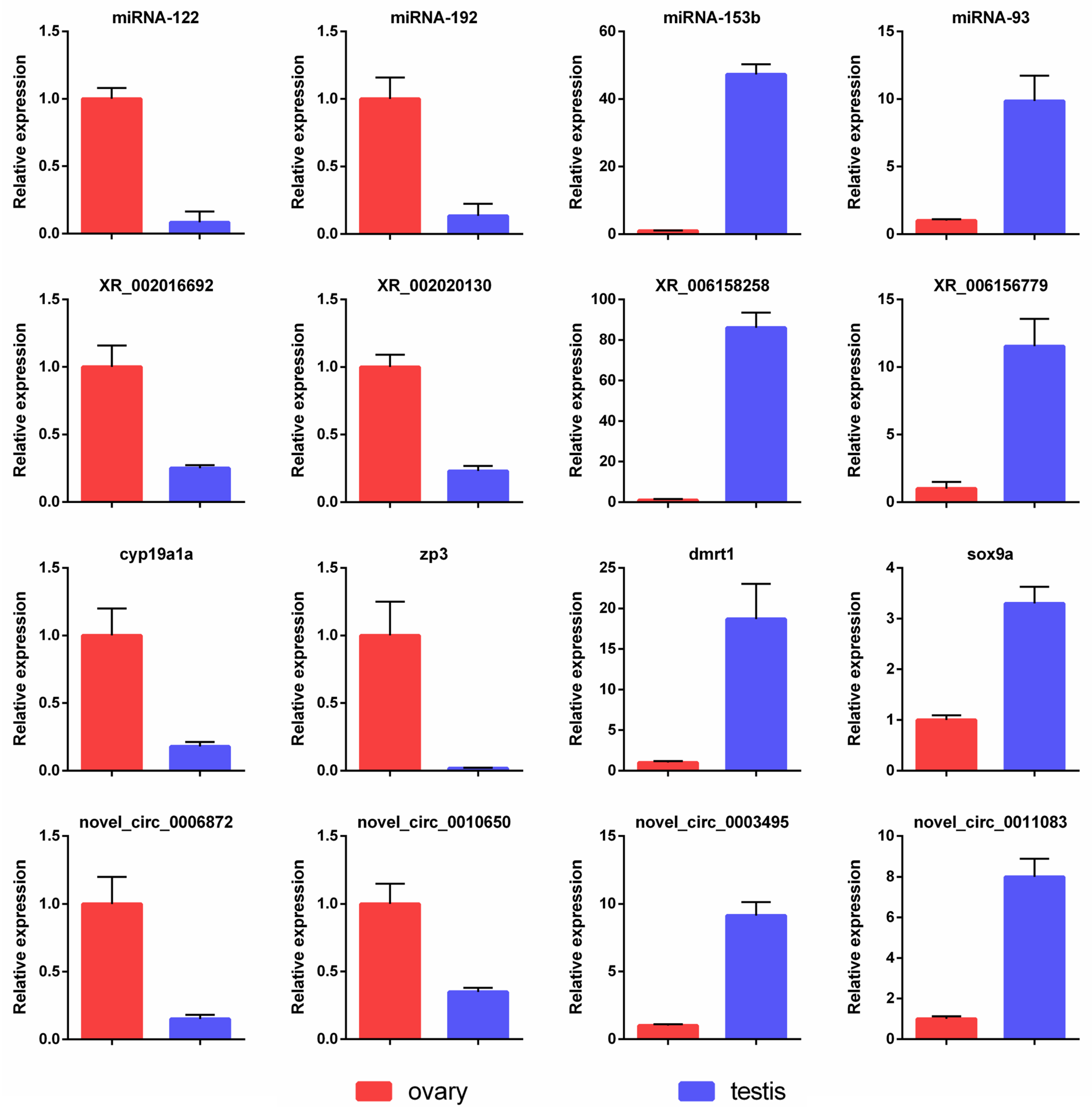
3.10. Validation of Expression Profiles by qPCR
3.11. Complex Interactions among mRNAs and ncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nagahama, Y.; Chakraborty, T.; Paul-Prasanth, B.; Ohta, K.; Nakamura, M. Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiol. Rev. 2021, 101, 1237–1308. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Gui, J.F. Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci. China Life Sci. 2015, 58, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Horne, C.R.; Hirst, A.G.; Atkinson, D. Selection for increased male size predicts variation in sexual size dimorphism among fish species. Proc. Biol. Sci. 2020, 287, 20192640. [Google Scholar] [CrossRef]
- Li, X.Y.; Mei, J.; Ge, C.T.; Liu, X.L.; Gui, J.F. Sex determination mechanisms and sex control approaches in aquaculture animals. Sci. China Life Sci. 2022, 65, 1091–1122. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.-S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Real, F.M.; Sekido, R.; Lupianez, D.G.; Lovell-Badge, R.; Jiménez, R.; Burgos, M. A microRNA (mmu-mir-124) prevents sox9 expression in developing mouse ovarian cells. Biol. Reprod. 2013, 89, 78. [Google Scholar] [CrossRef]
- Miao, N.; Wang, X.; Hou, Y.; Feng, Y.; Gong, Y. Identification of male-biased microRNA-107 as a direct regulator for nuclear receptor subfamily 5 group A member 1 based on sexually dimorphic microRNA expression profiling from chicken embryonic gonads. Mol. Cell Endocrinol. 2016, 429, 29–40. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Ryohei, S.; Robin, L.B. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008, 453, 930–934. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, H.; Xin, D.; Cheng, H.; Zhou, R. A novel ncRNA gene from mouse chromosome 5 trans-splices with Dmrt1 on chromosome 19. Biochem. Biophys. Res. Commun. 2010, 400, 696–700. [Google Scholar] [CrossRef]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef]
- Shen, Y.; Guo, X.; Wang, W. Identification and characterization of circular RNAs in zebrafish. FEBS Lett. 2017, 591, 213–220. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, X.; Chen, K.; Luo, H.; Yu, W.; Jia, S.; Li, Y.; Wang, Y.; Yang, P.; Zhu, Z.; et al. Production of YY supermale and XY physiological female common carp for potential eradication of this invasive species. J. World Aquacult. Soc. 2018, 49, 315–327. [Google Scholar] [CrossRef]
- Tessema, A.; Getahun, A.; Mengistou, S.; Fetahi, T.; Dejen, E. Reproductive biology of common carp (Cyprinus carpio Linnaeus, 1758) in Lake Hayq, Ethiopia. Fish. Aquat. Sci. 2020, 23, 16. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Zhou, Z.; Xiao, J.; Chen, B.; Huang, P.; Li, C.; Xue, Y.; Liu, R.; Bai, Y.; et al. Comparative transcriptome analysis of early sexual differentiation in the male and female gonads of common carp (Cyprinus carpio). Aquaculture 2023, 563, 738984. [Google Scholar] [CrossRef]
- Zhai, G.; Shu, T.; Chen, K.; Lou, Q.; Jia, J.; Huang, J.; Shi, C.; Jin, X.; He, J.; Jiang, D.; et al. Successful production of an all-female common carp (Cyprinus carpio L.) population using cyp17a1-deficient neomale carp. Engineering 2022, 8, 181–189. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Li, J.T.; Wang, Q.; Huang Yang, M.D.; Li, Q.-S.; Cui, M.S.; Dong, Z.J.; Wang, H.W.; Yu, J.H.; Zhao, Y.J.; Yang, C.R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Nan, P.; Zhang, W.; Wang, F.; Zhang, R.; Liang, T.; Ji, X.; Du, Q.; Chang, Z. Transcriptome analysis of three critical periods of ovarian development in Yellow River carp (Cyprinus carpio). Theriogenology 2018, 105, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, K.; Liu, F.; Jiang, M.; Chen, Z.; Chen, H.; Song, Y.; Tao, B.; Cui, X.; Li, Y.; et al. Dynamic gene expression and alternative splicing events demonstrate co-regulation of testicular differentiation and maturation by the brain and gonad in common carp. Front. Endocrinol. 2021, 12, 820463. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Sam, G.J.; Russell, J.G.; Stijn, V.D.; Alex, B.; Anton, J.E. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Zheng, Y.; Ji, B.; Song, R.; Wang, S.; Li, T.; Zhang, X.; Chen, K.; Li, T.; Li, J. Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res. 2016, 44, e123. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Bateman, A.; Finn, R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 2007, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, J.; Xu, T.; Yang, Q.; He, J.; Song, X. IRESfinder: Identifying RNA internal ribosome entry site in eukaryotic cell using framed k-mer features. J. Genet. Genom. 2018, 45, 403–406. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Riffo-Campos, A.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, S.; Yang, P.; Ma, Z.; Kang, L. Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biol. 2009, 10, R6. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Miyawaki, S.; Tachibana, M. Role of epigenetic regulation in mammalian sex determination. Curr. Top Dev. Biol. 2019, 134, 195–221. [Google Scholar] [CrossRef]
- Zhang, C.; Han, B.; Xu, T.; Li, D. The biological function and potential mechanism of long non-coding RNAs in cardiovascular disease. J. Cell Mol. Med. 2020, 24, 12900–12909. [Google Scholar] [CrossRef]
- Brown, C.; Hendrich, B.; Rupert, J.; Lafreniere, R.; Xing, Y.; Lawrence, J.; Willard, H. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Yousefi, H.; Maheronnaghsh, M.; Molaei, F.; Mashouri, L.; Aref, A.R.; Momeny, M.; Alahari, S.K. Long noncoding RNAs and exosomal lncRNAs: Classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2019, 39, 953–974. [Google Scholar] [CrossRef]
- Herpin, A.; Schartl, M. Plasticity of gene-regulatory networks controlling sex determination: Of masters, slaves, usual suspects, newcomers, and usurpators. EMBO Rep. 2015, 16, 1260–1274. [Google Scholar] [CrossRef]
- Capel, B. Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef]
- Salzburger, W. Understanding explosive diversification through cichlid fish genomics. Nat. Rev. Genet. 2018, 19, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of circRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Capel, B. Cell fate commitment during mammalian sex determination. Curr. Opin. Genet Dev. 2015, 32, 144–152. [Google Scholar] [CrossRef]
- Feng, K.; Cui, X.; Song, Y.; Tao, B.; Chen, J.; Wang, J.; Liu, S.; Sun, Y.; Zhu, Z.; Trudeau, V.; et al. Gnrh3 regulates PGC proliferation and sex differentiation in developing zebrafish. Endocrinology 2020, 161, bqz024. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.; Reinhart, B.; Slack, F.; Martindale, M.; Kuroda, M.; Maller, B.; Hayward, D.; Ball, E.; Degnan, B.; Muller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Loubalova, Z.; Fulka, H.; Horvat, F.; Pasulka, J.; Malik, R.; Hirose, M.; Ogura, A.; Svoboda, P. Formation of spermatogonia and fertile oocytes in golden hamsters requires piRNAs. Nat. Cell Biol. 2021, 23, 992–1001. [Google Scholar] [CrossRef]
- Xiao, J.; Zhong, H.; Zhou, Y.; Yu, F.; Gao, Y.; Luo, Y.; Tang, Z.; Guo, Z.; Guo, E.; Gan, X.; et al. Identification and characterization of microRNAs in ovary and testis of Nile tilapia (Oreochromis niloticus) by using solexa sequencing technology. PLoS ONE 2014, 9, e86821. [Google Scholar] [CrossRef]
- Houwing, S.; Kamminga, L.M.; Berezikov, E.; Cronembold, D.; Girard, A.; van den Elst, H.; Filippov, D.V.; Blaser, H.; Raz, E.; Moens, C.B.; et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in zebrafish. Cell 2007, 129, 69–82. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Liu, Q.; Li, B.; An, L.; Hao, R.; Zhao, J.; Liu, S.; Song, J. Coordinated microRNA and messenger RNA expression profiles for understanding sexual dimorphism of gonads and the potential roles of microRNA in the steroidogenesis pathway in Nile tilapia (Oreochromis niloticus). Theriogenology 2016, 85, 970–978. [Google Scholar] [CrossRef]
- Yu, Q.; Peng, C.; Ye, Z.; Tang, Z.; Li, S.; Xiao, L.; Liu, S.; Yang, Y.; Zhao, M.; Zhang, Y.; et al. An estradiol-17β/miRNA-26a/cyp19a1a regulatory feedback loop in the protogynous hermaphroditic fish, Epinephelus coioides. Mol. Cell. Endocrinol. 2020, 504, 110689. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Chen, K.; Jiang, M.; Jia, S.; Chen, J.; Tao, B.; Song, Y.; Li, Y.; Wang, Y.; Xiao, W.; et al. MiR-153b-3p regulates the proliferation and differentiation of male germ cells by targeting amh in common carp (Cyprinus carpio). Aquaculture 2021, 535, 736420. [Google Scholar] [CrossRef]
- Nishimura, T.; Sato, T.; Yamamoto, Y.; Watakabe, I.; Ohkawa, Y.; Suyama, M.; Kobayashi, S.; Tanaka, M. foxl3 is a germ cell–intrinsic factor involved in sperm-egg fate decision in medaka. Science 2015, 349, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Qi, S.; Wei, X.; Liu, X.; Li, Y.; Zhou, X.; Xiao, H.; Lu, B.; Wang, D.; Li, M. Germline sexual fate is determined by the antagonistic action of dmrt1 and foxl3/foxl2 in tilapia. Development 2021, 148, dev199380. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Tang, L.; Huang, F.; You, W.; Poetsch, A.; Nobrega, R.H.; Power, D.M.; Zhu, T.; Liu, K.; Wang, H.Y.; Wang, Q.; et al. ceRNA crosstalk mediated by ncRNAs is a novel regulatory mechanism in fish sex determination and differentiation. Genome Res. 2022, 32, 1502–1515. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, M.; Wang, Q.; Zhang, J.; Zhao, R.; Cao, Y.; Yu, S.; Wang, K.; Chen, Y.; Ma, Z.; Sun, X.; et al. Differential Expression of miRNAs, lncRNAs, and circRNAs between Ovaries and Testes in Common Carp (Cyprinus carpio). Cells 2023, 12, 2631. https://doi.org/10.3390/cells12222631
Hou M, Wang Q, Zhang J, Zhao R, Cao Y, Yu S, Wang K, Chen Y, Ma Z, Sun X, et al. Differential Expression of miRNAs, lncRNAs, and circRNAs between Ovaries and Testes in Common Carp (Cyprinus carpio). Cells. 2023; 12(22):2631. https://doi.org/10.3390/cells12222631
Chicago/Turabian StyleHou, Mingxi, Qi Wang, Jin Zhang, Ran Zhao, Yiming Cao, Shuangting Yu, Kaikuo Wang, Yingjie Chen, Ziyao Ma, Xiaoqing Sun, and et al. 2023. "Differential Expression of miRNAs, lncRNAs, and circRNAs between Ovaries and Testes in Common Carp (Cyprinus carpio)" Cells 12, no. 22: 2631. https://doi.org/10.3390/cells12222631