
Estrogen Actions in Placental Vascular Morphogenesis and Spiral Artery Remodeling: A Comparative View between Humans and Mice
 , ,
, ,
Abstract
:
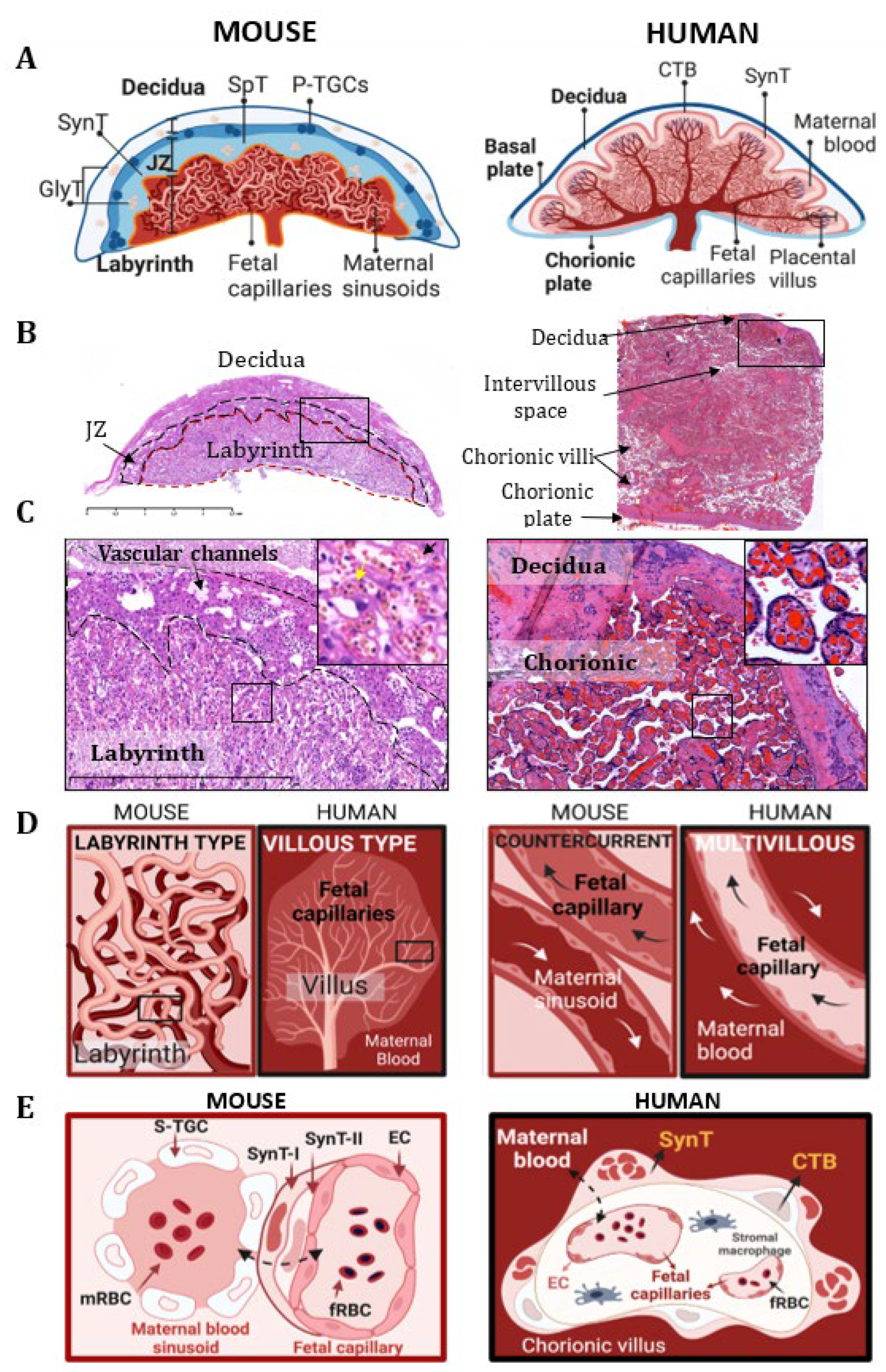
1. Comparative Placental Physiology and the Placental Vascular System between Human and Mouse Placenta
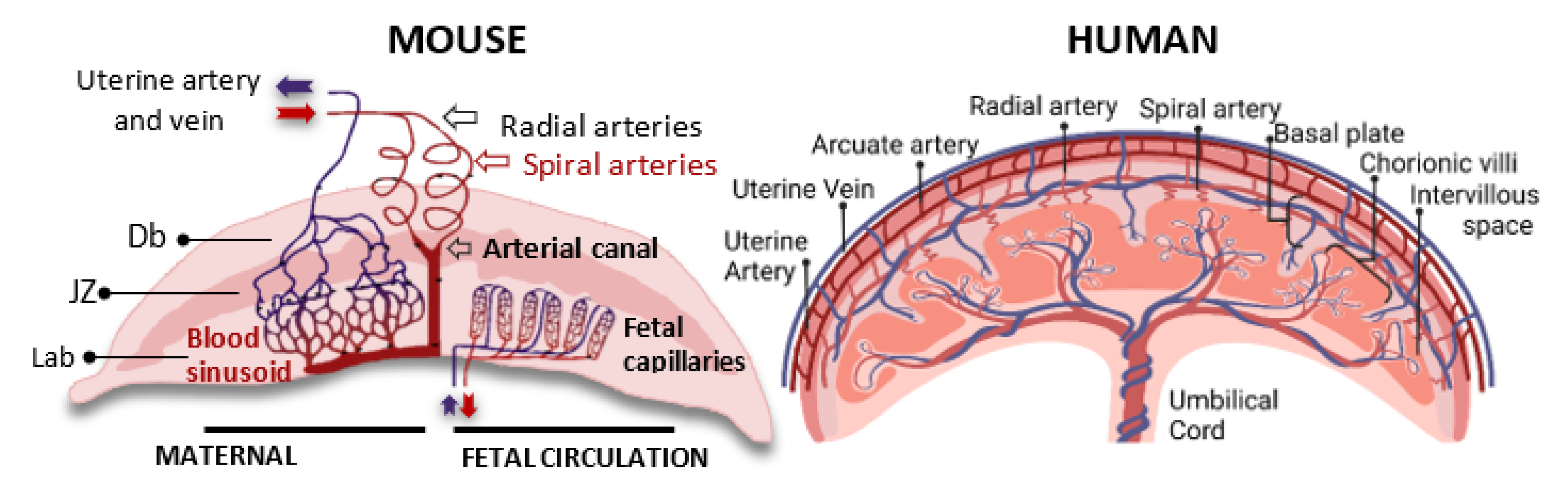
2. Comparative Anatomy of Fetal and Maternal Circulation in the Mouse and Human Placentas
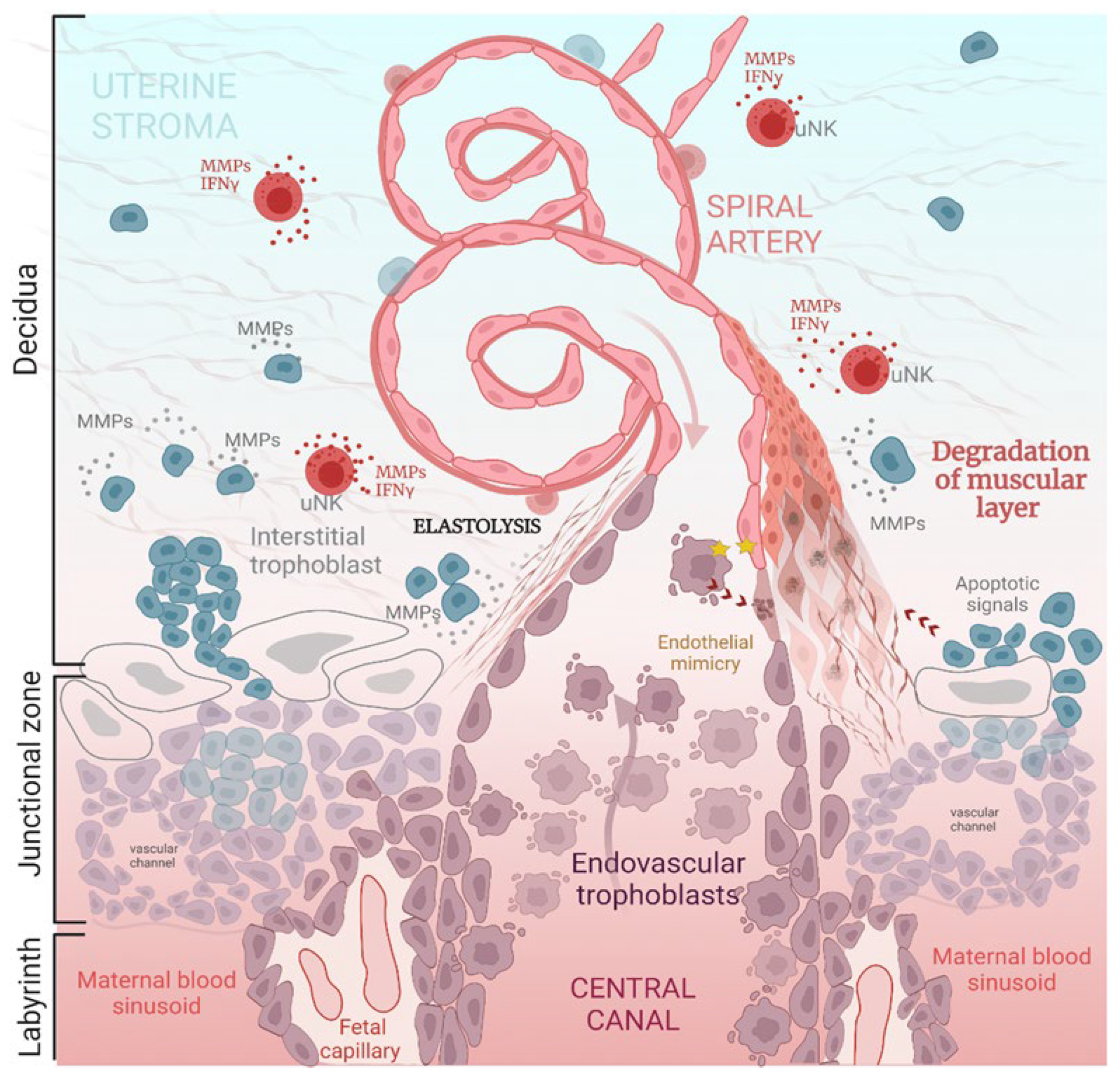
3. Brief Overview of Spiral Artery Remodeling (SAR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HUMAN | MOUSE | |
|---|---|---|
| Common features | ||
| Distinctive features | ||
| Cell-specific markers and secretory phenotype |
|
|
4. Overview of Estrogen Actions during Pregnancy
4.1. Estrogen Actions during Embryo Implantation and Uterine Angiogenesis
| Target | Function | Species | Implicated ER | Ref. | |
|---|---|---|---|---|---|
| Implantation | Uterine epithelium | Tissue responsiveness and functioning; | H, M | ERα | [80] |
| Uterine stroma | Epithelial growth; | H, M | ERα | [82,83] | |
| Pre-implantation remodeling; | H, M | ERα | [80,83,84] | ||
| Blastocyst | Attachment/implantation; | H, M | ERα | [77,78,79] | |
| Decidua | Decidual angiogenesis. | H, M, P | ERα | [51,74,85] | |
| Vascular remodeling | Trophoblast | Viability; | H | [83,86] | |
| Proliferation/differentiation; | H | ERα, ERβ | [87,88] | ||
| Invasion; | H, P | ERα, ERβ | [83,86] | ||
| ECM | MMP-dependent remodeling; | H | [89,90,91] | ||
| Vascular endothelium | NO and VEGFA synthesis; | H, M | ERα | [92,93] | |
| uNK | Migration and functional activity. | H | [36,94,95] | ||
| Systemic hemodynamics | Uterine blood flow Umbilical artery Myometrial artery Placental artery Mesenteric artery Aorta Heart | Increase arterial wall diameter; | H, M | EC/SMC ERα, ERβ | [51,96,97], |
| Relaxation; | H | EC ERα, ERβ | [98] | ||
| Relaxation; | H | EC ERβ | [99] | ||
| Relaxation; | H | SMC ERα | [99] | ||
| Relaxation; | R | EC and SMC ERα | [100] | ||
| Relaxation; | R | EC and SMC ERα | [72,100] | ||
| No major influence; | H, M | [97,101] | |||
| Cardio-vascular adaptation. | |||||
| Labor | Cervix | Softening and dilation; | H, M | [102,103,104] | |
| Myometrium | Contraction increase; | H, M | |||
| Ovary | Luteolysis. | H, M | |||
4.2. E2 and Hemodynamics
4.2.1. Role of Estrogens in Uterine Arterial Remodeling
4.2.2. Role of Estrogens in Uterine Vasodilation
4.2.3. Role of E2 in Relation to Myogenic Tone
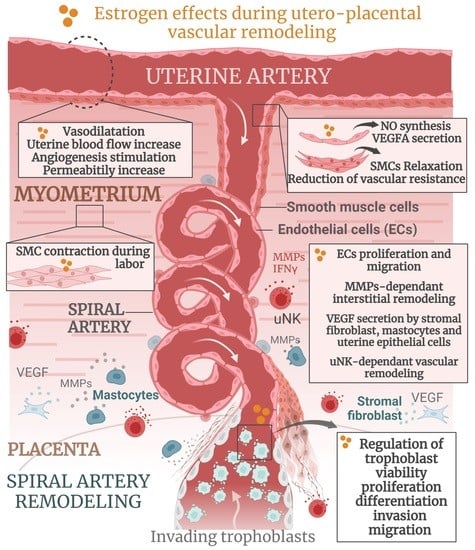
4.3. Estrogen Actions during Spiral Artery Remodeling
4.3.1. Role of Estrogens in SAR in Primates
4.3.2. Role of Estrogens in SAR in Humans
4.3.3. Role of Estrogens in SAR in Mice
5. Estrogens and Abnormal Placentation
6. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Georgiades, P.; Ferguson-Smith, A.; Burton, G. Comparative Developmental Anatomy of the Murine and Human Definitive Placentae. Placenta 2002, 23, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Adamson, S.; Lu, Y.; Whiteley, K.J.; Holmyard, D.; Hemberger, M.; Pfarrer, C.; Cross, J.C. Interactions between Trophoblast Cells and the Maternal and Fetal Circulation in the Mouse Placenta. Dev. Biol. 2002, 250, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Croy, B.A.; Yamada, A.; De Mayo, F.; Adamson, S.L. The Guide to Investigation of Mouse Pregnancy, 1st ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth—New Insights from Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.G.; Fortier, A.L.; Cross, J.C. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev. Biol. 2007, 304, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.; Cross, J. Development of Structures and Transport Functions in the Mouse Placenta. Physiology 2005, 20, 180–193. [Google Scholar] [CrossRef]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2019, 21, 27–43. [Google Scholar] [CrossRef]
- Cross, J.C.; Baczyk, D.; Dobric, N.; Hemberger, M.; Hughes, M.; Simmons, D.G.; Yamamoto, H.; Kingdom, J.C.P. Genes, Development and Evolution of the Placenta. Placenta 2003, 24, 123–130. [Google Scholar] [CrossRef]
- Wildman, D.E.; Chen, C.; Erez, O.; Grossman, L.I.; Goodman, M.; Romero, R. Evolution of the mammalian placenta revealed by phylogenetic analysis. Proc. Natl. Acad. Sci. USA 2006, 103, 3203–3208. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The Uterine Spiral Arteries in Human Pregnancy: Facts and Controversies. Placenta 2006, 27, 939–958. [Google Scholar] [CrossRef]
- Prefumo, F.; Sebire, N.; Thilaganathan, B. Decreased endovascular trophoblast invasion in first trimester pregnancies with high-resistance uterine artery Doppler indices. Hum. Reprod. 2004, 19, 206–209. [Google Scholar] [CrossRef] [Green Version]
- Simmons, D.G.; Rawn, S.; Davies, A.; Hughes, M.; Cross, J.C. Spatial and temporal expression of the 23 murine Prolactin/Placental Lactogen-related genes is not associated with their position in the locus. BMC Genom. 2008, 9, 352. [Google Scholar] [CrossRef]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: From spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef]
- Mandala, M.; Osol, G. Physiological Remodelling of the Maternal Uterine Circulation during Pregnancy. Basic Clin. Pharmacol. Toxicol. 2011, 110, 12–18. [Google Scholar] [CrossRef]
- Burton, G.; Woods, A.; Jauniaux, E.; Kingdom, J. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Croy, B.A.; Chen, Z.; Hofmann, A.P.; Lord, E.M.; Sedlacek, A.; Gerber, S.A. Imaging of Vascular Development in Early Mouse Decidua and Its Association with Leukocytes and Trophoblasts1. Biol. Reprod. 2012, 87, 125. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 207–212. [Google Scholar] [CrossRef]
- Cross, J.; Hemberger, M.; Lu, Y.; Nozaki, T.; Whiteley, K.; Masutani, M.; Adamson, S. Trophoblast functions, angiogenesis and remodeling of the maternal vasculature in the placenta. Mol. Cell Endocrinol. 2002, 187, 207–212. [Google Scholar] [CrossRef]
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef]
- Hu, D.; Cross, J.C. Ablation of Tpbpa-positive trophoblast precursors leads to defects in maternal spiral artery remodeling in the mouse placenta. Dev. Biol. 2011, 358, 231–239. [Google Scholar] [CrossRef]
- Whiteside, E.J.; Jackson, M.M.; Herington, A.C.; Edwards, D.R.; Harvey, M.B. Matrix Metalloproteinase-9 and Tissue Inhibitor of Metalloproteinase-3 Are Key Regulators of Extracellular Matrix Degradation by Mouse Embryos1. Biol. Reprod. 2001, 64, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Whitley, G.S.J.; Cartwright, J.E. Trophoblast-mediated spiral artery remodelling: A role for apoptosis. J. Anat. 2009, 215, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ashton, S.V.; Whitley, G.S.J.; Dash, P.; Wareing, M.; Crocker, I.P.; Baker, P.; Cartwright, J.E. Uterine Spiral Artery Remodeling Involves Endothelial Apoptosis Induced by Extravillous Trophoblasts Through Fas/FasL Interactions. Arter. Thromb. Vasc. Biol. 2005, 25, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Cross, J.C. Development of the hemochorial maternal vascular spaces in the placenta through endothelial and vasculogenic mimicry. Dev. Biol. 2014, 387, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Damsky, C.H.; Fitzgerald, M.L.; Fisher, S.J. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J. Clin. Investig. 1992, 89, 210–222. [Google Scholar] [CrossRef]
- Coan, P.; Conroy, N.; Burton, G.; Ferguson-Smith, A. Origin and characteristics of glycogen cells in the developing murine placenta. Dev. Dyn. 2006, 235, 3280–3294. [Google Scholar] [CrossRef]
- Huppertz, B. Traditional and New Routes of Trophoblast Invasion and Their Implications for Pregnancy Diseases. Int. J. Mol. Sci. 2019, 21, 289. [Google Scholar] [CrossRef]
- Moser, G.; Weiss, G.; Sundl, M.; Gauster, M.; Siwetz, M.; Lang-Olip, I.; Huppertz, B. Extravillous trophoblasts invade more than uterine arteries: Evidence for the invasion of uterine veins. Histochem. Cell Biol. 2016, 147, 353–366. [Google Scholar] [CrossRef]
- Albrecht, E.D.; Pepe, G.J. Regulation of Uterine Spiral Artery Remodeling: A Review. Reprod. Sci. 2020, 27, 1932–1942. [Google Scholar] [CrossRef]
- James, J.; Saghian, R.; Perwick, R.; Clark, A.R. Trophoblast plugs: Impact on utero-placental haemodynamics and spiral artery remodelling. Hum. Reprod. 2018, 33, 1430–1441. [Google Scholar] [CrossRef]
- Harris, L. Review: Trophoblast-Vascular Cell Interactions in Early Pregnancy: How to Remodel a Vessel. Placenta 2010, 31, S93–S98. [Google Scholar] [CrossRef]
- Varberg, K.M.; Soares, M.J. Paradigms for investigating invasive trophoblast cell development and contributions to uterine spiral artery remodeling. Placenta 2021, 113, 48–56. [Google Scholar] [CrossRef]
- Zhou, Y.; Fisher, S.J.; Janatpour, M.; Genbacev, O.; Dejana, E.; Wheelock, M.; Damsky, C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J. Clin. Investig. 1997, 99, 2139–2151. [Google Scholar] [CrossRef]
- Moffet-King, A. Natural killer cells and pregnancy. Nature Reviews Immunology 2002, 2, 656–663. [Google Scholar] [CrossRef]
- Huhn, O.; Zhao, X.; Esposito, L.; Moffett, A.; Colucci, F.; Sharkey, A.M. How Do Uterine Natural Killer and Innate Lymphoid Cells Contribute to Successful Pregnancy? Front. Immunol. 2021, 12, 607669. [Google Scholar] [CrossRef]
- Hanna, J.H.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; I Arnon, T.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef]
- Osol, G.; Mandala, M. Maternal Uterine Vascular Remodeling During Pregnancy. Physiology 2009, 24, 58–71. [Google Scholar] [CrossRef]
- Brosens, I.; Robertson, W.B.; Dixon, H.G. The physiological response of the vessels of the placental bed to normal pregnancy. J. Pathol. Bacteriol. 1967, 93, 569–579. [Google Scholar] [CrossRef]
- Moser, G.; Windsperger, K.; Pollheimer, J.; de Sousa Lopes, S.C.; Huppertz, B. Human trophoblast invasion: New and unexpected routes and functions. Histochem. Cell Biol. 2018, 150, 361–370. [Google Scholar] [CrossRef]
- Brosens, J.; Pijnenborg, R.; Brosens, I.A. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies. Am. J. Obstet. Gynecol. 2002, 187, 1416–1423. [Google Scholar] [CrossRef]
- Hu, D.; Cross, J.C. Development and function of trophoblast giant cells in the rodent placenta. Int. J. Dev. Biol. 2010, 54, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Redline, R.W.; Lu, C.Y. Localization of fetal major histocompatibility complex antigens and maternal leukocytes in murine placenta. Implications for maternal-fetal immunological relationship. Lab. Investig. J. Tech. Methods Pathol. 1989, 61, 27–36. [Google Scholar]
- Charalambous, F.; Elia, A.; Georgiades, P. Decidual spiral artery remodeling during early post-implantation period in mice: Investigation of associations with decidual uNK cells and invasive trophoblast. Biochem. Biophys. Res. Commun. 2012, 417, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Hiby, S.E.; Walker, J.J.; O’Shaughnessy, K.M.; Redman, C.W.; Carrington, M.; Trowsdale, J.; Moffett, A. Combinations of Maternal KIR and Fetal HLA-C Genes Influence the Risk of Preeclampsia and Reproductive Success. J. Exp. Med. 2004, 200, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Huppertz, B.; Frank, H.-G. The fibrinoids of the human placenta: Origin, composition and functional relevance. Ann. Anat. Anat. Anz. 1996, 178, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Croy, B.A.; Zhang, J.; Tayade, C.; Colucci, F.; Yadi, H.; Yamada, A.T. Analysis of Uterine Natural Killer Cells in Mice. Methods Mol. Biol. 2009, 612, 465–503. [Google Scholar] [CrossRef]
- Eick, G.N.; Thornton, J.W. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol. Cell Endocrinol. 2011, 334, 31–38. [Google Scholar] [CrossRef]
- Mesiano, S. The Endocrinology of Human Pregnancy and Fetal-Placental Neuroendocrine Development. Yen Jaffes Reprod. Endocrinol. 2014, 11, 243–271.e8. [Google Scholar] [CrossRef]
- Costa, M.A. The endocrine function of human placenta: An overview. Reprod. Biomed. Online 2016, 32, 14–43. [Google Scholar] [CrossRef]
- Wasada, T.; Akamine, Y.; Kato, K.-I.; Ibayashi, H.; Nomura, Y. Adrenal contribution to circulating estrogens in woman. Endocrinol. Jpn. 1978, 25, 123–128. [Google Scholar] [CrossRef]
- Das, A.; Mantena, S.R.; Kannan, A.; Evans, D.B.; Bagchi, M.K.; Bagchi, I.C. De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12542–12547. [Google Scholar] [CrossRef]
- Malassine, A.; Frendo, J.L.; Evain-Brion, D. A comparison of placental development and endocrine functions between the human and mouse model. Hum. Reprod. Updat. 2003, 9, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Devroey, P.; Camus, M.; Palermo, G.; Smitz, J.; Van Waesberghe, L.; Wisanto, A.; Wijbo, I.; Van Steirteghem, A.C. Placental production of estradiol and progesterone after oocyte donation in patients with primary ovarian failure. Am. J. Obstet. Gynecol. 1990, 162, 66–70. [Google Scholar] [CrossRef]
- Oakey, R. The Progressive Increase in Estrogen Production in Human Pregnancy: An Appraisal of the Factors Responsible. Vitam. Horm. 1971, 28, 1–36. [Google Scholar] [CrossRef]
- Abbassi-Ghanavati, M.; Greer, L.G.; Cunningham, F.G. Pregnancy and Laboratory Studies. Obstet. Gynecol. 2009, 114, 1326–1331. [Google Scholar] [CrossRef]
- Albrecht, E.D.; Pepe, G.J. Placental Steroid Hormone Biosynthesis in Primate Pregnancy. Endocr. Rev. 1990, 11, 124–150. [Google Scholar] [CrossRef]
- Peter, M.; Dörr, H.; Sippell, W. Changes in the Concentrations of Dehydroepiandrosterone Sulfate and Estriol in Maternal Plasma during Pregnancy: A Longitudinal Study in Healthy Women throughout Gestation and at Term. Horm. Res. 1994, 42, 278–281. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Siiteri, P.K.; Macdonald, P.C. Placental Estrogen Biosynthesis During Human Pregnancy1. J. Clin. Endocrinol. Metab. 1966, 26, 751–761. [Google Scholar] [CrossRef]
- Thomas, M.P.; Potter, B.V. The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Biol. 2013, 137, 27–49. [Google Scholar] [CrossRef]
- Falah, N.; Torday, J.; Quinney, S.K.; Haas, D.M. Estriol review: Clinical applications and potential biomedical importance. Clin. Res. Trials 2015, 1, 29–33. [Google Scholar] [CrossRef]
- Gérard, C.; Arnal, J.-F.; Jost, M.; Douxfils, J.; Lenfant, F.; Fontaine, C.; Houtman, R.; Archer, D.F.; Reid, R.L.; Lobo, R.A.; et al. Profile of estetrol, a promising native estrogen for oral contraception and the relief of climacteric symptoms of menopause. Expert Rev. Clin. Pharmacol. 2022, 15, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Estetrol: A Unique Steroid in Human Pregnancy. Available online: https://pubmed.ncbi.nlm.nih.gov/18462934/ (accessed on 13 December 2022).
- Chung, E.; Yeung, F.; Leinwand, L.A. Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J. Appl. Physiol. 2012, 112, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Devillers, M.M.; Petit, F.; Cluzet, V.; François, C.M.; Giton, F.; Garrel, G.; Cohen-Tannoudji, J.; Guigon, C.J. FSH inhibits AMH to support ovarian estradiol synthesis in infantile mice. J. Endocrinol. 2019, 240, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Maliqueo, M.; Echiburú, B.; Crisosto, N. Sex Steroids Modulate Uterine-Placental Vasculature: Implications for Obstetrics and Neonatal Outcomes. Front. Physiol. 2016, 7, 152. [Google Scholar] [CrossRef] [PubMed]
- Calzada-Mendoza, C.C.; Sánchez, E.C.; Campos, R.R.; Becerril, A.M.; Madrigal, E.B.; Sierra, A.R.; Mendez, E.B.; Ocharán, E.H.; Herrera, N.G. Differential aromatase (CYP19) expression in human arteries from normal and neoplasic uterus: An immunohistochemical and in situ hybridization study. Front. Biosci. 2006, 11, 389–393. [Google Scholar] [CrossRef]
- Bausero, P.; Ben-Mahdi, M.-H.; Mazucatelli, J.-P.; Bloy, C.; Perrot-Applanat, M. Vascular endothelial growth factor is modulated in vascular muscle cells by estradiol, tamoxifen, and hypoxia. Am. J. Physiol. Circ. Physiol. 2000, 279, H2033–H2042. [Google Scholar] [CrossRef]
- Napso, T.; Yong, H.E.J.; Lopez-Tello, J.; Sferruzzi-Perri, A.N. The Role of Placental Hormones in Mediating Maternal Adaptations to Support Pregnancy and Lactation. Front. Physiol. 2018, 9, 1091. [Google Scholar] [CrossRef]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Pastore, M.B.; Jobe, S.O.; Ramadoss, J.; Magness, R.R. Estrogen Receptor-α and Estrogen Receptor-β in the Uterine Vascular Endothelium during Pregnancy: Functional Implications for Regulating Uterine Blood Flow. Semin. Reprod. Med. 2012, 30, 46–61. [Google Scholar] [CrossRef]
- Mata, K.M.; Li, W.; Reslan, O.M.; Siddiqui, W.T.; Opsasnick, L.A.; Khalil, R.A. Adaptive increases in expression and vasodilator activity of estrogen receptor subtypes in a blood vessel-specific pattern during pregnancy. Am. J. Physiol. Circ. Physiol. 2015, 309, H1679–H1696. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Kirsch, J.D.; Kraft, K.C.; Knutson, D.L.; McClaflin, W.J.; Redmer, D.A. Time-Course of the Uterine Response to Estradiol-17β in Ovariectomized Ewes: Uterine Growth and Microvascular Development1. Biol. Reprod. 1998, 59, 606–612. [Google Scholar] [CrossRef]
- Albrecht, E.D.; Pepe, G.J. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int. J. Dev. Biol. 2010, 54, 397–408. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R. Local and systemic estradiol-17 beta: Effects on uterine and systemic vasodilation. Am. J. Physiol. Metab. 1989, 256, E536–E542. [Google Scholar] [CrossRef]
- Bs, K.C.; Zhang, L. Steroid Hormones and Uterine Vascular Adaptation to Pregnancy. Reprod. Sci. 2008, 15, 336–348. [Google Scholar] [CrossRef]
- Egashira, M.; Hirota, Y. Uterine receptivity and embryo-uterine interactions in embryo implantation: Lessons from mice. Reprod. Med. Biol. 2013, 12, 127–132. [Google Scholar] [CrossRef]
- Groothuis, P.; Dassen, H.; Romano, A.; Punyadeera, C. Estrogen and the endometrium: Lessons learned from gene expression profiling in rodents and human. Hum. Reprod. Updat. 2007, 13, 405–417. [Google Scholar] [CrossRef]
- Simon, C.; Dominguez, F.; Valbuena, D.; Pellicer, A.; Perilla, D.V. The role of estrogen in uterine receptivity and blastocyst implantation. Trends Endocrinol. Metab. 2003, 14, 197–199. [Google Scholar] [CrossRef]
- Winuthayanon, W.; Hewitt, S.; Korach, K.S. Uterine Epithelial Cell Estrogen Receptor Alpha-Dependent and -Independent Genomic Profiles That Underlie Estrogen Responses in Mice1. Biol. Reprod. 2014, 91, 110. [Google Scholar] [CrossRef]
- Pawar, S.; Laws, M.J.; Bagchi, I.C.; Bagchi, M.K. Uterine Epithelial Estrogen Receptor-α Controls Decidualization via a Paracrine Mechanism. Mol. Endocrinol. 2015, 29, 1362–1374. [Google Scholar] [CrossRef]
- Hewitt, S.; Goulding, E.H.; Eddy, E.; Korach, K. Studies Using the Estrogen Receptor α Knockout Uterus Demonstrate That Implantation but Not Decidualization-Associated Signaling Is Estrogen Dependent. Biol. Reprod. 2002, 67, 1268–1277. [Google Scholar] [CrossRef]
- Wang, H.; Bocca, S.; Anderson, S.; Yu, L.; Rhavi, B.S.; Horcajadas, J.; Oehninger, S. Sex Steroids Regulate Epithelial–Stromal Cell Cross Talk and Trophoblast Attachment Invasion in a Three-Dimensional Human Endometrial Culture System. Tissue Eng. Part C Methods 2013, 19, 676–687. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.T.; Greenwald, G.S. Evidence for a preimplantation rise in oestradiol-17 levels on day 4 of pregnancy in the mouse. Reproduction 1974, 41, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Isner, J.M. Estrogen and Angiogenesis. Arter. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [PubMed]
- He, W.-H.; Jin, M.-M.; Liu, A.-P.; Zhou, Y.; Hu, X.-L.; Zhu, Y.-M. Estradiol promotes trophoblast viability and invasion by activating SGK1. Biomed. Pharmacother. 2019, 117, 109092. [Google Scholar] [CrossRef] [PubMed]
- Bukovsky, A.; Caudle, M.R.; Cekanova, M.; Fernando, R.I.; Wimalasena, J.; Foster, J.S.; Henley, D.C.; Elder, R.F. Placental expression of estrogen receptor beta and its hormone binding variant—Comparison with estrogen receptor alpha and a role for estrogen receptors in asymmetric division and differentiation of estrogen-dependent cells. Reprod. Biol. Endocrinol. 2003, 1, 36. [Google Scholar] [CrossRef]
- Bonagura, T.W.; Pepe, G.J.; Enders, A.C.; Albrecht, E.D. Suppression of Extravillous Trophoblast Vascular Endothelial Growth Factor Expression and Uterine Spiral Artery Invasion by Estrogen during Early Baboon Pregnancy. Endocrinology 2008, 149, 5078–5087. [Google Scholar] [CrossRef]
- Ietta, F.; Bechi, N.; Romagnoli, R.; Bhattacharjee, J.; Realacci, M.; Di Vito, M.; Ferretti, C.; Paulesu, L. 17β-Estradiol modulates the macrophage migration inhibitory factor secretory pathway by regulating ABCA1 expression in human first-trimester placenta. Am. J. Physiol. Metab. 2010, 298, E411–E418. [Google Scholar] [CrossRef]
- Krivokuća, M.J.; Stefanoska, I.; Abu Rabi, T.; Al-Abed, Y.; Stošić-Grujičić, S.; Vićovac, L. Pharmacological inhibition of MIF interferes with trophoblast cell migration and invasiveness. Placenta 2015, 36, 150–159. [Google Scholar] [CrossRef]
- Dang, Y.; Li, W.; Tran, V.; Khalil, R.A. EMMPRIN-mediated induction of uterine and vascular matrix metalloproteinases during pregnancy and in response to estrogen and progesterone. Biochem. Pharmacol. 2013, 86, 734–747. [Google Scholar] [CrossRef]
- Van Der Heijden, O.W.; Essers, Y.P.; Fazzi, G.; Peeters, L.L.; De Mey, J.G.R.; Van Eys, G.J. Uterine Artery Remodeling and Reproductive Performance Are Impaired in Endothelial Nitric Oxide Synthase-Deficient Mice1. Biol. Reprod. 2005, 72, 1161–1168. [Google Scholar] [CrossRef]
- Rockwell, L.C.; Pillai, S.; Olson, C.E.; Koos, R.D. Inhibition of Vascular Endothelial Growth Factor/Vascular Permeability Factor Action Blocks Estrogen-Induced Uterine Edema and Implantation in Rodents1. Biol. Reprod. 2002, 67, 1804–1810. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.; Greaves, E.; Critchley, H.; Saunders, P. Estrogen-dependent regulation of human uterine natural killer cells promotes vascular remodelling via secretion of CCL2. Hum. Reprod. 2015, 30, 1290–1301. [Google Scholar] [CrossRef]
- Moffett, A.; Loke, C. Immunology of placentation in eutherian mammals. Nat. Rev. Immunol. 2006, 6, 584–594. [Google Scholar] [CrossRef]
- Mandalà, M. Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies. Int. J. Mol. Sci. 2020, 21, 2592. [Google Scholar] [CrossRef]
- Sanghavi, M.; Rutherford, J.D. Cardiovascular Physiology of Pregnancy. Circulation 2014, 130, 1003–1008. [Google Scholar] [CrossRef]
- de Sá, M.F.S.; Meirelles, R.S. Vasodilating Effect of Estrogen on the Human Umbilical Artery. Gynecol. Obstet. Investig. 1977, 8, 307–313. [Google Scholar] [CrossRef]
- Corcoran, J.J.; Nicholson, C.; Sweeney, M.; Charnock, J.C.; Robson, S.C.; Westwood, M.; Taggart, M.J. Human uterine and placental arteries exhibit tissue-specific acute responses to 17 -estradiol and estrogen-receptor-specific agonists. Mol. Hum. Reprod. 2013, 20, 433–441. [Google Scholar] [CrossRef]
- Leiberman, J.R.; Wiznitzer, A.; Glezerman, M.; Feldman, B.; Levy, J.; Sharoni, Y. Estrogen and progesterone receptors in the uterine artery of rats during and after pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1993, 51, 35–40. [Google Scholar] [CrossRef]
- Kulandavelu, S.; Qu, D.; Adamson, S.L. Cardiovascular Function in Mice During Normal Pregnancy and in the Absence of Endothelial NO Synthase. Hypertension 2006, 47, 1175–1182. [Google Scholar] [CrossRef]
- Challis, J.R.; Matthews, S.G.; Gibb, W.; Lye, S.J. Endocrine and Paracrine Regulation of Birth at Term and Preterm*. Endocr. Rev. 2000, 21, 514–550. [Google Scholar] [CrossRef]
- Lye, S.J.; Nicholson, B.J.; Mascarenhas, M.; MacKenzie, L.; Petrocelli, T. Increased expression of connexin-43 in the rat myometrium during labor is associated with an increase in the plasma estrogen: Progesterone ratio. Endocrinology 1993, 132, 2380–2386. [Google Scholar] [CrossRef]
- Welsh, T.; Johnson, M.; Yi, L.; Tan, H.; Rahman, R.; Merlino, A.; Zakar, T.; Mesiano, S. Estrogen receptor (ER) expression and function in the pregnant human myometrium: Estradiol via ERα activates ERK1/2 signaling in term myometrium. J. Endocrinol. 2011, 212, 227–238. [Google Scholar] [CrossRef]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Saoudi, Y.; Benharouga, M.; Graham, C.H.; Schaal, J.P.; Mazouni, C.; Feige, J.J.; Alfaidy, N. Role of EG-VEGF in human placentation: Physiological and pathological implications. J. Cell Mol. Med. 2009, 13, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Nien, J.K.; Espinoza, J.; Todem, D.; Fu, W.; Chung, H.; Kusanovic, J.P.; Gotsch, F.; Erez, O.; Mazaki-Tovi, S.; et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J. Matern. Neonatal Med. 2008, 21, 9–23. [Google Scholar] [CrossRef]
- Cullinan-Bove, K. Vascular endothelial growth factor/vascular permeability factor expression in the rat uterus: Rapid stimulation by estrogen correlates with estrogen-induced increases in uterine capillary permeability and growth. Endocrinology 1993, 133, 829–837. [Google Scholar] [CrossRef]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.; Sobel, M. Heparin-II Domain of Fibronectin Is a Vascular Endothelial Growth Factor-Binding Domain. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef]
- Pence, J.C.; Clancy, K.B.H.; Harley, B.A.C. The induction of pro-angiogenic processes within a collagen scaffold via exogenous estradiol and endometrial epithelial cells. Biotechnol. Bioeng. 2015, 112, 2185–2194. [Google Scholar] [CrossRef]
- Smith, S.K. Regulation of angiogenesis in the endometrium. Trends Endocrinol. Metab. 2001, 12, 147–151. [Google Scholar] [CrossRef]
- Hervé, M.A.J.; Meduri, G.; Petit, F.; Domet, T.S.; Lazennec, G.; Mourah, S.; Perrot-Applanat, M. Regulation of the vascular endothelial growth factor (VEGF) receptor Flk-1/KDR by estradiol through VEGF in uterus. J. Endocrinol. 2006, 188, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Zolton, J.R.; Sjaarda, L.A.; Mumford, S.L.; DeVilbiss, E.A.; Kim, K.; Flannagan, K.S.; Radoc, J.G.; Perkins, N.J.; Silver, R.M.; Wactawski-Wende, J.; et al. Circulating Vascular Endothelial Growth Factor and Soluble fms-Like Tyrosine Kinase-1 as Biomarkers for Endometrial Remodeling Across the Menstrual Cycle. Obstet. Gynecol. 2020, 137, 82–90. [Google Scholar] [CrossRef]
- Rajakumar, A.; Conrad, K.P. Expression, Ontogeny, and Regulation of Hypoxia-Inducible Transcription Factors in the Human Placenta1. Biol. Reprod. 2000, 63, 559–569. [Google Scholar] [CrossRef]
- Miyauchi, Y.; Sato, Y.; Kobayashi, T.; Yoshida, S.; Mori, T.; Kanagawa, H.; Katsuyama, E.; Fujie, A.; Hao, W.; Miyamoto, K.; et al. HIF1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16568–16573. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.-L.; Baban, D.; et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef]
- Khorram, O.; Garthwaite, M.; Magness, R.R. Endometrial and Myometrial Expression of Nitric Oxide Synthase Isoforms in Pre- and Postmenopausal Women. J. Clin. Endocrinol. Metab. 1999, 84, 2226–2232. [Google Scholar] [CrossRef]
- Tagawa, H.; Shimokawa, H.; Tagawa, T.; Kuroiwa-Matsumoto, M.; Hirooka, Y.; Takeshita, A. Short-Term Estrogen Augments Both Nitric Oxide-Mediated and Non-Nitric Oxide-Mediated Endothelium-Dependent Forearm Vasodilation in Postmenopausal Women. J. Cardiovasc. Pharmacol. 1997, 30, 481–488. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef]
- Krasinski, K.; Spyridopoulos, I.; Asahara, T.; Van Der Zee, R.; Isner, J.M.; Losordo, D.W. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation 1997, 95, 1768–1772. [Google Scholar] [CrossRef]
- Hunt, J.S.; Miller, L.; Vassmer, D.; Croy, B.A. Expression of the Inducible Nitric Oxide Synthase Gene in Mouse Uterine Leukocytes and Potential Relationships with Uterine Function during Pregnancy1. Biol. Reprod. 1997, 57, 827–836. [Google Scholar] [CrossRef]
- Robb, V.A.; Pepe, G.J.; Albrecht, E.D. Acute Temporal Regulation of Placental Vascular Endothelial Growth/Permeability Factor Expression in Baboons by Estrogen1. Biol. Reprod. 2004, 71, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Qi, Q.-R.; Li, Y.; Day, R.; Makhoul, J.; Magness, R.R.; Chen, D.-B. Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy. Int. J. Mol. Sci. 2020, 21, 4349. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.K.; Zamudio, S.; Coffin, C.; Parker, S.; Stamm, E.; Moore, L. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet. Gynecol. 1992, 80, 1000–1006. [Google Scholar] [PubMed]
- Khankin, E.V.; Ko, N.L.; Mandalà, M.; Karumanchi, S.A.; Osol, G. Normalization of wall shear stress as a physiological mechanism for regulating maternal uterine artery expansive remodeling during pregnancy. FASEB BioAdv. 2021, 3, 702–708. [Google Scholar] [CrossRef]
- Van Der Heijden, O.W.; Essers, Y.P.; Spaanderman, M.E.; De Mey, J.G.R.; Van Eys, G.J.; Peeters, L.L. Uterine Artery Remodeling in Pseudopregnancy Is Comparable to That in Early Pregnancy1. Biol. Reprod. 2005, 73, 1289–1293. [Google Scholar] [CrossRef]
- Orimo, A.; Inoue, S.; Ouchi, Y.; Orimo, H. Vascular Smooth Muscle Cells Possess Estrogen Receptor and Respond to Estrogena. Ann. N. Y. Acad. Sci. 1994, 748, 592–594. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Cox, B.E.; Roy, T.; Magness, R.R. Nitric oxide contributes to estrogen-induced vasodilation of the ovine uterine circulation. J. Clin. Investig. 1996, 98, 2158–2166. [Google Scholar] [CrossRef]
- Chen, D.-B.; Bird, I.M.; Zheng, J.; Magness, R.R. Membrane Estrogen Receptor-Dependent Extracellular Signal-Regulated Kinase Pathway Mediates Acute Activation of Endothelial Nitric Oxide Synthase by Estrogen in Uterine Artery Endothelial Cells. Endocrinology 2004, 145, 113–125. [Google Scholar] [CrossRef]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.-L.; Boudou, F.; Sautier, L.; Vessières, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2013, 111, E283–E290. [Google Scholar] [CrossRef]
- Rusidzé, M.; Faure, M.C.; Sicard, P.; Raymond-Letron, I.; Giton, F.; Vessieres, E.; Prevot, V.; Henrion, D.; Arnal, J.-F.; Cornil, C.A.; et al. Loss of function of the maternal membrane oestrogen receptor ERα alters expansion of trophoblast cells and impacts mouse fertility. Development 2022, 149, dev200683. [Google Scholar] [CrossRef]
- Kublickiene, K.-R.; Kublickas, M.; Lindblom, B.; Lunell, N.-O.; Nisell, H. A comparison of myogenic and endothelial properties of myometrial and omental resistance vessels in late pregnancy. Am. J. Obstet. Gynecol. 1997, 176, 560–566. [Google Scholar] [CrossRef]
- Stice, S.L.; Ford, S.P.; Rosazza, J.P.; Van Orden, D.E. Interaction of 4-Hydroxylated Estradiol and Potential- Sensitive Ca2+ Channels in Altering Uterine Blood Flow during the Estrous Cycle and Early Pregnancy in Gilts1. Biol. Reprod. 1987, 36, 369–375. [Google Scholar] [CrossRef]
- Kumar, P.; Kamat, A.; Mendelson, C.R. Estrogen Receptor α (ERα) Mediates Stimulatory Effects of Estrogen on Aromatase (CYP19) Gene Expression in Human Placenta. Mol. Endocrinol. 2009, 23, 784–793. [Google Scholar] [CrossRef]
- Babischkin, J.; Burleigh, D.; Mayhew, T.; Pepe, G.; Albrecht, E. Developmental Regulation of Morphological Differentiation of Placental Villous Trophoblast in the Baboon. Placenta 2001, 22, 276–283. [Google Scholar] [CrossRef]
- Aberdeen, G.W.; Bonagura, T.W.; Harman, C.R.; Pepe, G.J.; Albrecht, E.D. Suppression of trophoblast uterine spiral artery remodeling by estrogen during baboon pregnancy: Impact on uterine and fetal blood flow dynamics. Am. J. Physiol. Circ. Physiol. 2012, 302, H1936–H1944. [Google Scholar] [CrossRef]
- Schiessl, B.; Mylonas, I.; Hantschmann, P.; Kuhn, C.; Schulze, S.; Kunze, S.; Friese, K.; Jeschke, U. Expression of Endothelial NO Synthase, Inducible NO Synthase, and Estrogen Receptors Alpha and Beta in Placental Tissue of Normal, Preeclamptic, and Intrauterine Growth-restricted Pregnancies. J. Histochem. Cytochem. 2005, 53, 1441–1449. [Google Scholar] [CrossRef]
- Patel, S.; Kilburn, B.; Imudia, A.; Armant, D.R.; Skafar, D.F. Estradiol Elicits Proapoptotic and Antiproliferative Effects in Human Trophoblast Cells1. Biol. Reprod. 2015, 93, 74. [Google Scholar] [CrossRef]
- Bukovsky, A.; Cekanova, M.; Caudle, M.R.; Wimalasena, J.; Foster, J.S.; Henley, D.C.; Elder, R.F. Expression and localization of estrogen receptor-alpha protein in normal and abnormal term placentae and stimulation of trophoblast differentiation by estradiol. Reprod. Biol. Endocrinol. 2003, 1, 13. [Google Scholar] [CrossRef]
- De Loia, J.A.; Stewart-Akers, A.M.; Brekosky, J.; Kubik, C.J. Effects of exogenous estrogen on uterine leukocyte recruitment. Fertil. Steril. 2002, 77, 548–554. [Google Scholar] [CrossRef]
- Henderson, T.A.; Saunders, P.; Moffett-King, A.; Groome, N.P.; Critchley, H.O.D. Steroid Receptor Expression in Uterine Natural Killer Cells. J. Clin. Endocrinol. Metab. 2003, 88, 440–449. [Google Scholar] [CrossRef]
- Hofmann, A.P.; Gerber, S.A.; Croy, B.A. Uterine natural killer cells pace early development of mouse decidua basalis. Mol. Hum. Reprod. 2013, 20, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Borzychowski, A.; Chantakru, S.; Minhas, K.; Paffaro, V.; Yamada, A.; He, H.; Korach, K.; Croy, B. Functional Analysis of Murine Uterine Natural Killer Cells Genetically Devoid of Oestrogen Receptors. Placenta 2003, 24, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Hossain, N.; Paidas, M.J. Adverse Pregnancy Outcome, the Uteroplacental Interface, and Preventive Strategies. Semin. Perinatol. 2007, 31, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Placental Oxidative Stress: From Miscarriage to Preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, M.; Nakayama, T.; Sato, I.; Sato, N.; Izawa, N.; Mizutani, Y.; Furuya, K.; Yamamoto, T. Estrogen synthesis genes CYP19A1, HSD3B1, and HSD3B2 in hypertensive disorders of pregnancy. Endocrine 2012, 42, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wu, Y.; Chen, X.; Chang, X.; Zhou, Q.; Zhou, J.; Ying, H.; Zheng, J.; Duan, T.; Wang, K. Decreased Maternal Serum 2-Methoxyestradiol Levels are Associated with the Development of Preeclampsia. Cell Physiol. Biochem. 2014, 34, 2189–2199. [Google Scholar] [CrossRef]
- Wuu, J.; Hellerstein, S.; Lipworth, L.; Wide, L.; Xu, B.; Yu, G.-P.; Kuper, H.; Lagiou, P.; Hankinson, S.E.; Ekbom, A.; et al. Correlates of pregnancy oestrogen, progesterone and sex hormone-binding globulin in the USA and China. Eur. J. Cancer Prev. 2002, 11, 283–293. [Google Scholar] [CrossRef]
- Farhi, J.; Ben Haroush, A.; Andrawus, N.; Pinkas, H.; Sapir, O.; Fisch, B.; Ashkenazi, J. High serum oestradiol concentrations in IVF cycles increase the risk of pregnancy complications related to abnormal placentation. Reprod. Biomed. Online 2010, 21, 331–337. [Google Scholar] [CrossRef]
- Ma, W.-G.; Song, H.; Das, S.K.; Paria, B.C.; Dey, S.K. Estrogen is a critical determinant that specifies the duration of the window of uterine receptivity for implantation. Proc. Natl. Acad. Sci. USA 2003, 100, 2963–2968. [Google Scholar] [CrossRef]
- Palomba, S.; Russo, T.; Falbo, A.; Di Cello, A.; Tolino, A.; Tucci, L.; La Sala, G.B.; Zullo, F. Macroscopic and microscopic findings of the placenta in women with polycystic ovary syndrome. Hum. Reprod. 2013, 28, 2838–2847. [Google Scholar] [CrossRef]
- Maliqueo, M.; Poromaa, I.S.; Vanky, E.; Fornes, R.; Benrick, A.; Akerud, H.; Stridsklev, S.; Labrie, F.; Jansson, T.; Stener-Victorin, E. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Maliqueo, M.; Benrick, A.; Johansson, J.; Shao, R.; Hou, L.; Jansson, T.; Wu, X.; Stener-Victorin, E. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am. J. Physiol. Metab. 2012, 303, E1373–E1385. [Google Scholar] [CrossRef]
- Gould, D.A.; Moscoso, G.J.; Young, M.P.A.; Barton, D.P.J. Human First Trimester Fetal Ovaries Express Oncofetal Antigens and Steroid Receptors. J. Soc. Gynecol. Investig. 2000, 7, 131–138. [Google Scholar] [CrossRef]
- Starzyk, K.A.; Salafia, C.M.; Pezzullo, J.C.; Lage, J.M.; Parkash, V.; Vercruysse, L.; Hanssens, M.; Pijnenborg, R. Quantitative differences in arterial morphometry define the placental bed in preeclampsia. Hum. Pathol. 1997, 28, 353–358. [Google Scholar] [CrossRef]
- Açıkgöz, S.; Bayar, U.O.; Can, M.; Güven, B.; Mungan, G.; Doğan, S.; Sümbüloğlu, V. Levels of Oxidized LDL, Estrogens, and Progesterone in Placenta Tissues and Serum Paraoxonase Activity in Preeclampsia. Mediat. Inflamm. 2013, 2013, 862982. [Google Scholar] [CrossRef]
- Kadyrov, M.; Schmitz, C.; Black, S.; Kaufmann, P.; Huppertz, B. Pre-eclampsia and Maternal Anaemia Display Reduced Apoptosis and Opposite Invasive Phenotypes of Extravillous Trophoblast. Placenta 2003, 24, 540–548. [Google Scholar] [CrossRef]
- Sosa, S.E.Y.; Flores-Pliego, A.; Espejel-Nuñez, A.; Medina-Bastidas, D.; Vadillo-Ortega, F.; Zaga-Clavellina, V.; Estrada-Gutierrez, G. New Insights into the Role of Matrix Metalloproteinases in Preeclampsia. Int. J. Mol. Sci. 2017, 18, 1448. [Google Scholar] [CrossRef]
- Merchant, S.J.; Davidge, S.T. The role of matrix metalloproteinases in vascular function: Implications for normal pregnancy and pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 2004, 111, 931–939. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Chiu, K.; Roberts, J.M.; Fisher, S.J. Preeclampsia is associated with abnormal expression of adhesion molecules by invasive cytotrophoblasts. J. Clin. Investig. 1993, 91, 950–960. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef]
- Kanasaki, K.; Palmsten, K.; Sugimoto, H.; Ahmad, S.; Hamano, Y.; Xie, L.; Parry, S.; Augustin, H.G.; Gattone, V.H.; Folkman, J.; et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008, 453, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Jin, J.; Shan, X. The effects of estradiol on inflammatory and endothelial dysfunction in rats with preeclampsia. Int. J. Mol. Med. 2020, 45, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusidzé, M.; Gargaros, A.; Fébrissy, C.; Dubucs, C.; Weyl, A.; Ousselin, J.; Aziza, J.; Arnal, J.-F.; Lenfant, F. Estrogen Actions in Placental Vascular Morphogenesis and Spiral Artery Remodeling: A Comparative View between Humans and Mice. Cells 2023, 12, 620. https://doi.org/10.3390/cells12040620
Rusidzé M, Gargaros A, Fébrissy C, Dubucs C, Weyl A, Ousselin J, Aziza J, Arnal J-F, Lenfant F. Estrogen Actions in Placental Vascular Morphogenesis and Spiral Artery Remodeling: A Comparative View between Humans and Mice. Cells. 2023; 12(4):620. https://doi.org/10.3390/cells12040620
Chicago/Turabian StyleRusidzé, Mariam, Adrien Gargaros, Chanaëlle Fébrissy, Charlotte Dubucs, Ariane Weyl, Jessie Ousselin, Jacqueline Aziza, Jean-François Arnal, and Françoise Lenfant. 2023. "Estrogen Actions in Placental Vascular Morphogenesis and Spiral Artery Remodeling: A Comparative View between Humans and Mice" Cells 12, no. 4: 620. https://doi.org/10.3390/cells12040620