

Intrahippocampal Inoculation of Aβ1–42 Peptide in Rat as a Model of Alzheimer’s Disease Identified MicroRNA-146a-5p as Blood Marker with Anti-Inflammatory Function in Astrocyte Cells

 , , ,
, , ,  , ,
, ,
Abstract
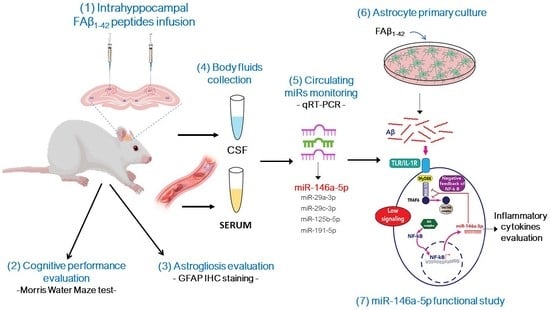
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Amyloid-Β1–42 Peptide for In Vivo Use
2.3. Model of Alzheimer Disease Generated by Intracranial Infusion of FAβ1–42
2.4. Morris Water Maze Test
2.5. Body Fluid Collection and Sampling
2.5.1. Blood Collection and Serum Separation
2.5.2. Cerebrospinal Fluid Collection and Preparation
2.6. Analysis of Circulating miRNAs Expression in Animals
2.6.1. Total RNA Extraction from Body Fluids
2.6.2. Reverse Transcription Reaction (RT)
2.6.3. Real Time Quantitative RT-PCR (qRT-PCR)
2.7. Immunofluorescence Staining
2.8. Primary Culture, FAβ1–42 Treatment, qRT-PCR, and ELISA
2.8.1. Primary Astrocytes Preparation and Culture
2.8.2. Treatment of Primary Astrocytes with FAβ, LPS, BMS345541, and miRNA Inhibitor
2.8.3. Cell Viability Assay
2.8.4. miRNA and mRNA Quantification from Primary Astrocytes Culture
2.8.5. Sandwich Enzyme-Linked Immunosorbent Assay
2.9. Statistical Analysis
3. Results
3.1. Cognitive Impairment in Rats Inoculated with 1 µg/µL of FAβ1–42 Peptides Solutions
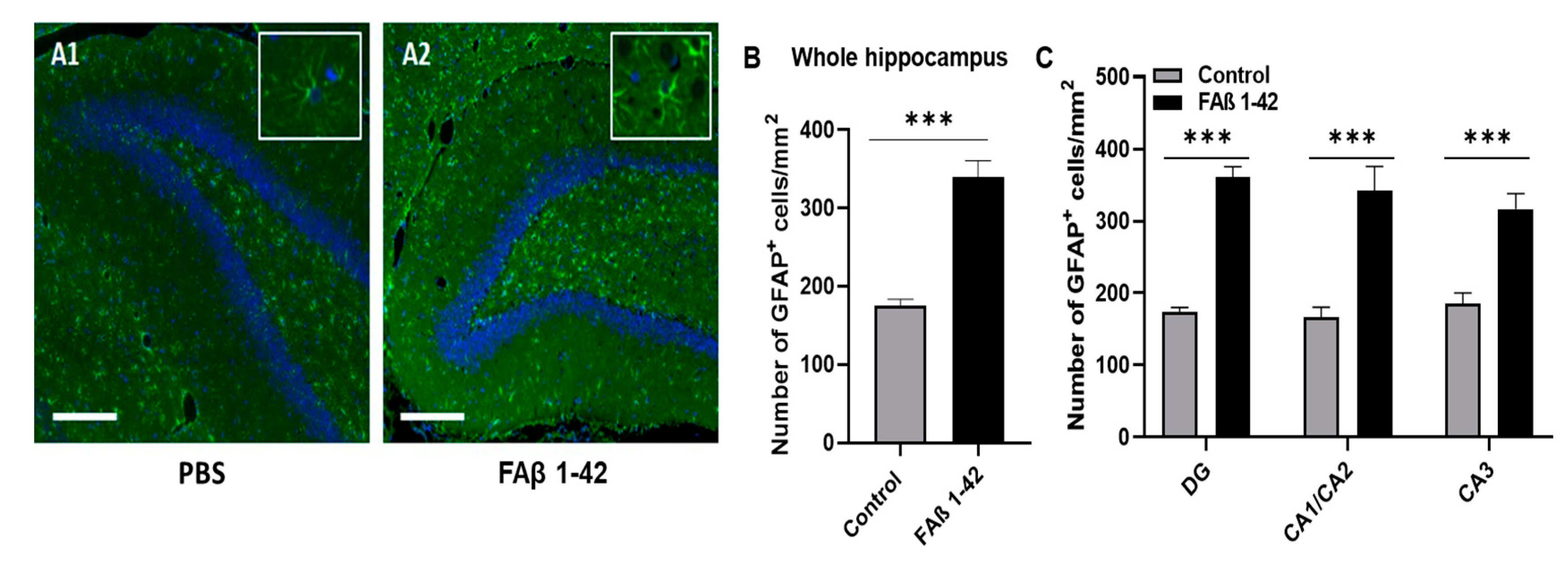
3.2. FAβ1–42 Infusion Leads to an Inflammatory Response in the Hippocampus of Rats
3.3. Intrahippocampal Injection of FAβ1–42 Leads to Dysregulation of Circulating miRNAs
3.4. Increasing Amount of Infused FAβ1–42 Peptides in Hippocampus of Rats Further Dysregulates the Relative Abundance of Circulating miRNAs in Serum Samples
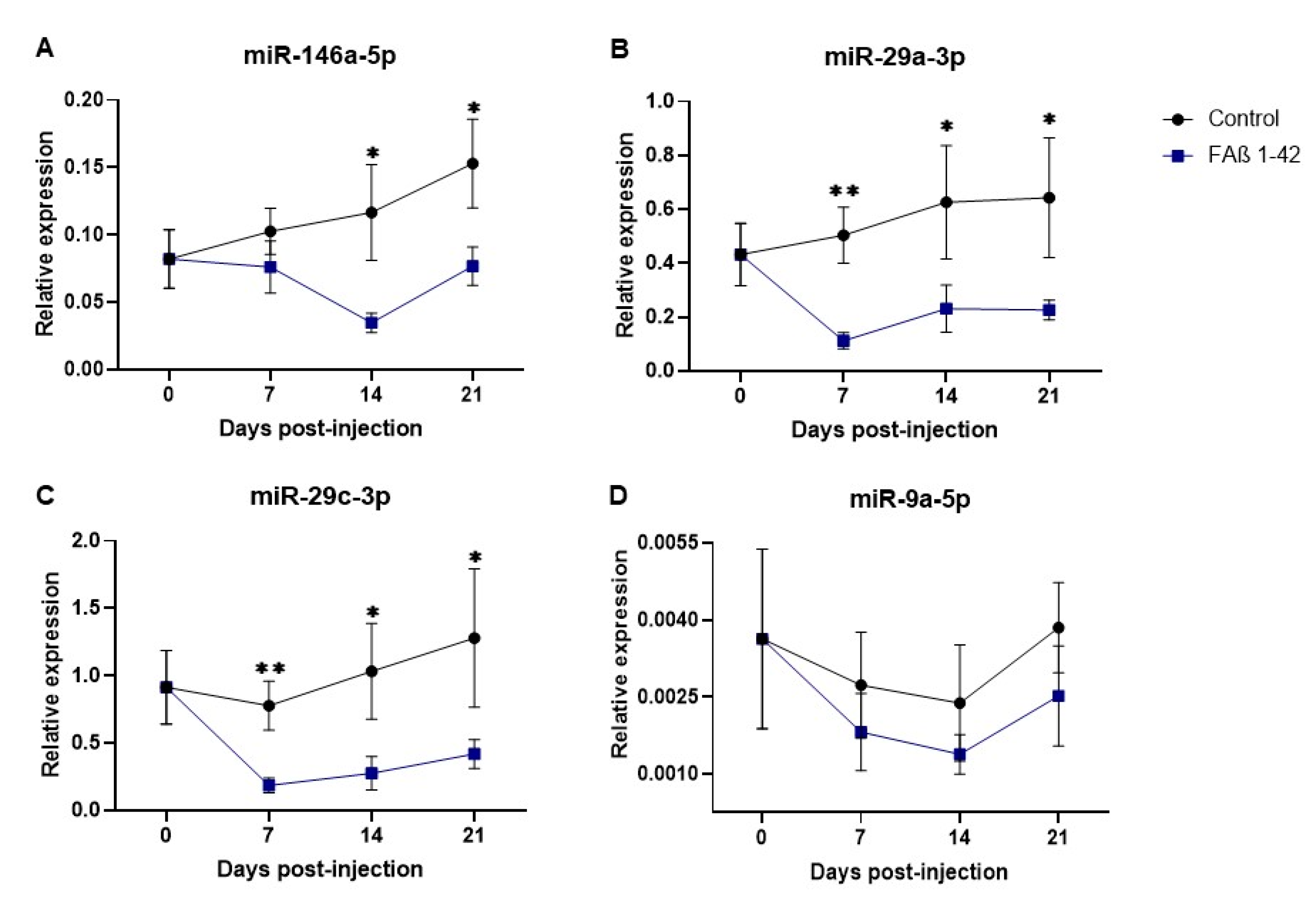
3.5. Kinetics of Circulating miRNA Expression Detected in Rats Infused with FAβ1–42 Peptides Solution Used at 2.5 µg/µL
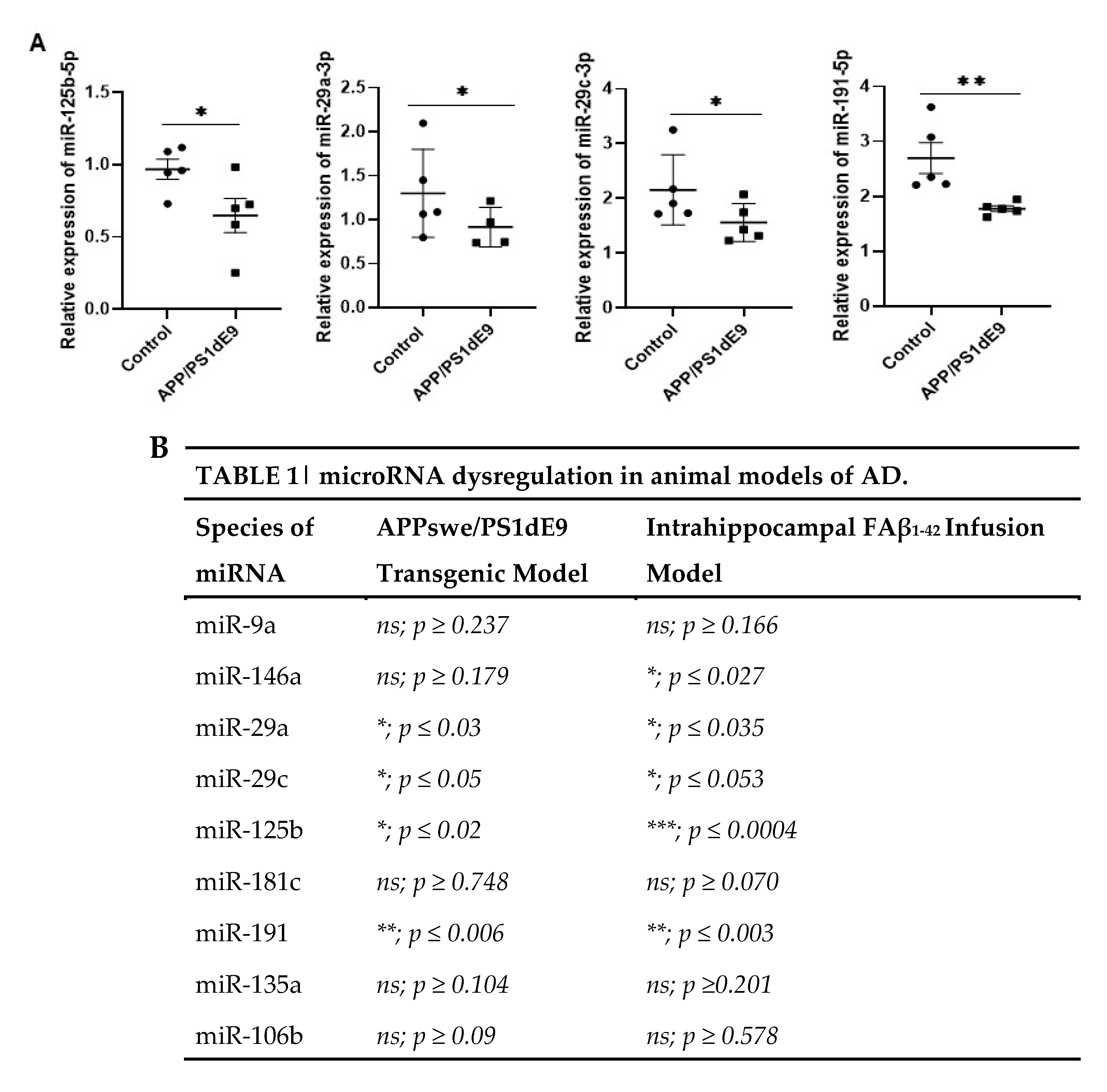
3.6. Common miRNAs Dysregulated in APPswe/PS1dE9 Transgenic Mice Model and FAβ-Brain Infused Rat Model
3.7. Intrahippocampal Inoculation of FAβ1–42 Leads to Increased Expression of miR-146a in CSF
3.8. FAβ1–42 Peptides Induces the Expression of miRNA-146a-5p in Primary Astrocytes through the NF-κB Cell Signaling Pathway but Without Inducing Production of Pro-Inflammatory Cytokines
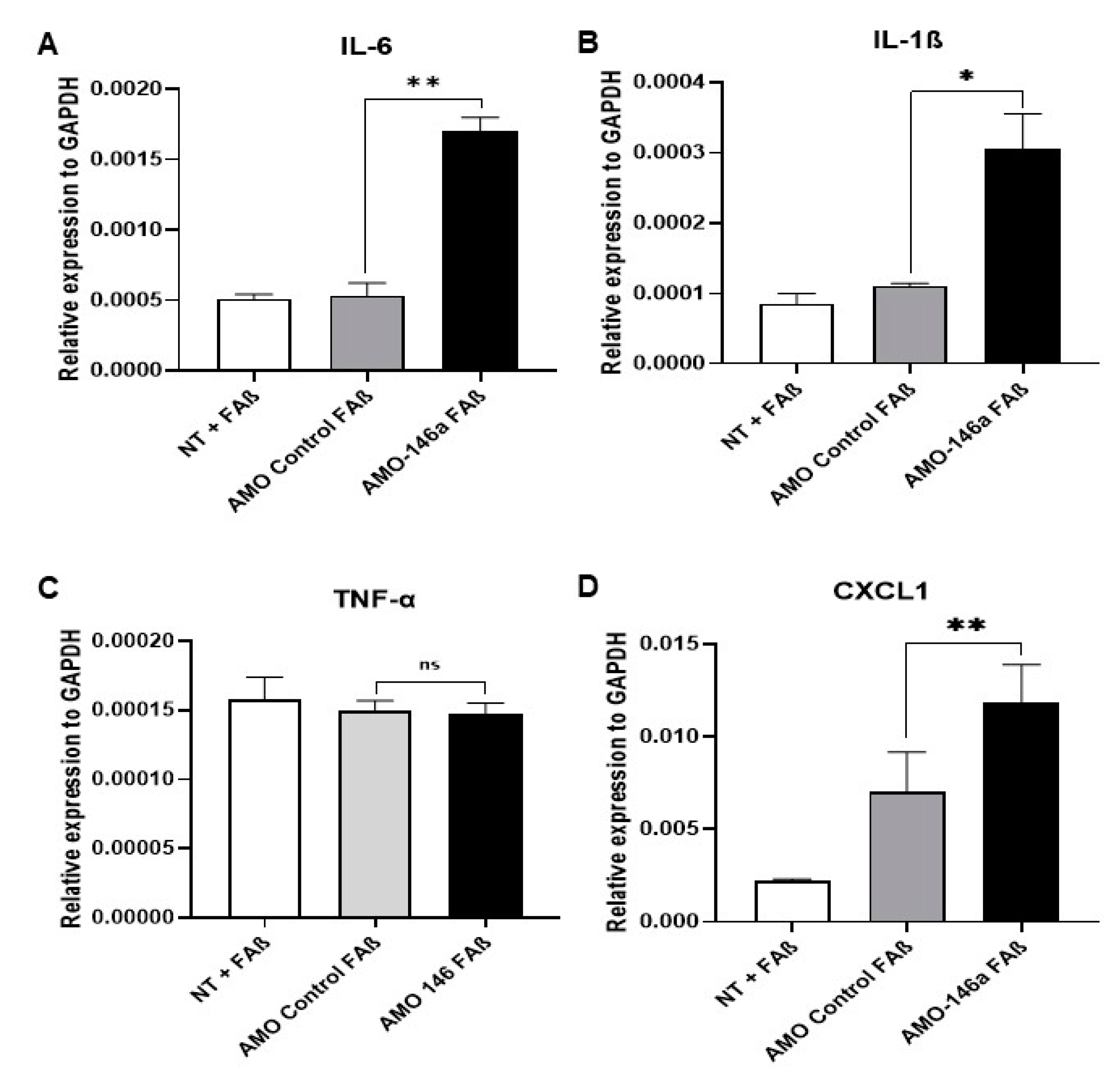
3.9. Anti-miRNA-146a-5p Oligonucleotides (AMO-146a) Rescue the Expression of IRAK-1 and TRAF-6 as Well as Pro-Inflammatory Cytokines in FAβ1–42-Treated Astrocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 63–75.e62. [Google Scholar] [CrossRef]
- Mullard, A. Alzheimer amyloid hypothesis lives on. Nat. Rev. Drug Discov. 2016, 16, 3–5. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [Green Version]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Mantile, F.; Prisco, A. Vaccination against β-Amyloid as a Strategy for the Prevention of Alzheimer’s Disease. Biology 2020, 9, 425. [Google Scholar] [CrossRef]
- Wong, R.S.; Cechetto, D.F.; Whitehead, S.N. Assessing the Effects of Acute Amyloid β Oligomer Exposure in the Rat. Int. J. Mol. Sci. 2016, 17, 1390. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, F.; Cechova, K.; Valis, M.; Kuca, K.; Zhang, B.; Hort, J. MicroRNAs in Alzheimer’s Disease: Diagnostic Markers or Therapeutic Agents? Front. Pharmacol. 2019, 10, 665. [Google Scholar] [CrossRef]
- Shaffer, J.L.; Petrella, J.R.; Sheldon, F.C.; Choudhury, K.R.; Calhoun, V.D.; Coleman, R.E.; Doraiswamy, P.M. Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology 2013, 266, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Gilad, S.; Meiri, E.; Yogev, Y.; Benjamin, S.; Lebanony, D.; Yerushalmi, N.; Benjamin, H.; Kushnir, M.; Cholakh, H.; Melamed, N.; et al. Serum microRNAs are promising novel biomarkers. PLoS ONE 2008, 3, e3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edsbagge, M.; Andreasson, U.; Ambarki, K.; Wikkelsø, C.; Eklund, A.; Blennow, K.; Zetterberg, H.; Tullberg, M. Alzheimer’s Disease-Associated Cerebrospinal Fluid (CSF) Biomarkers do not Correlate with CSF Volumes or CSF Production Rate. J. Alzheimer’s Dis. 2017, 58, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogianni, D.P.; Kalligosfyri, P.M.; Kyriakou, I.K.; Christopoulos, T.K. Advances in microRNA analysis. Anal. Bioanal. Chem. 2018, 410, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Wang, M.; Qin, L.; Tang, B. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Bryan, K.J.; Lee, H.G.; Perry, G.; Smith, M.A.; Casadesus, G. Transgenic Mouse Models of Alzheimer’s Disease: Behavioral Testing and Considerations Methods of Behavior Analysis in Neuroscience; Taylor & Francis Group©, LLC.: Boca Raton, FL, USA, 2009. [Google Scholar]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Lane-Donovan, C.; Herz, J. ApoE, ApoE Receptors, and the Synapse in Alzheimer’s Disease. Trends Endocrinol. Metab. 2017, 28, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Lecanu, L.; Papadopoulos, V. Modeling Alzheimer’s disease with non-transgenic rat models. Alzheimer’s Res. Ther. 2013, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, A.; Wani, W.Y.; Anand, R.; Gill, K.D. Spontaneous and induced nontransgenic animal models of AD: Modeling AD using combinatorial approach. Am. J. Alzheimer’s Dis. Other Dement. 2013, 28, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Faucher, P.; Mons, N.; Micheau, J.; Louis, C.; Beracochea, D.J. Hippocampal Injections of Oligomeric Amyloid β-peptide (1-42) Induce Selective Working Memory Deficits and Long-lasting Alterations of ERK Signaling Pathway. Front. Aging Neurosci. 2015, 7, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthick, C.; Nithiyanandan, S.; Essa, M.M.; Guillemin, G.J.; Jayachandran, S.K.; Anusuyadevi, M. Time-dependent effect of oligomeric amyloid-β (1-42)-induced hippocampal neurodegeneration in rat model of Alzheimer’s disease. Neurol. Res. 2019, 41, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Garza-Manero, S.; Arias, C.; Bermúdez-Rattoni, F.; Vaca, L.; Zepeda, A. Identification of age- and disease-related alterations in circulating miRNAs in a mouse model of Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, A.; Jimenez-Mateos, E.M.; Calero, M.; Medina, M.; Engel, T. Detecting Circulating MicroRNAs as Biomarkers in Alzheimer’s Disease. Methods Mol. Biol. 2018, 1779, 471–484. [Google Scholar] [CrossRef]
- Kumar, S.; Vijayan, M.; Bhatti, J.S.; Reddy, P.H. MicroRNAs as Peripheral Biomarkers in Aging and Age-Related Diseases. Prog. Mol. Biol. Transl. Sci. 2017, 146, 47–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Z.; Ong, K.L.; Seeher, K.; Armstrong, N.J.; Thalamuthu, A.; Brodaty, H.; Sachdev, P.; Mather, K. Circulating microRNAs as Biomarkers of Alzheimer’s Disease: A Systematic Review. J. Alzheimer’s Dis. 2016, 49, 755–766. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhang, L.; Dong, Y.; Ji, H.; Shen, L. The Potential Markers of Circulating microRNAs and long non-coding RNAs in Alzheimer’s Disease. Aging Dis. 2019, 10, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Borbély, E.; Horváth, J.; Furdan, S.; Bozsó, Z.; Penke, B.; Fülöp, L. Simultaneous changes of spatial memory and spine density after intrahippocampal administration of fibrillar aβ1-42 to the rat brain. BioMed Res. Int. 2014, 2014, 345305. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xu, J.; Xu, J.; Cheng, J.; Jiao, D.; Zhou, C.; Dai, Y.; Chen, Q. Lower Serum Levels of miR-29c-3p and miR-19b-3p as Biomarkers for Alzheimer’s Disease. Tohoku J. Exp. Med. 2017, 242, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 1998. [Google Scholar]
- Wenk, G.L. Assessment of spatial memory using the radial arm maze and Morris water maze. Curr. Protoc. Neurosci. 2004, 26, 5–8. [Google Scholar] [CrossRef]
- Vigneron, N.; Meryet-Figuière, M.; Guttin, A.; Issartel, J.P.; Lambert, B.; Briand, M.; Louis, M.H.; Vernon, M.; Lebailly, P.; Lecluse, Y.; et al. Towards a new standardized method for circulating miRNAs profiling in clinical studies: Interest of the exogenous normalization to improve miRNA signature accuracy. Mol. Oncol. 2016, 10, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Beeton, C.; Garcia, A.; Chandy, K.G. Drawing blood from rats through the saphenous vein and by cardiac puncture. J. Vis. Exp. 2007, 7, 266. [Google Scholar] [CrossRef]
- Pegg, C.C.; He, C.; Stroink, A.R.; Kattner, K.A.; Wang, C.X. Technique for collection of cerebrospinal fluid from the cisterna magna in rat. J. Neurosci. Methods 2010, 187, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Duy, J.; Koehler, J.W.; Honko, A.N.; Minogue, T.D. Optimized microRNA purification from TRIzol-treated plasma. BMC Genom. 2015, 16, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simion, V.; Henriet, E.; Juric, V.; Aquino, R.; Loussouarn, C.; Laurent, Y.; Martin, F.; Midoux, P.; Garcion, E.; Pichon, C.; et al. Intracellular trafficking and functional monitoring of miRNA delivery in glioblastoma using lipopolyplexes and the miRNA-ON RILES reporter system. J. Control. Release Off. J. Control. Release Soc. 2020, 327, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Sobilo, J.; Clemoncon, R.; Natkunarajah, S.; Ezzine, S.; Abdallah, F.; Lerondel, S.; Pichon, C.; Baril, P. Positive radionuclide imaging of miRNA expression using RILES and the human sodium iodide symporter as reporter gene is feasible and supports a protective role of miRNA-23a in response to muscular atrophy. PLoS ONE 2017, 12, e0177492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Faraldi, M.; Gomarasca, M.; Sansoni, V.; Perego, S.; Banfi, G.; Lombardi, G. Normalization strategies differently affect circulating miRNA profile associated with the training status. Sci. Rep. 2019, 9, 1584. [Google Scholar] [CrossRef] [Green Version]
- Reverchon, F.; de Concini, V.; Larrigaldie, V.; Benmerzoug, S.; Briault, S.; Togbé, D.; Ryffel, B.; Quesniaux, V.F.J.; Menuet, A. Hippocampal interleukin-33 mediates neuroinflammation-induced cognitive impairments. J. Neuroinflamm. 2020, 17, 268. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Galland, F.; Seady, M.; Taday, J.; Smaili, S.S.; Gonçalves, C.A.; Leite, M.C. Astrocyte culture models: Molecular and function characterization of primary culture, immortalized astrocytes and C6 glioma cells. Neurochem. Int. 2019, 131, 104538. [Google Scholar] [CrossRef]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, R.; Grabert, K.; Davies, C.L.; Alfieri, A.; Antel, J.P.; Healy, L.M.; McColl, B.W. Corrigendum: Divergent Neuroinflammatory Regulation of Microglial TREM Expression and Involvement of NF-κB. Front. Cell. Neurosci. 2017, 11, 256. [Google Scholar] [CrossRef]
- Ezzine, S.; Vassaux, G.; Pitard, B.; Barteau, B.; Malinge, J.M.; Midoux, P.; Pichon, C.; Baril, P. RILES, a novel method for temporal analysis of the in vivo regulation of miRNA expression. Nucleic Acids Res. 2013, 41, e192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverchon, F.; Mortaud, S.; Sivoyon, M.; Maillet, I.; Laugeray, A.; Palomo, J.; Montécot, C.; Herzine, A.; Meme, S.; Meme, W.; et al. IL-33 receptor ST2 regulates the cognitive impairments associated with experimental cerebral malaria. PLoS Pathog. 2017, 13, e1006322. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kappaB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A primary neural cell culture model to study neuron, astrocyte, and microglia interactions in neuroinflammation. J. Neuroinflamm. 2020, 17, 155. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zheng, M.M.; Ma, Y.; Han, X.J.; Ma, X.Q.; Qu, C.Q.; Du, Y.F. Soluble oligomers and fibrillar species of amyloid β-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem. Biophys. Res. Commun. 2012, 429, 125–130. [Google Scholar] [CrossRef]
- Nell, H.J.; Whitehead, S.N.; Cechetto, D.F. Age-Dependent Effect of β-Amyloid Toxicity on Basal Forebrain Cholinergic Neurons and Inflammation in the Rat Brain. Brain Pathol. 2015, 25, 531–542. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Sofroniew, M.V.; Messing, A.; deLanerolle, N.C.; Rempe, D.; Rodríguez, J.J.; Nedergaard, M. Neurological diseases as primary gliopathies: A reassessment of neurocentrism. ASN Neuro 2012, 4, e00082. [Google Scholar] [CrossRef] [Green Version]
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. Beta-sheet breaker peptide prevents Abeta-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Delay, C.; Hébert, S.S. MicroRNAs and Alzheimer’s Disease Mouse Models: Current Insights and Future Research Avenues. Int. J. Alzheimer’s Dis. 2011, 2011, 894938. [Google Scholar] [CrossRef] [Green Version]
- Delay, C.; Mandemakers, W.; Hébert, S.S. MicroRNAs in Alzheimer’s disease. Neurobiol. Dis. 2012, 46, 285–290. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef] [Green Version]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Chen, W.D.; Wang, Y.D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front. Immunol. 2014, 5, 578. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [Green Version]
- McLarnon, J.G.; Ryu, J.K. Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer’s disease brain. Curr. Alzheimer Res. 2008, 5, 475–480. [Google Scholar] [CrossRef]
- Huang, H.; Nie, S.; Cao, M.; Marshall, C.; Gao, J.; Xiao, N.; Hu, G.; Xiao, M. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. Age 2016, 38, 303–322. [Google Scholar] [CrossRef] [Green Version]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztal, T.E.; Stainier, D.Y.R. Transcriptional adaptation: A mechanism underlying genetic robustness. Development 2020, 147, dev186452. [Google Scholar] [CrossRef]
- Bai, Y.; Su, X.; Piao, L.; Jin, Z.; Jin, R. Involvement of Astrocytes and microRNA Dysregulation in Neurodegenerative Diseases: From Pathogenesis to Therapeutic Potential. Front. Mol. Neurosci. 2021, 14, 556215. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 2008, 9, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Li, Y.Y.; Cui, J.G.; Zhao, Y.; Kruck, T.P.; Percy, M.E.; Tarr, M.A.; Lukiw, W.J. Characterization of an NF-kappaB-regulated, miRNA-146a-mediated down-regulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J. Inorg. Biochem. 2009, 103, 1591–1595. [Google Scholar] [CrossRef]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Gohda, J.; Akiyama, T.; Inoue, J.I. TNF receptor-associated factor 6 (TRAF6) plays crucial roles in multiple biological systems through polyubiquitination-mediated NF-κB activation. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 97, 145–160. [Google Scholar] [CrossRef]
- Mattsson, N.; Rosén, E.; Hansson, O.; Andreasen, N.; Parnetti, L.; Jonsson, M.; Herukka, S.K.; van der Flier, W.M.; Blankenstein, M.A.; Ewers, M.; et al. Age and diagnostic performance of Alzheimer disease CSF biomarkers. Neurology 2012, 78, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.; Fan, W.; Wang, Y.; Cai, Y.; Li, X.; Chen, F.; Chen, X.; Yang, J.; Tang, P.; Chen, H.; et al. Intranasal Administration of miR-146a Agomir Rescued the Pathological Process and Cognitive Impairment in an AD Mouse Model. Mol. Ther. Nucleic Acids 2019, 18, 681–695. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquino, R.; de Concini, V.; Dhenain, M.; Lam, S.; Gosset, D.; Baquedano, L.; Forero, M.G.; Menuet, A.; Baril, P.; Pichon, C. Intrahippocampal Inoculation of Aβ1–42 Peptide in Rat as a Model of Alzheimer’s Disease Identified MicroRNA-146a-5p as Blood Marker with Anti-Inflammatory Function in Astrocyte Cells. Cells 2023, 12, 694. https://doi.org/10.3390/cells12050694
Aquino R, de Concini V, Dhenain M, Lam S, Gosset D, Baquedano L, Forero MG, Menuet A, Baril P, Pichon C. Intrahippocampal Inoculation of Aβ1–42 Peptide in Rat as a Model of Alzheimer’s Disease Identified MicroRNA-146a-5p as Blood Marker with Anti-Inflammatory Function in Astrocyte Cells. Cells. 2023; 12(5):694. https://doi.org/10.3390/cells12050694
Chicago/Turabian StyleAquino, Ruth, Vidian de Concini, Marc Dhenain, Suzanne Lam, David Gosset, Laura Baquedano, Manuel G. Forero, Arnaud Menuet, Patrick Baril, and Chantal Pichon. 2023. "Intrahippocampal Inoculation of Aβ1–42 Peptide in Rat as a Model of Alzheimer’s Disease Identified MicroRNA-146a-5p as Blood Marker with Anti-Inflammatory Function in Astrocyte Cells" Cells 12, no. 5: 694. https://doi.org/10.3390/cells12050694