

CRISPR-Cas System: The Current and Emerging Translational Landscape
by
 ,
,
Narendranath Bhokisham
*,
Ethan Laudermilch
,
Lindsay L. Traeger
,
Tonya D. Bonilla
,
Mercedes Ruiz-Estevez
and
Jordan R. Becker
Corporate Research Material Labs, 3M Center, 3M Company, Maplewood, MN 55144, USA
*
Author to whom correspondence should be addressed.
Cells 2023, 12(8), 1103; https://doi.org/10.3390/cells12081103
Submission received: 1 January 2023
/
Revised: 3 April 2023
/
Accepted: 4 April 2023
/
Published: 7 April 2023
(This article belongs to the Special Issue CRISPR-Based Genome Editing in Translational Research)
Abstract
:CRISPR-Cas technology has rapidly changed life science research and human medicine. The ability to add, remove, or edit human DNA sequences has transformative potential for treating congenital and acquired human diseases. The timely maturation of the cell and gene therapy ecosystem and its seamless integration with CRISPR-Cas technologies has enabled the development of therapies that could potentially cure not only monogenic diseases such as sickle cell anemia and muscular dystrophy, but also complex heterogenous diseases such as cancer and diabetes. Here, we review the current landscape of clinical trials involving the use of various CRISPR-Cas systems as therapeutics for human diseases, discuss challenges, and explore new CRISPR-Cas-based tools such as base editing, prime editing, CRISPR-based transcriptional regulation, CRISPR-based epigenome editing, and RNA editing, each promising new functionality and broadening therapeutic potential. Finally, we discuss how the CRISPR-Cas system is being used to understand the biology of human diseases through the generation of large animal disease models used for preclinical testing of emerging therapeutics.
1. Introduction
This review will focus on the transformation of human medicine brought about by the CRISPR-Cas system. Since its inception in 2012, the Cas9 variant of the CRISPR-Cas system has generated enormous interest due to the ease with which any DNA sequence in the genome can be targeted for alteration. The CRISPR-Cas9 system comprises two elements: an RNA-guided DNA endonuclease called Cas9 isolated from Streptococcus pyogenes and its guiding RNA called guide RNA or gRNA [1]. Cas9 in combination with gRNA can be engineered to target genomic sequences that are complementary to the sgRNA and catalyze a double-stranded break (DSB) in the DNA backbone. DSBs are then primarily repaired by either the error-prone non-homologous end-joining (NHEJ) pathway [2], or by error-free homology-directed repair (HDR) [3,4]. These mechanisms can be exploited to either disrupt (knock-out) genes or introduce new sequences (knock-ins) into the host genome [5]. The versatility and ease of use of the CRISPR-Cas system represents a transformative improvement upon previous gene editing technologies (e.g., Transcription Activator-Like Effector Nucleases, TALEN) [6]. The ability to easily target any region of the genome and create knock-ins or knock-outs is valuable in multiple fields including human medicine [7], agriculture [8], industrial biotechnology [9], among others.
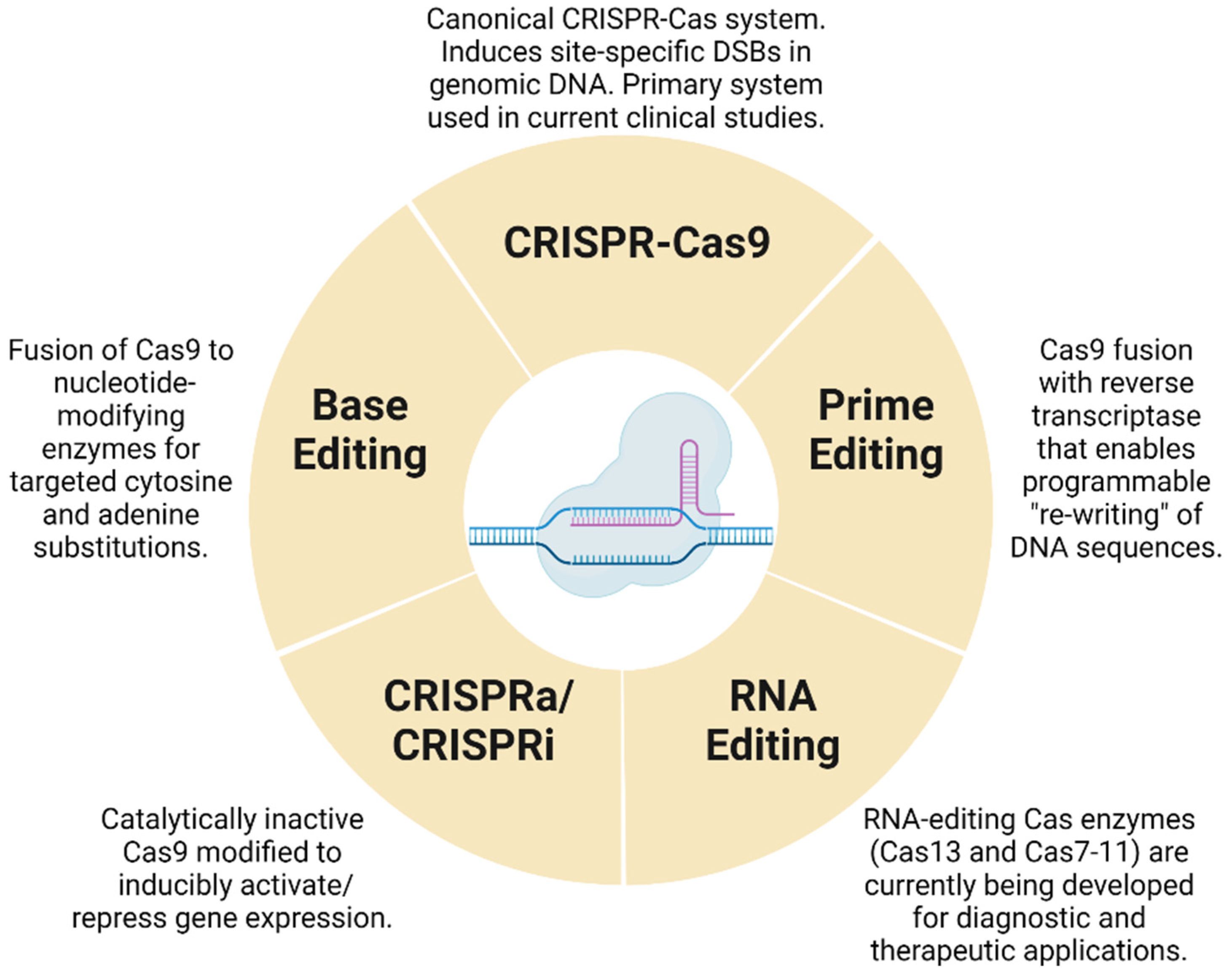
While CRISPR-Cas systems are revolutionizing medicine, these systems are themselves undergoing an evolution. The single greatest liability of the Cas9-based gene editing is the formation of off-target double-stranded breaks in the genome which have the potential to generate mutations, large chromosomal aberrations such as translocations, inversions, etc. [10]. To overcome this limitation, Cas9-based base editors and prime editors [11] have been developed that operate without inducing a double-stranded break, thereby reducing the risk of chromosomal rearrangements. In this article, we will cover the current state of the art pertaining to base and prime editing in the field of human medicine. In addition to base and prime editors, Cas9 has been mutated to create an enzymatically deactivated Cas9 or dead Cas9 (dCas9) that has no endonuclease activity yet retains the ability to specifically locate and bind onto a target DNA sequence [12]. Standalone dCas9s as well as dCas9 fusions with various transcriptional activators [13] have enabled the modulation of transcriptional activity. Though dCas9 is not part of any therapeutic product that is currently under development, dCas9 serves as a tool of academic interest enabling the screening of signaling pathways and helping further the understanding of human disease biology [14]. Further, newer Cas proteins are being evaluated that provide differentiated editing performance over the S. pyogenes Cas9, such as the ones that selectively allow the editing of RNA instead of DNA [15,16]. In our article, we collectively refer to all Cas variants as the CRISPR-Cas system, and we cover some of the relevant topics surrounding the use of Cas9, dCas9s and other novel Cas proteins in the context of translational medicine (Figure 1). Finally, another important aspect of translational medicine is the availability of disease models in animals to test the efficacy of various therapies prior to clinical trials in humans. CRISPR-Cas systems have enabled the creation of monogenic disease models in animals that can then act as test beds to study various therapies, including CRISPR-Cas-derived cell and gene therapies [17,18,19]. In this article, we will cover some of the latest advances in the creation of animal models using the CRISPR-Cas system-based gene editing tools.
Within its first decade [1], the Cas9-based CRISPR-Cas system has altered the limits of human medicine, with several ongoing clinical trials leveraging this technology. In the coming sections, we will discuss how various CRISPR-Cas systems are not only used to treat monogenic disorders such as sickle cell anemia and Duchenne muscular dystrophy but also complex heterogenous diseases such as cancer, HIV-AIDS, and diabetes. CRISPR-Cas systems have seamlessly integrated into the gene and cell therapy ecosystem that has been under development for over the three decades, enabling ex vivo gene editing in the form of cell therapy and in vivo gene editing in the form of gene therapy [20,21]. In this article, we will detail the status of several completed and ongoing clinical trials being carried out with various CRISPR-Cas systems for treating various human diseases.
2. Current Use of CRISPR-Cas Systems in Clinical Trials
As of 15 December 2022, we identified 71 different clinical trials in the clinicaltrials.gov database that were directly associated with various CRISPR-Cas systems. Of the 71 trials, we identified 45 ongoing or completed trials that are directed towards human therapeutic product development. Of these, 23 trials were on various forms of cancer; 14 trials were on blood-related disorders including sickle cell disease and β-thalassemia; 3 were trials on HIV infection; and 1 trial each was on HPV, viral keratitis (HSV-1), Leber congenital amaurosis, hereditary angioedema, and Duchenne muscular dystrophy. In the coming sections, we will discuss how CRISPR-Cas systems are being leveraged for therapeutic development for each of these conditions.
2.1. CRISPR-Cas Technologies in the Treatment of Cancers
All clinical trials registered so far for cancer treatment use the Cas9 variant of the CRISPR-Cas system and involve the knock-out (KO) of one or more selected genes of interest. A small number of trials use the CRISPR-Cas9 system to knock-in specified sequences at target loci, and to our knowledge, there is one trial (NCT05397184 [22]) employing the base editing technology. In the upcoming section, we will survey the current range of clinical trials that involve knock-in/out of genes using CRISPR-Cas9. The lone clinical trial related to base editing for cancer is covered in the later sections of the paper.
2.1.1. Cellular Immunotherapies
CRISPR-Cas9 technology is being increasingly used for engineering T lymphocytes, a type of immune cell that plays a central role in the fight against cancer. In the last decade, a new class of therapies referred to as adoptive cell therapy or cellular immunotherapy has gained prominence, and this involves harvesting and reprogramming a patient’s own T cells and employing them to treat cancer [23,24]. One of the foremost approaches in the cellular immunotherapy field is the CAR-T therapy, where T cells from cancer patients are isolated, engineered with chimeric antigen receptors (CAR), and injected back into patients [25] (Figure 2). CARs are designed to specifically target select antigens that are presented on the cancer cells. As of December 2022, six CAR-T therapies have been approved by the FDA to target CD19 and BCMA antigens in B cell-related cancers [26]. In addition to CAR-T, other adoptive cell therapy approaches include engineering a synthetic T cell receptor (TCR) into T cells and using them for treating cancer, referred to as TCR Therapy or TCR-T [27]. Tumor-infiltrating lymphocytes (TILs) therapy is another approach where lymphocytes that have infiltrated the tumors are isolated, enriched with or without genetic modifications and later injected back into cancer patients [28]. Although most current clinical trials involving the CRISPR-Cas9 technology are focused towards CAR-T therapy, we will also highlight a few TCR-T- and TIL-based therapies that involve CRISPR-Cas9 engineering in the sections below (See Table 1 and Table 2).
CAR-T therapies have proven to be very effective against B cell malignancies by enabling complete remissions in >80% of patient populations; however, the median event-free survival rate after 12 months was only in the range of 40–60% [29,30], indicating the occurrence of cancer relapse in patients who had complete remission earlier. Some of the drawbacks of the current class of CAR-T therapies include antigen escape by the cancer cells where CAR-specific antigens are lost in the cancer cells due to the selective pressure posed by the CAR-T, eventually leading to the relapse of cancer cells without the targeted antigen [31]. Beyond B cell malignancies, CAR-T treatments have proven to be less effective against solid cancers due to a variety of reasons including the presence of strong immunosuppressive microenvironments in solid cancers, inability of the engineered T cells to infiltrate into them and a lack of uniform presentation of cancer-specific target antigens across all cells in a solid tumor [32,33]. Due to the onset of exhaustion or dysfunction in T cells that is mediated by internal signaling mechanisms, CAR-T cells have also been unable to display anti-tumor activity for longer periods of time [34]. In the case of T cell malignancies, CAR-T have proven to be more ineffective due to the difficulty in separating a healthy T cell from a tumor-infected T cell. In addition, due to the presence of CAR-specific antigens in both healthy T cells and CAR-T cells themselves, T cell aplasia and fratricide are some of the risks associated with employing the T cell therapy towards T cell malignancies [35]. A variety of approaches are being pursued to alleviate many of the drawbacks listed above and are detailed in many reviews [36,37]. In this article, we will focus specifically on clinical trials where the CRISPR-Cas9 technology has been employed to alleviate some of the drawbacks of the current generation of T cell therapies.
2.1.2. Role of CRISPR-Cas9 Tools to Mediate Immune Check Point Inhibition in CAR-T Therapy
Persistent antigen stimulation in T cells coupled with immunosuppressive microenvironments in cancers lead to a loss of anti-tumor activity in T cells, referred as T cell exhaustion or dysfunction [34]. One strategy to increase the anti-tumor activities of T cells and delay the exhaustion of T cell function is to use anti-programmed cell death protein (PD1) therapy [38]. PD1 protein (coded by PDCD1) expressed on the surface of the T cells and is a key immune check point regulator. Upon PD1′s interaction with its ligand, the programmed death ligand (PDL1) that is expressed on surfaces of cancer cells, PD1 mediates a negative regulatory role in the anti-tumor activity of T cells [39]. Blocking PD1′s interaction with PDL1 ligand via the anti-PD1 monoclonal antibody therapy has proven to be effective in combination with various T cell therapies including CAR-T therapy [40].
With the advent of facile gene editing technologies, CRISPR-Cas9 has been used for disruption of the PDCD1 gene encoding for PD1, and this has resulted in the enhancement of anti-tumor effects of CAR-T in animal models [41,42]. Consequently, several clinical trials [43,44,45,46,47,48,49,50] have been registered that involve using the CRISPR-Cas9 system for disruption of PDCD1 in CAR-T and TCR-T cells. So far, the results from three phase 1 trials have been published [51,52,53], but the results are inconclusive for the enhanced anti-tumor effect produced by the disruption of PDCD1. In one trial [51], T cells carried disruptions in PDCD1 and endogenous TCR loci (TRAC and TRBC). In addition, these cells were also inserted with exogenous TCRs specific to NY-ESO-1 tumor antigen. These CRISPR-Cas9-engineered T cells were found to be persisting at peak levels for 4 months after infusion and displayed anti-tumor activity even for up to 9 months post infusion in one of the patients in the trial. Persistence of engineered T cells in this study was longer than in previous trials where similar cells without disruptions to PDCD1 and endogenous TCR loci peaked after a week of injection and had a half-life of ~1 week [54,55,56]. The reasons for prolonged persistence in this study could be attributed to disruption in PDCD1 and/or endogenous TCR genes, or other external factors such as exogenous T cell receptor design, etc. The exact reasons for enhanced survival of engineered T cells needs to be studied further.
In another trial [53], mesothelin-directed CAR-T cells were used to target mesothelin-positive tumors, and here, these cells had disruptions in the PDCD1 locus as well as in the TRAC locus of TCR. Contrary to the previous study discussed above [51], in this study, PD1-disrupted CAR-T cells did not display prolonged persistence and enhanced anti-tumor activity in patients. Levels of engineered T cells peaked a week after injection and became undetectable after 1 month. In a similar study involving the mesothelin-directed CAR-T cells with intact PDCD1, when used in conjunction with the anti-PD1 therapy, they displayed prolonged persistence [57]. The reasons for the lack of prolonged persistence in this study could be multifactorial, but an interesting observation that the authors point to was that a minority of injected cell populations that had their endogenous TCR intact were found to be persisting at the end of the study, hinting at the possibility of the importance of TCR signaling playing a role in prolonged persistence of these engineered cells. In the third study [52], while PD1-disrupted cells persisted in patients for 4 four weeks after infusion, no objective conclusions could be made on the effect of PDCD1 disruption on anti-tumor activity due to the small trial size and short duration of study.
While PD1 is presented on the surface of T cells and can be accessed easily by anti-PD1 therapies, other non-surface regulators of T cell function are harder to be reached and exploited via antibody-based therapy. With the emergence of the CRISPR-Cas9 system, these internal regulators can be easily accessed to study for their role in enhanced anti-tumor T cell functions. One such internal regulator, the cytokine-inducible SH2-containing protein (CISH), has been knocked out using the CRISPR-Cas9 system and has been demonstrated to enhance reactivity towards cancer antigens in TIL therapy [58]. Two ongoing trials [59,60] are currently studying the use of TILs with disruptions in CISH for non-small cell lung and gastrointestinal cancers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of autologous cellular immunotherapies using the CRISPR-Cas9-based gene editing system.
Table 1.
List of autologous cellular immunotherapies using the CRISPR-Cas9-based gene editing system.
| Conditions Targeted | Targets Knocked out via CRISPR | Targets Knocked in (via Lentivirus or CRISPR) | Sponsor | Clinical Trial ID |
|---|---|---|---|---|
| Advanced hepatocellular carcinoma | PD1 | Central South University | NCT04417764 [43] | |
| Advanced esophageal squamous cell carcinoma | PD1 | Hangzhou Cancer Hospital | NCT03081715 [44] | |
| Metastatic gastrointestinal cancers | CISH | Intima Bioscience, Inc. | NCT04426669 [60] | |
| Metastatic non-small cell lung cancer | NCT05566223 [59] | |||
| Metastatic non-small cell lung cancer | PD1 | Sichuan University | NCT02793856 [45] | |
| EBV+ malignancies | PD1 | Nanjing University | NCT03044743 [46] | |
| CD5+ relapsed/refractory T cell malignancies | CD5 | CD5-CAR (via lentivirus) | Huazhong University | NCT04767308 [61] |
| Acute lymphocytic leukemia | HPK-1 | CD19-CAR (via lentivirus) | Xijing Hospital | NCT04037566 [62] |
| Multiple solid tumors | PD1 and TRAC | Mesothelin-CAR (via lentivirus) | Chinese PLA General Hospital | NCT03545815 [47] |
| Mesothelin-positive multiple solid tumors | NCT03747965 [48] | |||
| Advanced EGFR-positive solid tumors | TGF-β receptor Ⅱ | EGFR-CAR (via lentivirus) | NCT04976218 [63] | |
| Multiple myeloma | PD1, TRAC and TRBC | NY-ESO-1-TCR (via lentivirus) | University of Pennsylvania | NCT03399448 [49] |
| Acute myeloid leukemia | TRBC and TRAC | Wilms Tumor 1-TCR (via CRISPR) | Intellia Therapeutics | NCT05066165 [64] |
2.1.3. Role of CRISPR-Cas9 Tools in Design of Allogenic ”Off-the-Shelf” T Cell Therapies
All the six CAR-T therapies approved so far are autologous in nature with each patient-specific CAR-T cell needing to be manufactured separately, placing a heavy burden on the supply chain. Autologous therapies pose further challenges for late-stage cancer patients who have undergone several rounds of treatments, as their T cells cannot be scaled up to the desired dose levels [65]. Creating a stockpile of allogenic CAR-T cells would ease manufacturing challenges and give the late-stage patients a shot at this therapy. However, there are challenges in creating effective allogenic CAR-T therapies, including mismatches in the donor and the recipient’s human leukocyte antigens (HLA) leading to the onset of graft vs. host disease (GVHD) and rejection of the allogenic CAR-T cells by the host. Mechanistically, endogenous TCRs and major histocompatibility class molecules (MHC Classes I and II) are the mediators of self- and non-self-discrimination in T cells, and CRISPR-Cas9 tools have been used to disrupt these molecules to create allogenic therapies [65,66,67] (Figure 3). A number of registered clinical trials are focused towards disrupting the TRAC loci of the TCR [50,68,69,70,71,72,73,74] and β2-microglobulin gene [68,71,72,73,74], an MHC Class I molecule.
So far, phase 1 safety data [66] from one trial [70] has been published recently involving the use of CRISPR-Cas9-engineered allogenic CD19-specific CAR-T cells for the treatment of children with refractory B cell leukemia. In this study, CRISPR-Cas9 was used to target an endogenous TCR locus (TRAC) as well as the gene coding for CD52. Results from this trial indicated that allogenic CAR-T cells disappeared four weeks after infusion, and four out of six patients exhibited complete remission, indicating evidence of anti-tumor activity by the allogenic T cells. Two patients did not display any expansion of the therapeutic T cells leading to progression in disease. Given the allogenic nature of these cells, there is a high risk of GVHD, and hence, this study was performed under high doses of lymphodepleting drugs such as cyclophosphamide, and yet, two patients developed GVHD. Given the high doses of lymphodepleting drugs, there were eight instances of viral infections as well. The results from this trial highlighted some of the promises and challenges in designing an effective allogenic cancer CAR-T therapy.
Using the CRISPR-Cas9 system to disrupt one or more MHC class I and II molecules could reduce the risks of GVHD and mediate better tolerance of these cells by the host, eventually paving the way for these therapies to be performed at lower doses of lymphodepleting drugs with lower risks of viral infections. Another ongoing trial [50], performed by Caribou Biosciences, to treat relapsed or refractory B cell non-Hodgkin lymphoma involves an allogenic T cell product (CB-010), where healthy donor T cells are taken, and the endogenous TCR was disrupted at the TRAC loci. To extend the anti-tumor activity of these cells, PDCD1 was interrupted as well. In addition, an CD19-specific CAR cassette was precisely inserted at the TRAC loci using the CRISPR-Cas9 system to regulate the expression of CAR [75]. The preliminary results [76] from this study indicated that all six patients who had received this treatment showed a complete response (CR) to the treatment. After 6 months, three of the six patients maintained CR; after 12 months, two of six patients maintained CR; and after 18 months, one patient maintained CR. More details on side effects such as GVHD, viral infections, etc., are unavailable at the time of writing of this paper. In summary, CRISPR-Cas9 tools have provided the flexibility of combining many strategies such as disruptions of PD1 and other immune check point regulators along with the removal of endogenous TCR and MHC class molecules to create a T cell therapeutic that is both safe and enduring as well as more sustainable from a manufacturing and cost perspective.
Table 2.
List of allogenic cellular immunotherapies using the CRISPR-Cas9-based gene editing system.
Table 2.
List of allogenic cellular immunotherapies using the CRISPR-Cas9-based gene editing system.
| Conditions Targeted | Targets Knocked out via CRISPR | Targets Knocked in (via Lentivirus or CRISPR) | Sponsor | Clinical Trial ID |
|---|---|---|---|---|
| Acute myeloid leukemia | CD33 | None | Vor Biopharma | NCT05309733 [77] |
| Relapsed or refractory CD19+ leukemia and lymphoma | TRAC and β2M | CD19 CAR (via lentivirus) | Chinese PLA General Hospital | NCT03166878 [68] |
| Relapsed or refractory leukemia and lymphoma | CD19+ CD20 CAR or CD19+ CD22 CAR (via lentivirus) | NCT03398967 [69] | ||
| B cell acute lymphoblastic leukemia | TRAC and CD52 | CD19 CAR (via lentivirus) | Great Ormond Street Hospital | NCT04557436 [70] |
| Elapsed/refractory B cell non-Hodgkin lymphoma | TRAC and PD1 | CD19 CAR at TRAC loci (via CRISPR) | Caribou Biosciences, Inc. | NCT04637763 [50] |
| Relapsed or refractory T or B cell malignancies | TRAC, β2M and CD70 | CD70 CAR at TRAC loci (via CRISPR) | CRISPR Therapeutics AG | NCT04502446 [71] |
| Renal cell carcinoma | NCT04438083 [72] | |||
| B cell malignancy | TRAC and β2M | CD19 CAR at TRAC loci (via CRISPR) | NCT04035434 [73] | |
| Relapsed or refractory multiple myeloma | TRAC and β2M | BCMA CAR at TRAC loci (via CRISPR) | NCT04244656 [74] |
2.1.4. Using CRISPR-Cas9 Tools for Precise Insertion of CARs in T Cell Therapies
CARs in CAR-T therapy as well as engineered TCRs in TCR-T therapy are inserted into T cells via lentivirus and other retroviral vectors. These viruses insert the engineered receptor cassettes randomly in the genome and often at multiple locations leading to mutations in non-desired genes and differential expression of these cassettes within the population [78]. Using the CRISPR-Cas9 system, precise insertion of CARs under promoters of endogenous TCR genes have enabled controlled expression of these cassettes and have been shown to delay T cell exhaustion and differentiation with improved anti-tumor activity [75]. In recently launched clinical trials [50,64,71,72,73,74], CRISPR-Cas9 has been used instead of lentivirus to specifically knock-in CARs under the TRAC loci of TCR to control the CAR expression. The interim results from one such trial conducted by Caribou Biosciences involving the product CB-010 are discussed in the previous section. This trial and several others are still ongoing, and complete results are yet to be published.
2.1.5. Role of CRISPR-Cas9 Tools to Mediate Effective CAR-T Therapy towards T Cell Malignancies
CAR-T therapies have been more successful against B cell-related malignancies than T cell-related malignancies. One of the reasons for CAR-T being ineffective against T cell-related cancers is due to the fratricide effects mediated by CAR-T cells where these cells target their fellow CAR-T cells carrying the same antigens that they are engineered to recognize and fight against. One trial [61] is focusing on removing CD5 in CAR-T cells to make them effective against T cell malignancies. While there is a baseline level of expression of CD5 in thymocytes, T, and B1 cells, CD5 expression is elevated in over 85% of T cell-related malignancies. Engineering a CAR-T therapy against CD5 molecule to target T cell malignancies might lead to fratricide due to the presence of the same CD5 molecule in engineered CAR-T cells as well. In preclinical studies [79], CRISPR-Cas9 tools have helped in mitigating fratricide effects by selectively removing such antigens in CAR-T cells and focusing the anti-cancer activity towards cancer cells.
In addition to fratricide effects, engineering an autologous T cell therapy for T cell malignancies risks injecting a lymphoblastic T cell into the patients. To overcome such risks, an allogenic T cell therapy from healthy donors can negate the risk of injection of lymphoblastic T cells in patients. Two ongoing trials [71,72] conducted by CRISPR Therapeutics are focused on evaluating an allogenic CAR therapy (referred as CTX130™) for treating CD70+ relapsed or refractory T or B cell malignancies and renal cell carcinomas. Here, T cells are taken from healthy donors, CD70 loci and the two loci for TCR (TRAC and TRBC) are disrupted using the CRISPR-Cas9 system, and a CD70-specific CAR is precisely inserted into TRAC loci using the CRISPR-Cas9 system. Initial results indicated that in patients who had high doses (>300 million cells), there was a complete response in 29% of patients, and progression of disease was prevented in 100% of patients. There were no side effects such as cytokine release syndrome (grade 3 or higher) in these high dose group of patients in the trial [80]. This trial is still ongoing, and complete results are yet to be published.
2.1.6. CRISPR-Cas9 Tools to Reduce Side Effects of CAR-T-Based Therapies
CRISPR-Cas9 systems have also helped in studying ways to mitigate other side effects of CAR-T therapies including cytokine release syndrome (CRS), prevalent in adult B cell acute lymphoblastic leukemia (BLL) patients when treated with the CD19-specific CAR-T cell therapy. Pre-clinical studies have indicated that CRISPR-Cas9-mediated decreased expression of HPK-1 in CAR-T cells mitigated the CRS effects while maintaining an effective anti-tumor capability [81]. These findings are being validated in a phase 1 clinical trial [62] in patients and results indicated that 72.7% of patients (8 out of 11) had complete remissions of cancer with no patient experiencing CRS (grade 3 or higher) or neurological events [82].
2.1.7. Role of CRISPR-Cas9 Tools in TCR Therapies
Beyond the CAR-T- and TIL-based therapy described above, adoptive cell therapies also include therapies based on T cells with engineered TCRs. In this approach, exogenous TCRs that are designed to target a specific antigen are engineered into T cells. To prevent the mispairing and/or competing of exogenous TCRs with endogenous TCRs, both the α and β chains of the endogenous receptor are removed by knocking out TRAC and TRBC using CRISPR-Cas9 tools. Two clinical trials have used this approach to target multiple myeloma [49] and acute myeloid leukemia [64]. The NCT03399448 [49] trial targeting multiple myeloma [49] is one of the first trials to publish data on safety and efficacy of CRISPR-Cas9-engineered T cells where, in addition to removing α and β chains of the endogenous TCR, the immune checkpoint regulator PD1 was also removed to enhance the anti-tumor activity of these cells with engineered TCRs [51].
Recently, Intellia Therapeutics initiated a trial [64] for evaluating their autologous TCR therapy for treatment of acute myeloid leukemia. Their product, named NTLA-5001, comprised T cells that had their TRBC and TRAC loci disrupted sequentially using CRISPR-Cas9, and later, an exogenous TCR specific to Wilms Tumor 1-specific antigen was inserted into the TRAC loci using CRISPR-Cas9 [83]. As of October 2022, Intellia therapeutics has indicated that this trial has been stopped, and a similar allogenic product is under preclinical development. In future, CRISPR-Cas9 tools are expected to play a significant role in the advancement of engineered TCR-based therapies as well.
2.1.8. Gene Editing Efficiencies and Safety Profiles of CRISPR-Cas9-Edited T Cell Therapies
In the multiple clinical studies (NCT03399448 [49], NCT03747965 [48], NCT02793856 [45], NCT04557436 [70]) that have been published so far on CRISPR-Cas9-mediated gene editing in T cell therapies, gene-edited products were found to be clinically acceptable and safe. One of the major risks associated with CRISPR-Cas9-mediated gene editing has been the off-target editing. While all four studies cited above identified off-target edits, their frequencies were low between 0.05–4% in the overall gene-edited populations. The first published trial involving CRISPR-Cas9 editing in T cell therapies (NCT03399448 [49]) involved simultaneous editing of three different genes (TRAC, TRBC, and PDCD1) via electroporation of Cas9 with three different sgRNAs, one for each target. As a result, the cell population had TRAC edited in only 45% of cells, TRBC in 15%, and PDCD1 in 20% of cells. More importantly, only 10% of cells had edits at all the three loci, with over 30% of cells having no edits in any of the loci. These statistics indicate that the overall low editing efficiency of the CRISPR-Cas9 system used in this study. Though there were off-target edits for each of the locus, the vast majority of the cells had on-target edits, with average on-target editing frequency across three patients in the trial was 99.4% for the PDCD1 locus, 98.6% for the TRAC locus, and 95.% for the TRBC locus. The risk of translocation exists during simultaneous editing of multiple loci, and translocations were observed in all manufactured products; however, the translocation frequencies were low and close to the limits of detection of the qPCR assay used for measurement. In general, translocations decreased over time during manufacturing as well as post infusion, indicating no positive selection for these translocations. Subsequently, after four months of infusion, barring one large 9.3 kb deletion, all other translocations were not detected [51].
Another study [52] used electroporated plasmids containing genes for Cas9 and sgRNAs to edit PDCD1 locus in T cells and observed a median editing efficiency of 5.8% in cell products across 12 patients. Off-target effects were measured at 18 different potential off-target sites, and the median off-target frequency was 0.05% across all patients with the majority of these effects occurring in the intergenic and intronic regions. In another trial [48], published by Han et al. [53], CRISPR-Cas9 was used to simultaneously target two genes, TRAC and PDCD1, via electroporation of Cas9 and two sgRNAs and reported a mean editing efficiency of 87.6% in the PDCD1 locus and 95.7% in the TRAC locus. To measure possible off-target effects, 26 different potential off-target sites were genotyped, and no off-target edits were observed. Since two sites were edited simultaneously, translocation frequencies were measured as well and were found to be similar to previous CRISPR-based [51] and TALEN-based multi-gene editing studies [84].
Interestingly, Intellia Therapeutics unveiled their gene editing approach [83] for their NTLA-5001 product evaluated in a clinical trial [64]. Here, they employed a sequential editing approach to KO first the TRBC locus of TCR receptor using a lipid nanoparticle (LNP) containing Cas9 and a sgRNA. Later, in a subsequent round of gene editing, the TRAC locus was targeted followed by the precise insertion of CAR at the TRAC locus. The DNA template coding for engineered TCR was delivered via an AAV6 virus instead of electroporation or lipid-based transfection of a double-stranded DNA. Sequential gene editing at different targets avoided risks of translocation, and they reported endogenous TCR KOs in 98% of cells with precise exogenous TCR insertion at the TRAC locus in over 50% of cells. This clinical trial was initially designed to be an autologous therapy, but later, it was discontinued and is now under development to be an allogenic product. Currently, the impediment in assessing the safety of these products is that in studies that are completed so far, the size of the trials are small and have employed products with low gene editing efficiencies. As the editing efficiency improves, a fresh set of assessments for safety and efficacy of various approaches described above would be required. The outcome of many of ongoing clinical trials may provide the future directions for this field.
2.2. CRISPR-Cas9 Therapies for Other Non-Infectious Diseases
Information related to these subsequent clinical trials can be found in Table 3.
2.2.1. Cell Therapy for Sickle Cell Disease (SCD) or Transfusion-Dependent β-Thalassemia (TDT)
Sickle cell anemia is a genetic disease caused by homozygous mutations in the β-globin gene, resulting in misshaped and rigid red blood cells that are prone to clog small blood vessels and deprive tissue of oxygen (vaso-occlusive crisis); further, in sickle cell disease, red blood cells are fragile and die easily, leading to a shortage of red blood cells (anemia). Similarly, β-thalassemia is a genetic blood disease in which the body fails to produce adequate hemoglobin due to deleterious mutations in β-globin resulting in anemia; patients with the most severe disease require regular blood transfusions to survive (transfusion-dependent β-thalassemia). In both diseases, increased expression of fetal hemoglobin over adult hemoglobin is associated with better patient outcomes. Several groups are developing treatments that have demonstrated promise as a functional cure for one or both diseases and are described below.
A collaboration between CRISPR Therapeutics and Vertex Pharmaceuticals produced the CRISPR-Cas9 therapeutic exagamglogene autotemcel (“exa-cel”, formerly CTX001) to functionally cure both sickle cell disease (SCD) and transfusion-dependent β-thalassemia (TDT) [85,86,87,88,89,90]. In this therapy, CD34+ hematopoietic stem and progenitor cells (HSPCs) are isolated from a patient, modified with exa-cel to knock-out the expression of a repressor to fetal hemoglobin (BCL11A), thereby increasing expression of fetal hemoglobin in the blood to compensate for the loss of adult hemoglobin (refer to Khosravi et al. [91] for a clear illustration of how BCL11A facilitates the switch from fetal to adult hemoglobin). The edited cells are then returned to the patient. The therapeutic goal is for patients to require fewer blood transfusions (β-thalassemia) and experience fewer vaso-occlusive crises (sickle cell disease). Early study results on the first two patients to receive exa-cel reported the editing efficiency of the exa-cel therapeutic to be 68.9% for patient one (received exa-cel for β-thalassemia), and for patient two, there was 82.6 and 78.7% editing efficiency (patient two received two lots of exa-cel for sickle cell disease) [92]. Subsequent early study results with 31 patients with sickle cell disease reported that after a single exa-cell infusion, all patients in the trial experienced no adverse events related to therapy and no vaso-occlusive crises for 2–32 months after exa-cell therapy (for reference, patients experienced 2–9.5 severe vaso-occlusive crises in the two years preceding exa-cel) [93]; at six months post infusion, the average percentage of bone marrow CD34+ HSPCs or peripheral blood mononuclear cells (PBMCs) with an edited BCL11A allele was 86.6% or 76.0%, respectively. Additionally, early results from a β-thalassemia trial indicated that out of the 44 individuals in the trial, 42 of them did not require RBC transfusion post therapy in the 1–36 months post exa-cel; the 2 patients that had not stopped RBC transfusion had reduced the transfusion volume by ~80% (for reference, these 44 individuals had received between 15 and 71 RBC transfusions in the two years preceding exa-cell therapy) [94]; editing efficiencies were somewhat reduced in this study compared to the aforementioned trial for sickle cell disease, with the average percentage of bone marrow CD34+ HSPCs or PBMCs containing an edited BCL11A allele was 74.3% or 63.4%, respectively
EdiGene and Bioray Laboratories used a similar approach as in exa-cel in their respective therapies, ET-01 and BRL-101; both therapies use CRISPR-Cas9-mediated KO of BCL11A expression in autologous CD34+ HSPCs in β-thalassemia patients [95,96,97]. Preliminary safety and efficacy results on a single patient were published for ET-01, demonstrating ~80% editing efficiency in the ET-01 therapeutic and after infusion, editing frequency at 52 weeks of ~60 and ~80% in peripheral blood and bone marrow, respectively [98]; the patient saw in increase in fetal hemoglobin and after 87 days post ET-01, the patient stopped requiring blood transfusions, staying transfusion-free for at least 15 months. Likewise, initial data from a study investigating BRL-101 in two children with β-thalassemia demonstrated engraftment of CRISPR-Cas9-modified HSPCs and increases in fetal hemoglobin expression, and both patients did not require a blood transfusion for >18 months post treatment [99]. This study demonstrated high editing rates in the BRL-101 therapeutic (97.17 and 98.22% for patient 1 and 2, respectively), and post therapy, editing efficiencies of PBMCs were in the range of 61.72–79.55% over an 18-month period.
Table 3.
List of CRISPR-Cas9-based gene editing therapies for other non-infectious diseases.
| Conditions Targeted | Gene Target | Edit Type | Therapeutic | Sponsor | Clinical Trial ID |
|---|---|---|---|---|---|
| Sickle cell disease or β-thalassemia | BCL11A | KO (NHEJ) | exa-cel | CRISPR Therapeutics and Vertex Pharmaceuticals | NCT03655678 [85] |
| NCT05477563 [86] | |||||
| NCT03745287 [87] | |||||
| NCT05356195 [88] | |||||
| NCT05329649 [89] | |||||
| NCT04208529 [90] | |||||
| ET-01 | EdiGene (GuangZhou) Inc. | NCT04925206 [95] | |||
| NCT04390971 [96] | |||||
| BRL-101 | Bioray Laboratories | NCT05577312 [97] | |||
| β-globin | HDR | nula-cel | Graphite Bio, Inc | NCT04819841 [100] | |
| CRISPR_SCD001 | UCLA, UC Berkeley | NCT04774536 [101] | |||
| iHSCs with corrected β-globin | ALLIFE Medical Science and Technology | NCT03728322 [102] | |||
| β-thalassemia | γ-globin promoter | KO (NHEJ) | BRL-101 | Bioray Laboratories | NCT04211480 [103] |
| EDIT-301 | Editas Medicine, Inc. | NCT05444894 [104] | |||
| Sickle cell disease | NCT04853576 [105] | ||||
| Type 1 diabeties | proprietary | VCTX210A | CRISPR Therapeutics and ViaCyte | NCT05210530 [106] | |
| Leber congenital amaurosis 10 | CEP290 | EDIT-101 | Editas Medicine, Inc. | NCT03872479 [107] | |
| Hereditary angioedema | KLKB1 (liver) | NTLA-2002 | Intellia Therapeutics | NCT05120830 [108] | |
| Duchenne muscular dystrophy | Dp427c | Exon skipping | CRD-TMH-001 | Cure Rare Diseases, Inc | NCT05514249 [109] |
Several other companies are also developing therapies using CRISPR-Cas9 gene editing therapies for TDT and SCD. Editas Medicine is testing EDIT-301, an autologous cell therapy comprising CD34+ hematopoietic stem cells edited to promote fetal hemoglobin expression (NCT05444894 and NCT04853576) [104,105]. It was previously observed that some mutations within the distal CCAAT-box region of fetal hemoglobin (γ-globin) promoters resulted in persistence of fetal hemoglobin in adults; EDIT-301 mimics naturally occurring mutations through Cas12a RNP-mediated disruption of this region to promote the expression of gamma globin (and thus the formation of fetal hemoglobin) [110,111]. Early clinical data from the first two patients that received EDIT-301 for sickle cell disease had positive initial findings with successful hematopoietic engraftment and an increase in fetal hemoglobin levels; further, both patients did not have vaso-occlusive crises in the 1.5 or 5 months following treatment [112].
Finally, two separate teams are using homology-directed repair to replace the SNP that causes sickle cell disease. Graphite Bio is evaluating their drug nulabeglogene autogedtemcel (nula-cel, formerly GPH101), which uses a high-fidelity Cas9 and non-integrating template DNA delivered from AAV6 to correct the β-globin gene through homology-directed repair (HDR) in autologous CD34+ HSPCs cells [113]. A press release from Graphite Bio indicated the first patient had received nula-cel [114], though the trial has subsequently and voluntarily been paused pending an adverse event that occurred in the first patient [115]. UCLA and UC Berkley researchers developed CRISPR-SCD001, which similarly corrects the β-globin gene through HDR, though in this case, the CRISPR-Cas9 system is delivered ex vivo via electroporation [101]. No additional information on this study was available to date.
2.2.2. Type 1 Diabetes
A few years ago, ViaCyte published early clinical results demonstrating successful implantation of a visualizable device containing pancreatic progenitor cells differentiated from a proprietary human pluripotent stem cell line (CyT49) for the purposes of treating type 1 diabetes [116,117]. These studies showed that implanted pancreatic progenitor cells survived and matured into beta cells which produced insulin in a meal-responsive manner. However, while promising, patients in the study had to take immunosuppressive drugs to avoid allogenic cell rejection, and several adverse events were observed in the study that were attributed to immunosuppression itself.
In an effort to avoid allogenic cell rejection while providing a functional cure for type 1 diabetes, ViaCyte joined efforts with CRISPR Therapeutics to create a combination therapy called VCTX210, which comprised the same transplantable device containing pancreatic beta-cell precursors derived from CyT49 cells transgenically modified to prevent triggering an immune reaction using a proprietary CRISPR-Cas9 scheme [106]. The first patient was transplanted with VCTX210 in February 2022 [118], though no preliminary results updates were found to date.
2.2.3. Leber Congenital Amaurosis 10 (LCA10)
Editas Medicine developed EDIT-101 for the treatment of Leber congenital amaurosis (LCA10), a severe retinal disease that leads to blindness that is caused by mutations in the CEP290 gene, the most common of which is an intronic A-to-G mutation causing a splice donor site and corresponding cryptic exon insertion [119]. EDIT-101 is a subretinal injection of AAV5 containing DNA for Cas9 and two guide RNAs; the gRNAs target two regions flanking the pathogenic intronic mutation in CEP290 (IVS26), excising or inverting it to restore function [120]. The results from 14 patients treated with EDIT-101 indicated 3/14 patients responded to treatment with improvements in retinal sensitivity and/or visual tests; of these, 2 patients were identified as homozygous for the IVS26 mutation [121]; subsequently, Editas has paused the clinical trial in search of a collaborative partner to work with on this therapy.
2.2.4. Hereditary Angioedema
Hereditary angioedema (HAE) is a genetic disorder caused by an autosomal dominant mutation in KLKB1, encoding for prekallikrein, which is produced in the liver and circulates in the blood, where it is converted to kallikrein by the protease Factor XII. It is characterized by severe and reoccurring swelling (HAE attacks) in various regions of the body, causing pain or threatening life (e.g., in the case of throat swelling). Intellia Therapeutics developed NTLA-2002, an in vivo therapy for hereditary angioedema, which is delivered intravenously and comprises LNPs containing Cas9 mRNA and sgRNA specific to KLKB1 [108]. Upon intravenous delivery, NTLA-2002 is taken up by liver cells where it causes indels in KLKB1, knocking out function [122]. The interim clinical study results for 10 patients receiving NTLA-2002 reported that the therapeutic was well tolerated by patients, who experienced a dose-dependent reduction in plasma kallikrein and large reduction in HAE attacks over the study period, with the first three patients being attack free for 5–10 months post treatment (for reference, this patient cohort suffered about 1–7 attacks per month prior to treatment) [123].
2.2.5. Duchenne Muscular Dystrophy
As of 15 December 2022, the ClinicalTrials.gov database contains only one clinical study involving the use of a CRISPR therapeutic intervention aimed at altering gene expression (NCT05514249, Table 4) [109]. The phase 1 study, sponsored by Cure Rare Diseases, Inc in collaboration with UMass Chan Medical School, enrolled a single patient with Duchenne muscular dystrophy (DMD) who was treated with a single intravenous dose of the investigational new drug (IND) CRD-TMH-001. DMD is a rare, X-linked recessive disorder that affects ~1 in 3500 males worldwide [124]. In DMD, most affected individuals lack the dystrophin protein due to mutations that result in frameshift errors. The dystrophin gene spans 2.6 Mb and contains 79 exons and is the largest gene in the human genome. The Cure Rare Diseases therapeutic relied on CRISPR-mediated exon skipping, targeting one or more exons that were causing frameshift errors to produce an upregulated isoform of dystrophin. Exon skipping typically involves targeting antisense oligonucleotides to introns that flank the exon of interest in pre-mRNA to induce splicing of the aberrant exon. Similarly, CRISPR-Cas can be used to induce alternative splicing. Cure Rare Diseases describes using CRISPR components regulated by a muscle-specific promoter, packaged in a patient-matched AAV serotype with low immunogenicity, that targets delivery to muscle cells.
2.3. CRISPR-Cas9 Therapies for Viral Infections
2.3.1. Human Immunodeficiency Virus (HIV-1)
Excision BioTheraputics evaluated dosages and long-term safety of the in vivo biologic EBT-101 for the treatment of HIV-1 infection [125,126]. EBT-101 is an AAV9-delivered CRISPR-Cas9 therapeutic given intravenously to target and excise HIV-1 proviral DNA (the viral form that has integrated into the genome of infected cells and is impervious to standard treatments). EBT-101 gRNAs target HIV-1 proviral DNA and create multiple dsDNA breaks to cut out a large section of the proviral genome, which, when repaired through NHEJ, renders the HIV-1 proviral genome incomplete and unable to replicate, effectively removing it from reservoir cells and tissues. In vivo preclinical studies targeting simian immunodeficiency virus (SIV) in rhesus macaques demonstrated the therapy was well tolerated, with successful excision of HIV-1 proviral DNA from a wide-range of tissue reservoirs, including CD4+ T cells, brain, and lymph nodes, among others [127]. Recently, the first human patient was dosed with EBT-101 (July 2022), with the first press release reporting the treatment had been well tolerated and the clinical trial was moving forward as planned [128].
Ex vivo cell therapy approaches have also been proposed for the prevention or treatment of HIV. One clinical trial from Affiliated Hospital to Academy of Military Medical Sciences in Beijing aims to investigate the safety of transplanted allogenic CD4+ cells that have had KO of CCR5, a cell coreceptor for HIV entry [129]; indeed, non-functional CCR5 alleles have been observed in naturally HIV-resistant humans [130]. A case report was published for a single patient who was diagnosed with an HIV infection and, shortly thereafter, T cell acute lymphoblastic leukemia (T-ALL) [131]. Following antiviral therapy that resulted in undetectable virus in serum and chemotherapy for T-ALL, the patient received allogenic hematopoietic stem cell transplantation. Prior to transplantation, CD34+ HSPCs were isolated and edited to KO the CCR5 gene with the CRISPR-Cas9 approach. Transplanted cells were successfully engrafted, with cells with a modified CCR5 gene persisting for at least 19 months. However, post therapy, the proportion of circulating bone marrow cells that had a disrupted CCR5 gene was low (<8%), which was insufficient to cure HIV-1 infection, highlighting that improvements in CRISPR-Cas9 editing efficiency in this system are needed.
2.3.2. Human Papilloma Virus (HPV)
A clinical trial sponsored by First Affiliated Hospital at Sun Yat-Sen University in Guangzhou, China proposes to use two in vivo approaches to treat persistent human HPV infection and related cervical malignant neoplasia: a TALEN-based and a CRISPR-Cas9-based therapy [132]. HPV, once integrated into the host genome, is particularly difficult to treat. Each therapy would be encoded in a plasmid administered in a gel and would target two HPV genes (the E6 and E7 oncogenes) to destroy the integrated viral genome, causing apoptosis or growth inhibition of infected cells. The status of this study is unknown and was last updated in 2017. In the interim, the team has been publishing results from their efforts to improve editing efficiency of the HPV E6/E7 oncogenes through a method called gene knock-out chain reaction (PMID: 35036522) [133], and exploration of a CRISPR/Cas13a system to knock-out HPV E6/E7 mRNAs in vivo cell culture [134].
Table 4.
List of CRISPR-Cas-based gene editing therapies for infectious diseases.
| Condition Targeted | Gene Target | Edit Type | Therapeutic | Sponsor | Clinical Trial ID |
|---|---|---|---|---|---|
| HIV-1 | HIV proviral DNA | Viral genome split (NHEJ) | EBT-101 | Excision Biotherapeutics | NCT05144386 [125] |
| NCT05143307 [126] | |||||
| HPV | E6/E7 genes of HPV16/18 | Viral genome split (NHEJ) | Talen: TALEN-HPV16 E6/E7 or TALEN-HPV18 E6/E7; CRISPR-Cas9: CRISPR/Cas9-HPV16 E6/E7T1 or CRISPR/Cas9-HPV18 E6/E7T2 | First Affiliated Hospital, Sun Yat-sen University | NCT03057912 [132] |
| Viral keratitis | HSV-1 genome | Viral genome split (NHEJ) | CRISPR/Cas9 mRNA | Shanghai BDgene Co., Ltd. | NCT04560790 [135] |
2.3.3. Viral Keratitis, Herpes Simplex Virus 1 (HSV-1)
BDgene Co investigated safety and dosing of an in vivo CRISPR-Cas9 mRNA therapy (BD111) for corneal inflammation (keratitis) caused by HSV-1 infection [135]. BD111 is delivered by lentivirus into the cornea. In preclinical research, this therapy demonstrated transport from the cornea into neurons (trigeminal ganglia, which are viral reservoirs) with a short life span of Cas9 activity (three days post injection); following injection, viral replication is inhibited by guiding CRISPR-Cas9 activity to specific regions of the HSV-1 genome, causing a large genome excision subsequent INDEL formation [136]. Clinical investigation of BD111 is primed to move forward having received FDA orphan drug designation in June 2022 [137].
3. Base Editing
3.1. Principles of Base Editing
One of the most promising advancements utilizing the CRISPR system is that of base editing. Developed by David Liu’s lab in 2016 [138], the first base-editing-based therapeutics made their way into phase I clinical trials in 2022, with many more in the pipeline. Base editing works by directly converting one or more DNA bases to another (for example, C to T) without the initiation of a double-strand break or the use of repair templates. Early efforts in base editing used either an inactive Cas9 or a Cas9 nickase fused to a cytidine deaminase to directly convert cytidine to uridine, resulting in a C-to-T substitution [138,139]. Thus, the Cas9 effectively serves as a programmable guide to direct the deaminase to the desired chromosomal location. When the guide RNA binds to the target DNA sequence and forms an RNA:DNA hybrid, this leaves a stretch of single-stranded DNA (ssDNA) directly adjacent to the PAM sequence. Since ssDNA is the substrate for deaminase enzymes, the ssDNA in this bubble can then be targeted by the base editor. The nucleotides closest to the PAM are obstructed by the Cas9 and are unable to be edited, but the nucleotides furthest from the PAM are accessible to the deaminase, facilitating base editing in a narrow window at a set distance from the PAM sequence. Since the initial invention of base editing, much work has been conducted to improve the fidelity of the approach and improve the scope of the base conversions that are possible [11,140]. Using classical base editing, it is possible to make four nucleotide conversions (C to T, A to G, T to C, G to A) that represent almost one-third of pathogenic mutations in humans. A more recent advance, prime editing [141], can make the remaining nucleotide conversions and will be discussed in more detail later.
3.2. Clinical Applications of Base Editing
Since the invention of base editing in 2016, several companies have formed around base editing technology, with various therapeutics in the pipeline. In general, base editing therapeutics can be divided into the type of delivery method that is used (Figure 4). Some therapies are delivered directly to patients using either LNPs or a viral vector, while others are first delivered ex vivo to hematopoietic stem cells (HSCs) or T cells, typically through electroporation, and the edited cells are then reintroduced to the patient. Current base editors that have entered clinical trials are discussed below.
3.2.1. In Vivo Delivery
An attractive feature of base editing is that it raises the potential to directly modify cells in tissues of patients with high efficiency and low probability of off-target effects. The first in vivo-delivered base editor to enter phase I clinical trials is sponsored by Verve Therapeutics and uses LNPs for delivery of the base editing machinery [142]. This therapeutic, named VERVE-101, is designed to treat heterozygous familial hypercholesterolemia (HeFH). HeFH is a commonly inherited genetic disorder affecting around 1 in 250 people globally and is caused by mutations in the low-density lipoprotein receptor gene (LDLR) that result in abnormally high LDL levels [143]. Rare forms of familial hypercholesterolemia can also be caused by gain-of-function mutations in PCSK9, which is preferentially expressed in the liver [144]. Naturally occurring PCSK9 loss-of-function variants are also common (2–3% in some populations) and result in lowered LDL levels [145,146], suggesting PCSK9 as a target for treating HeFH. The siRNA-based therapeutic inclisiran inhibits PCSK9 expression and lowers LDL levels for several months [147], raising the possibility that an irreversible editing of PCSK9 might confer longer-term or permanent lowering of LDL levels. VERVE-101 uses an adenine base editor to target and effectively knock-out PCSK9 through the induction of a single base edit. While the first patient in the VERVE-101 clinical trial was just enrolled in July 2022, and therefore, no data are available to assess the safety or efficacy. Verve and collaborators have conducted several preclinical studies in animal models that delivered promising results [148,149,150]. In a nonhuman primate model, the base editor was delivered to cynomolgus monkeys using an LNP-based delivery approach. The targeting of PCSK9 was very efficient, with 70% of liver PCSK9 edited at the highest dose, resulting in an 83% reduction in blood PCSK9 levels. As a result, LDL-C levels were reduced by up to 69%, with durable effects lasting at least through the duration of the study, which was over 1 year [148].
3.2.2. Ex Vivo Delivery
Base editing also offers the ability to modify cells that have been extracted from patients, primarily HSCs or T cells, in cell therapy applications. There are two candidates for such applications that have advanced to clinical trials. The first to be delivered to patients, in May 2022, is a phase I trial sponsored by the Great Ormond Street Hospital for Children (NCT05397184 [22]). For this therapy, named BE CAR-7, T cells were collected from healthy donors to make allogeneic CAR-T cells for later infusion into patients with relapsed/refractory T cell acute lymphoid leukemia. After collecting the donor T cells, the base editor designed to modify three genes (TRBC, CD52, CD7) was delivered via electroporation, followed by delivery of a chimeric antigen receptor with a lentiviral vector [151,152]. While no peer-reviewed studies have been reported about the outcomes for this trial, news reports have hinted at promising results by reporting which showed that the first patient enrolled in this trial is leukemia free six months after she received the edited CAR-T cells and a second bone marrow transplant [153].
The second candidate is sponsored by Beam Therapeutics and is named BEAM-101 (NCT05456880). In November 2022, Beam successfully enrolled its first patient in its phase I/II clinical trial for BEAM-101, which is designed to treat severe SCD and β-thalassemia. Beam’s approach is modeled after individuals with natural genetic variants that cause continued expression of fetal hemoglobin, which can prevent or alleviate SCD symptoms [154]. Accordingly, BEAM-101 is designed to modify HSCs ex vivo with a base editor to activate the expression of fetal hemoglobin before transfusion of the HSCs back into the patient [155]. Beam also has a base editing-based therapeutic named BEAM-102 in pre-clinical development that similarly aims to treat SCD. In this case, however, Beam seeks to induce a base edit in HSCs that will revert the SNP that causes SCD to the normal hemoglobin sequence.
4. Prime Editing
As discussed previously, cytosine and adenine base editors are not able to make all the possible nucleotide changes. Therefore, while the potential applications of base editing are vast, they are also limited by the enzymes tethered to the Cas9. In 2019, Liu’s lab also developed a technique termed prime editing [141], which has overcome some of the limitations of base editing, expanding the potential scope of this class of CRISPR-based applications. As in base editing, prime editing covalently attaches an enzyme to a Cas9 nickase to target a desired genomic locus. However, instead of a deaminase, prime editing uses a modified reverse transcriptase (RT). In addition, prime editing uses a modified version of the guide RNA termed the prime editing guide RNA (pegRNA), consisting of a typical guide RNA that also contains an RT template sequence with the desired edit. The Cas9 nickase first makes a single strand cut, and the resulting 3′ hydroxyl group from the cleaved DNA serves as the primer to initiate reverse transcription of the template sequence. After reverse transcription, cellular repair processes then incorporate the newly synthesized DNA into the target site, and the permanent editing of both strands is accomplished by cellular DNA repair machinery. The result is that any nucleotide change can be induced with prime editing, and short indels can also be made without inducing double-strand breaks. Importantly, prime editing can also induce changes at distances further from the PAM than first-generation base editors, which should provide more flexibility in edits and lower reliance on the precise location of the PAM sequence. While no clinical trials based on prime editing are underway, the scope of DNA edits afforded by prime editing makes this a promising future modality. However, compared with base editing, prime editing is less efficient and induces more indels [11], so improvements are needed for prime editing to reach its full potential.
5. CRISPR and Gene Regulation
5.1. CRISPR Interference/CRISPR Activation (CRISPRi/CRISPRa)
CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) rely on the use of catalytically inactive dCas9, fused to effector domains that either repress or activate gene transcription (CRISPRi or CRISPRa, respectively) [156,157]. While dCas9 lacks endonuclease activity, it retains the ability to interact with guide RNA and bind target DNA loci, functioning as an RNA-guided DNA binding protein [12]. For CRISPRi in mammalian systems, dCas9 is fused with transcriptional repressor domains, most notably the Krüppel-associated box (KRAB) domain, one of the most potent transcriptional repressors in the human genome [12,156,158]. KRAB-based transcriptional repression relies on recruitment of KAP1 and on KAP1 recruitment of additional co-repressors (e.g., HP1, SETDB1, histone deacetylases) that propagate heterochromatin [156,159]. In CRISPRa, dCas9 is fused with transcriptional activator domains that promote gene expression. In eukaryotic cells, activator domains used include VP64 (four tandem repeats of Herpes simplex virus VP16 domain) fused with additional activation domains including Rta (Epstein–Barr virus R transactivator), p65 (subunit of NFκB), and HSF1 [156,157,160,161]. The multidomain construct VPR (fusion of VP64, p65, and Rta) is often used in mammalian systems to recruit and stabilize transcription factors and activate transcription [162].
5.2. CRISPR Epigenetic Editors
Catalytically inactive dCas9 can also be fused to catalytic epigenetic effector domains to alter DNA methylation and histone modifications to induce targeted gene silencing or activation. DNA methylation effectors successfully used for CRISPR-targeted gene silencing in human cells include the de novo methyltransferase DNMT3A and DNMT3L domains, and DNA demethylation domains successfully used for CRISPR-targeted gene activation include TET1 [163,164,165,166]. Histone modifiers have also been fused to dCas9 to induce targeted acetylation or methylation of H3K27, and methylation of H3K4, H3K9, and H3K79 in human cells [167,168]. Combinatorial fusions that incorporate both transcriptional and repressor epigenetic domains, for example, KRAB and DNMT3A, have demonstrated synergistic silencing [169].
5.3. Applications of CRISPR Gene Regulation in Models of Human Disease
5.3.1. Retinitis Pigmentosa
Retinitis pigmentosa (RP) is a rare genetic disease that leads to progressive vision loss and affects ~1 in 4000 people globally [170]. In a mouse model of RP where mice carry a spontaneous mutation of the rod-phosphodiesterase gene (rd10 mice), a dCas9-KRAB system was targeted to the Nrl gene, which regulates rod versus cone photoreceptor determination through activation of Nr2e3, a transcription factor that represses transcription of multiple cone-specific genes [171,172]. Repression of Nrl by dCas9-KRAB resulted in reprogramming of rods into cone-like cells which are resistant to rod photoreceptor retinitis pigmentosa-specific mutations and prevented secondary cone loss [173,174]. In another study, Böhm et al. used dCas9-VPR to transactivate expression of a rhodopsin homolog from Opn1nw in a rhodopsin-deficient (Rho+/−) mouse model for RP [175]. The leading cause of RP is mutations in the RHO photoreceptor gene, which encodes for the most abundant protein in rod cells of the retina [176,177]. However, phototransduction molecules in rods and cones are encoded by several distinct but functionally equivalent genes [178]. Activation of Opn1nw in Rho+/− mice using dCas9-VPR resulted in amelioration of retinal degeneration and improved retinal function [175].
5.3.2. Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral muscular dystrophy (FSHD) is a rare genetic disease that affects ~1 in 20,000 males and females of all ages, and leads to progressive muscle degeneration in the face, shoulders, and upper arms [179]. FSHD is linked to contractions or loss of methylation of the D4Z4 macrosatellite repeat array at 4q35, which allows aberrant full-length DUX4 expression (DUX4-FL) in skeletal muscle leading to muscle atrophy [180,181]. Epigenetic dysregulation of the FSHD locus is proposed to also contribute to DUX-FL expression and pathogenesis, since FSHD onset, progression, and severity is highly variable. Himeda et al. used lentiviral delivery of dCas9-KRAB to the DUX4 promoter or exon 1 in FSHD myocytes derived from patient bicep muscle and demonstrated reduced expression of DUX4-fl to ~45% of endogenous levels in FSHD myocytes [182].
5.3.3. Cancer
Altered gene expression is a hallmark of cancer, from down regulation of tumor suppressor genes (e.g., PTEN, BRCA1, CDKN2A, RASSF1, HIC1) to upregulation of oncogenes (e.g., GRN, FHL2, CNKSR1) [183,184,185,186,187,188,189,190]. For example, the loss of expression of tumor suppressor genes on chromosome 10, notably, the phosphate and tensin homolog (PTEN) tumor suppressor gene, is a common feature of numerous cancers that arises from somatic mutations, indels, and transcriptional and post-transcriptional alternations [191]. Epigenetic silencing through methylation of the PTEN promoter region has been reported in numerous cancers including melanoma, endometrial, breast, gastric, and colorectal cancers [192,193,194,195,196]. Using dCas9-VPR (VP64-p65-Rta), PTEN expression can be reactivated in melanoma cell lines, resulting in the repression of AKT, mTOR, and MAPK oncogenic pathways and increased sensitivity to B-Raf and P13K/mTOR inhibitors [197]. In primary breast myoepithelial cells, Saunderson et al. demonstrated that transient transfection with dCas9 DNMT3A-3L induced a state of hypermethylation leading to a p16 repression-driven hyper-proliferation, preventing senescence, and potentially leading to early tumorigenesis [198,199]. This study demonstrated that “hit-and-run” epigenetic alterations can induce heritable altered cellular processes.
5.3.4. Imprinting Diseases
Epigenetic editors may also help rare imprinting diseases, changing methylation in imprinting control centers where imprinted genes are inappropriately silenced or overexpressed, to treat these and restore proper imprinted gene expression. Approximately 100 genes in the human genome have been identified to be imprinted, and 9 different imprinting diseases have been described [200,201]. They are typically found in gene clusters, regulated by imprinting control centers (ICRs) that are either methylated (inactive) or unmethylated (active) under coordinated epigenetic control. During maternal oogenesis, ICRs typically become hypermethylated, while most ICRs in male germ cells are fully unmethylated. The ICRs control expression of long antisense transcripts that silence expression of the protein coding genes they overlap [202]. The E6-AP ubiquitin ligase expressed from UBE3A gene is imprinted in neurons, requiring a functional maternal copy for expression [203]. The paternal copy is silenced by a long non-coding RNA (UBE3A-ATS) of which the promotor/exon 1 region completely lacks methylation (maternal copy is fully CpG methylated). Angelman syndrome is a rare neurological disorder caused by deficiency in UBE3A expression, most often due to a 5- to 7 Mb maternal deletion in chromosomal region 15q11.2-q13, uniparental disomy, or translocation in the maternal copy [204]. Wolter et al. demonstrated that targeting sgRNA-Cas9 to Snord115 (a small nucleolar RNA gene located in the 3′ region of the UBE3A-ATS) resulted in the activation of paternal UBE3A in cultured human and mouse neurons. Using this same strategy, they used AAV to target neurons in a mouse model of AS during embryonic and early postnatal stages, which resulted in the expression of paternal UBE3A for at least 17 months and rescue of normal anatomical and behavioral phenotypes [205]. Approaches using dCas epigenome editors targeted to methylate CpG islands at the UBE3A-ATS transcriptional start site have also been proposed as a means of activating UBE3A expression [206].
5.3.5. HIV
The major barrier to curing HIV-1 infection is viral latency [207]. One approach for a cure is to administer latency-reversing agents to purge cells harboring harboring latent viral genomes, then treat with antiretroviral therapy. Another approach is to promote a permanent latent state by inhibiting HIV-1 transcription factors. In line with the second approach, da Costa et al. used sgRNAs targeting the HIV-1 proviral genome (LTR1-LTR5) and CRISPR/dCas9-KRAB to block HIV-1 reactivation in latently infected T cells and myeloid cells treated with latency-reversing agents such as PKC agonists and HDAC inhibitors [208]. They found that targeting the LTR enhancer region provided the best repression, and one of their CRISPR constructs inhibited latency reactivation by 160×.
5.4. Challenges and Future Perspectives
Of the clinical trials listed at the ClinicalTrials.gov database as of 15 December 2022, there are no published studies relying on use of therapeutics using CRISPRi, CRISPRa, or CRISPR-epigenetic editing. These newer CRISPR technologies offer numerous benefits, most notably, less concern for introducing off-target permanent genomic alterations with associated error-prone repair [10]. However, many unknowns remain, which include unknown off-target events and their long-term effects, dosing requirements, and duration of heritability. Even though dCas9 is catalytically inactive, off-target effects can be problematic for certain genomic loci and can lead not only to genome-wide changes in gene expression, but also unintended indel mutations [209,210,211]. To optimize specificity, important considerations in construct design include: sgRNA that incorporates structural elements in addition to sequence specificity, precise positioning of sgRNA relative to transcription start sites, and incorporation of distal regulatory elements (e.g., enhancers and locus control regions) [212]. Additionally, the use of engineered dCas9 or Cas9 orthologues that rely on rarer PAM sequences have been proposed as alternatives [213]. Lastly, in native tissues, the gold standard for gene delivery is the use of recombinant adeno-associated viral (rAAV) vectors; however, dCas9 fusions (e.g., 5.8 kb dCas9-VPR) often exceed AAV genome packaging capacity (~5 kb) [214]. Optimized strategies for efficient and targeted payload delivery have used split rAAV systems or lentivirus vectors, but challenges with serotype matching and tissue tropism remain [215,216]. Directed evolution of viral vectors, the use of functionalized LPNs, or the use of viral-like particles (VLPs) for packaging and delivery are other viable options [217,218,219].
Using sgRNA-dCas systems to alter gene expression has vast potential in therapeutic development for numerous human diseases, especially those that may require a modularized precision strategy that can address multiple pathogenic features as in the case of cancer. For example, using multiplexed sgRNAs, gene expression patterns contributing to cell proliferation, evasion of growth inhibition, apoptosis escape, immune evasion, drug resistance, and sustained angiogenesis could be all be addressed in a single in vivo cancer therapeutic [212,220]. In instances where transient ectopic expression is sufficient in achieving the therapeutic goal, where constitutive dCas9 expression is not required, CRISPRi/CRISPRa approaches may serve as important tools. iPSC differentiation or cellular reprogramming to restore pluripotency are potential ex vivo application targets for CRISPRi/CRISPRa, and potential in vivo applications include reversal of retrovirus latency allowing for subsequent anti-retroviral treatment [162,221].
CRISPR-epigenetic editing provides additional important functionality for gene regulation-based therapeutics—inducing changes in DNA methylation and/or histone modification that are heritable and providing stable transcriptional changes over hundreds of cell divisions [222]. Importantly, diseases caused by aberrant epigenetic programming (e.g., functional allele is available on the other parent-of-origin chromosome but silenced, improper biallelic expression) such as rare imprinting diseases can be directly impacted by epigenetic editing, with curative potential if administered at appropriate developmental timing [206]. Other important applications of CRISPR-epigenomic editing include the ability to transactivate functionally equivalent genes (with or without combined disruption of a pathogenic allele) in diseases caused by a heterozygous dominant haploinsufficiency. To this end, there are three biotech companies working on CRISPR-epigenomic editing that launched this year: Chroma Medicine, founded by David Lui; Tune Therapeutics, co-founded by Charlei Gerlash and Fyodor Urnov; and Epic Bio, led by Amber Salzman, based on Stanley Qi’s research. Of the three companies, Epic Bio is the only one that has disclosed their development pipeline, which relies on their Gene Expression Modulation System (GEMS) platform. Therapeutics in the research phase at Epic Bio include treatments for: facioscapulohumeral muscular dystrophy, targeting re-methylation the D4Z4 region to suppress DUX4 expression; Retinitis Pigmentosa 4, suppressing endogenous mutated RHO expression and producing normal RHO expression; Retinitis Pigmentosa 11, restoring PRPF3 expression to normal physiological levels; Alpha-1 Antitrypsin Deficiency, suppressing endogenous mutated A1AT expression and producing normal A1AT; and finally, heterozygous familial hypercholesterolemia, targeting pathways known to reduce cholesterol.
6. Emerging Applications of CRISPR-Cas in RNA Editing
6.1. RNA Editing
The therapeutic potential of genome editing for the treatment of genetic disease is considerable and still emerging. However, unresolved concerns regarding irreversible genetic changes at “off-target” loci may hinder some clinical applications. Recent developments in the identification of RNA-targeting Cas enzymes may offer a path to transient therapies that leverage the precision of CRISPR-Cas technology without the liabilities of permanent/heritable genetic alteration [223].
Enzymatic RNA editing as a biological process was first described in the 1980s–1990s with the discovery and characterization of two main RNA editing protein families, ADAR (adenosine deaminases acting on RNA) and APOBEC (apolipoprotein B mRNA editing catalytic polypeptide-like) [224,225]. The ADAR class of enzymes target specific adenosines in double-stranded RNA (dsRNA) for deamination, thereby converting them to inosines [226]. Inosine base pairs with cytidine and is recognized by translational and splicing machinery as guanosine, resulting in an effective A-to-G substitution [227]. Similarly, the APOBEC family of proteins target cytidines in single-stranded RNA (ssRNA) and ssDNA for deamination to uracil, effectively causing a C-to-T substitution. ADAR editing of coding sequences in pre-mRNAs in the nervous system and dsRNAs that feed into interfering RNA (RNAi) pathways provide important functional diversity beyond what is encoded in the genome [226]. APOBEC1 editing of endogenous mRNAs has been demonstrated to regulate functionally consequential changes to the sequences of encoded proteins, while cytidine deamination within the genomes of RNA and ssDNA viruses by APOBEC3 family members serves as a part of the innate immune response to viral infection [228].
By the mid-1990s, companies and academic groups were already exploring approaches to harness RNA editing for therapeutic benefit. In 1995, Ribozyme Pharmaceuticals demonstrated efficient and targeted reversion of a premature stop codon in dystrophin by endogenous ADAR in Xenopus embryos and mammalian cell extracts through the delivery of exogenous RNA oligos [229]. However, due to the imprecise nature of ADAR editing among other considerable technical challenges, RNA base editing as a therapeutic approach has hitherto failed to make significant progress toward advancing into the clinic. In the meantime, the development of zinc finger nucleases, TALENs, and, ultimately, CRISPR has led to an explosion of DNA editing approaches being applied to the development of new therapies and their clinical applications [230]. Other RNA-based approaches such as RNAi and mRNA have also moved rapidly into clinic with multiple blockbuster drugs approved [231]. Nonetheless, the potential to target RNA-mediated diseases and concerns regarding irreversible off-target effects of genomic DNA editing by CRISPR highlights the value that efficient RNA editing technologies could have for clinical and diagnostic use.
6.2. Cas RNA Endonucleases
In 2015, the RNA editing field received a jump-start when Cas13 (formerly known as C2c2) from Leptotrichia shahii was first described as a programmable RNA-guided RNA endonuclease [232,233]. The Cas13 family of proteins is targeted to ssRNA by sequence-specific 60–66-nucleotide-long CRISPR RNAs (crRNAs), whereupon it cleaves the target and then proceeds to indiscriminately cleave other ssRNA molecules. This activity has already been leveraged for the development of RNA-based COVID-19 and circulating tumor RNA/DNA diagnostics [234]. Base editing technologies relying on fusion of catalytically dead Cas13 (dCas13) to ADAR have also been developed and used to correct disease associated mutations at the RNA level in cellular models [15,235]. However, the in vivo therapeutic potential of Cas13 is limited by considerable collateral cleavage of off-target ssRNAs, which provides a desirable signal amplification for diagnostic purposes, but it is cytotoxic in multiple mammalian cell types [236]. As a result, there is a recognized need for more specific RNA editing tools.
The discovery of Type III-E CRISPR-Cas systems, first characterized in two 2021 publications, appears to be the breakthrough that addresses many of these short-comings [16,237]. Alternately referred to as Cas7-11 by Özcan and colleagues, and gRAMP (giant repeat-associated mystery protein) by van Beljouw et al., these fusions of four Cas7 subunits to a single Cas11 subunit result in the largest CRISPR-Cas effectors yet identified at 1300 to 1900 amino acids. Unlike Cas13, Cas7-11 exhibits highly specific and non-cytotoxic RNA targeting in cells [16]. In initial studies, Cas7-11 supports robust transcript knock-out in mammalian cells and, by fusion of catalytically dead dCas7-11 to ADAR2, efficient RNA base editing [16]. If the prohibitive size of these enzymes can be overcome (some progress in this regard has already been made [238]), this technology opens several new doors for therapeutic applications that could have a significant impact on the rapidly evolving RNA therapy space.
6.3. Therapeutic Applications of RNA-Cleaving Cas Enzymes
In addition to many of the same diagnostic approaches to which Cas13 has already been applied, the specificity of Cas7-11 lends itself to in vivo applications for the treatment of cancer, rare genetic diseases, and viral infections (Figure 5). Early studies indicate that Cas7-11 could be effectively used in the same manner as RNAi to negatively regulate gene expression through transcript destruction [16]. Modified Cas7-11 fusion proteins and other approaches that promote transcript stabilization or modulate splice sites can also be envisioned as future variations. Transcript regulation in this manner has broad potential for cancer treatment by modulating the expression of therapeutic targets in a tunable and temporally restricted manner that does not rely on the development of small molecule ligands. This technology could equally be applied in the treatment of rare genetic diseases to control the production of toxic proteins as in Huntington’s Disease, or in modulating any other RNA-mediated process including (but not limited to) hypertension, pain, and cell–cell communication [239,240,241]. The fusion of Cas7-11 to ADAR2 has already been used for efficient base editing in mammalian cells, suggesting additional applications in the treatment of genetic diseases caused by discrete heritable mutations [16]. Importantly, early research indicates that Cas711 may have fewer off-target effects than other RNAi methods which, if it is a general feature of this system across different RNA targets and cell types, could indicate an advantage over current clinical technologies for the modulation of transcript stability [16]. Outside of targeting endogenously encoded RNAs, RNA-cleaving Cas enzymes could have massive potential in combating RNA viruses and retroviruses such as SARS-CoV-2 and HIV.
While significant hurdles including the large size of the Cas7-11 protein, how to package it into a therapy, and how to deliver it effectively to target tissues will need to be addressed in coming years, RNA-cleaving Cas enzymes as therapeutic and research tools are an emerging area at the forefront of CRISPR technology with transformative potential for how diseases are studied and treated.
7. Development of Large Animal Models of Human Diseases
CRISPR and Preclinical Development
The use of CRISPR technology has revolutionized not only the treatment of human diseases but also the field of gene editing in large animal models. Genetically modified large animal models are of significant importance because they are used in the development of new therapies and as research models of human diseases. They are used to demonstrate the feasibility of in vivo CRISPR editing of somatic and germline cells as a therapeutic approach, and to evaluate pre-clinical efficacy and safety of new drugs prior to the initiation of clinical trials [242].
Small animals such as rodents are the predominant models used to study human disease, but often they do not fully recapitulate the pathological changes or symptoms that a disease produces in humans. These differences are due to high anatomical and physiological divergence between species and have led to the failure of many drugs in clinical trials that were only screened in small animal models [243]. Large animal models of human diseases can more accurately recapitulate the characteristics of the diseases, potentially making treatments developed in large animal models more likely to work in humans. Additionally, imaging tools for the screening of the disease or the treatment itself can be tuned in large animal models because of their similarities with humans.
The main large animal models currently used to mimic human diseases are non-human primates, pigs, sheep, goats, and dogs [242] (Figure 6). Non-human primates are the preferred model, not only due to their similarity in physiology and genetics, but also because they display cognition and social behaviors as well. The handicap is that they have long pregnancies and, in general, are expensive to maintain. Pigs are also extensively used as animal models and have the following advantages over non-human primates: early sexual maturity, short reproductive cycle, and high number of offspring per litter [244].
The first monkey genetically modified using CRISPR was reported in 2014 by Niu et al. [245]. Here, two genes were modified simultaneously (peroxisome proliferator-activated receptor gamma, PPARG, and recombination-activating gene 1, RAG1) by injecting Cas9 mRNA and sgRNAs against the two targets into one-cell-stage embryos. Following this report, Chen et al. [246] showed that CRISPR-genetically modified monkeys could transmit the modification through the germline, and that it was possible to create a model of Duchenne muscular dystrophy by disrupting the dystrophin gene in monkeys [247]. They showed that these monkeys displayed muscle degeneration and other characteristics of the disease in humans. Examples of CRISPR-edited monkeys created to resemble human diseases are the early-onset Parkinson’s disease (PD) model created by Yang et al. [248,249] targeting two exons in the PINK1 gene, the acute monkey PD models developed by Li et al. [250], Sun et al. [251], and Yang et al. [252], and the model for human adrenal hypoplasia congenita (AHC) and hypogonadotropic hypogonadism (HH) reported by Kang et al. [253] after knocking out the DAX1 gene. Genetically modified monkey models of human diseases have been also used to evaluate the efficacy of experimental drugs. An example is the study of Tu et al. [254], where they found that the use of the antidepressant fluoxentine treatment alleviated the abnormal brain activities of the autism spectrum disorder monkey model [255].
The first CRISPR-genetically modified pig was reported in 2014 [256] and was created by microinjection of the CRISPR-Cas9 system into the zygotes, making the modification germline transmitted. Another way of creating genetically modified pigs is by somatic cell nuclear transfer (SCNT), where pig fibroblasts can be edited in single or several loci at the same time using CRISPR and then fused into enucleated oocytes to create an edited zygote [257,258,259]. This technology avoids mosaic mutations and detectable off-target effects (see below). Examples of pig models that recapitulate human diseases are phenylketonuria (PKU) [260], Huntington disease (HD) [261], Neurofibromatosis type I [262], and type II collagenopathy [263]. Due to the similarity of organ size between pigs and humans, significant efforts have been invested in the creation of pigs carrying organs that can be transplanted into humans (xenotransplantation). There are two problems associated with this: (1) the immunological compatibility between the two species, or the necessity to immunocompromise the recipient, and (2) the transmission of pig viruses (porcine endogenous retroviruses or PERVs) into humans. To overcome these problems, several KO pig models lacking the rejection antigens [264,265,266] or carrying inactivated PERVs have been developed [267,268], all mediated by CRISPR. These breakthroughs have led to the first transplants of pig heart and kidney into humans [269,270].
The CRISPR-gene-edited sheep and goats that have been produced are related to the agriculture and pharmaceutical fields. Those two species are of high value because of their meat, milk, fibers, and other bio-products, so the gene modification efforts (zygote injection of CRISPR-Cas9, or SCNT with CRISPR-modified fibroblasts) have been applied preferentially into those. Despite that, there are several sheep models for human diseases such as cystic fibrosis, created by disrupting the CFTR gene [271], human hypophosphatasia, developed by knocking out the ALPL gene [272], and human deafness, achieved by editing the OTOF gene [273].
There are few groups working to create genetically modified dogs that resemble human diseases using the CRISPR/Cas9 system. One prominent dog study used CRISPR-Cas9 (delivered by adeno-associated viruses AAV) to restore the expression of dystrophin in a dog model of Duchenne muscular dystrophy, accompanied by improved muscle histology and amelioration of the disease [274]. Several models have been created by knocking genes into the genome of large animals. For example, a non-human primate model with OCT4-hrGFP (octamer-binding transcription factor 4-humanized recombinant green fluorescent protein) was achieved by using CRISPR-Cas9-assisted HR [275]. The pig model of HD mentioned above was developed by Yan et al. [261] using CRIPSR-Cas9 to insert a large CAG repeat into the pig HTT gene in fibroblast followed by SCNT. The brain of this pig model showed neurodegeneration in the medium spiny neurons like affected patients do. One example of sheep created by knocking in using CRISPR is the turbo GFP sheep [276].
There are several advantages of using CRISPR-Cas9 to create large animal models. The first is that this system is specific and can target any gene in the genome. The second is that several genes can be edited at the same time by co-injecting/delivering different gRNAs in one-step, which makes the development of human multigenic disease models possible. The third advantage is that the creation of the homozygous mutant founders of a colony is much faster because CRISPR can edit both alleles, so investigators do not have to mate heterozygous founders to obtain the KO animals. This advantage is critical because it saves years of mating large animals to establish your model of interest. However, some technological challenges remain. One is the production of off-target effects. If the gRNA sequence has high similarity with another part of the genome than the intended target, off-target editing can cause undesirable effects in the model. If off-target effects happen, they could be diluted over generations, but it can take years in the case of large animals due to their long gestational period and time to reach sexual maturity. The risk of off-target effects can be reduced using publicly available bioinformatic tools to optimize gRNA design. Another challenge is the possibility of mosaicism or having an animal model with a mix of cells with different genotypes. This issue could happen for several reasons: (a) injection of Cas9/gRNA is too late, when the zygote has already started dividing, (b) the translation of the Cas9 mRNA is delayed and does not happen until the embryo has divided already, or (c) the expression of Cas9 mRNA is prolonged. Although mosaic animals can help us understand the dosage effect in some diseases, it is undesired in most studies. To overcome it, there are several strategies than can be implemented: (a) inject the CRISPR components in a more efficient format, like Cas9/gRNA ribonucleoproteins, so they are ready to act because they do not need to be translated, (b) use electroporation to deliver the components into early one-cell zygotes before the first division happens [277], and (c) shorten the half-life of the Cas9 enzyme [278].
In terms of the market, there are five main companies producing large animal models: Recombinetics Inc. (Eagan, MN, USA) (with its four subsidiaries, Surrogen, Makana Therapeutics, Acceligen, and Regenevida), Precision BioSciences (Durham, NC, USA) (partnering with Agrivida, Inc., Woburn, MA, USA), eGenesis, Genus plc (Cambridge, MA, USA) (together with Caribou Biosciences, San Diego, CA, USA), and Synthetic Genomics/Lung Biotechnology. They also focus on editing the pig genome in the way they can produce human transplantable organs.
More recently, CRISPR-Cas systems have been modified to generate missense mutations and early stop codons. One example is the third-generation base editor (BE3), which has been used by several investigators to create large animal models that resemble different human diseases. Li et al. (2018) used BE3 and SCNT to develop one pig model for the ablepharon macrostamia syndrome (AMS) in humans, which is caused by a point mutation in the TWIST2 gene, and another pig model for the oculocutaneous albinism type 1 (OCA1), caused by mutation of the TYR gene [279]. Both models reproduced very accurately the phenotypes of human diseases. Another example of the use of BE to create an animal model was reported by Wang et al. [280], where they showed the first Hutchinson-Gilford progeria syndrome (HGPS) monkey model that was achieved by mutating the LMNA gene via microinjection into zygotes. Other groups have used BEs to prove that different point mutations in several genes can be achieved in one animal model. A group of researchers used cytosine base editors (CBEs) delivered by embryo injection or SCNT to create pigs carrying point mutations in three different genes at the same time (DMD, TYR, and LMNA; RAG1, RAG3, and IL2RG) [281]. In this study, they also achieved the edition of multiple copies of the PERV in porcine embryos and fibroblasts. Yuan et al. [282] also utilized BE3 and successfully produced pigs that had three-point mutations in the genes GGTA1, B4galNT2, and CMAH.
8. Conclusions
Ever since its inception, the CRISPR-based genome editing technology has rapidly transformed human medicine. In the field of cancer, FDA approvals of multiple adoptive cell therapies in the last decade have heralded a paradigm shift in the way cancer is treated. The co-emergence of CRISPR technologies and its coupling to cell therapy has enabled rapid improvements in the cell therapy space. Using CRISPR-Cas9, precision-tailored and multiplexed gene editing is enabling the generation of durable and efficacious off-the-shelf allogenic therapies for several cancers without side effects from fratricides, CRS, etc.
Many of the CRISPR-Cas9-edited cell therapy products are still in early-stage clinical trials, and given the small number and limited duration of the current clinical trials where therapeutic products with low gene editing efficiencies were tested, the need of the hour is for longer-duration trials testing therapeutic products with higher gene editing efficiencies to better understand the safety of the CRISPR-edited cell therapy products. Forthcoming results from several in-progress clinical trials will provide critical insights for development of future CRISPR therapeutics. Beyond cancers, the power of the CRISPR-Cas system has facilitated the development of precision tools for the in vivo demolition of genome-integrated viruses such as HIV, HPV, and HSV-1 in tissues. Likewise, it shows promise for those suffering from life-long, debilitating, and/or degenerative diseases such as sickle cell disease and β-thalassemia, type 1 diabetes, and Duchenne muscular dystrophy, opening the door for the previously impossible: not just a treatment for, but a functional cure for these diseases.
The application of CRISPR-Cas technology to generate large animal models which mimic human diseases is of significant importance, as they facilitate the safety and efficacy of testing prior to human application. Several different diseases (cystic fibrosis, Duchenne muscular dystrophy, and Parkinson and Huntington diseases, to name a few) have been modeled using monkeys, pigs, sheep, goats, and dogs, and they have the advantage of having short reproductive cycles and similar physiology and organ size to humans, thus enabling accelerated testing during therapeutic product development.
Novel approaches for therapeutic development that rely on CRISPR-epigenome editing are still in the research phase and offer the promise of inducing heritable changes in gene expression without relying on DNA cutting and error-prone repair. In addition, other in vivo therapies are being developed that directly modulate or bypass disease-causing alleles, sometimes in a tissue-specific manner, which show great promise but suffer from the similar concerns with off-target effects. Emerging technologies using RNA-targeting Cas enzymes may be an additional approach to leverage the specificity and versatility of CRISPR-Cas in a reversible manner that obviates the need to deliver gene-edited material. Further development of this technology will likely be necessary before it is clinically impactful.
Taken together, the CRISPR-Cas system has transformed within a decade into a product and technology platform with many different tools in its arsenal. As detailed in this paper, each of these tools has a unique role to play, all the way from lab bench to bedside, and one can only predict that the CRISPR-Cas system will evolve to be better and become more potent in its second decade of invention.
Author Contributions
All the authors (N.B., E.L., L.L.T., T.D.B., M.R.-E. and J.R.B.) contributed towards the conceptualization, data curation, writing and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We would like to thank Sue Woulfe and Guy Joly for reviewing this manuscript and providing their comments and suggestions. All the figures were created with Biorender.com.
Conflicts of Interest
All authors are employees of 3M Company and declare no further conflict of interest.
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and Regulation of Human Non-Homologous DNA End-Joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in Genome Editing through Control of DNA Repair Pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Fellmann, C.; Gowen, B.G.; Lin, P.-C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR–Cas in Drug Discovery and Therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The Emerging and Uncultivated Potential of CRISPR Technology in Plant Science. Nat. Plants 2019, 5, 778–794. [Google Scholar] [CrossRef]
- Donohoue, P.D.; Barrangou, R.; May, A.P. Advances in Industrial Biotechnology Using CRISPR-Cas Systems. Trends Biotechnol. 2018, 36, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR-Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-Guided Gene Activation by CRISPR-Cas9–Based Transcription Factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Özcan, A.; Krajeski, R.; Ioannidi, E.; Lee, B.; Gardner, A.; Makarova, K.S.; Koonin, E.V.; Abudayyeh, O.O.; Gootenberg, J.S. Programmable RNA Targeting with the Single-Protein CRISPR Effector Cas7-11. Nature 2021, 597, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Shrock, E.; Güell, M. Chapter Six—CRISPR in Animals and Animal Models. In Progress in Molecular Biology and Translational Science; Torres-Ruiz, R., Rodriguez-Perales, S., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 152, pp. 95–114. [Google Scholar] [CrossRef]
- Tu, Z.; Yang, W.; Yan, S.; Guo, X.; Li, X.-J. CRISPR/Cas9: A Powerful Genetic Engineering Tool for Establishing Large Animal Models of Neurodegenerative Diseases. Mol. Neurodegener. 2015, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.R.Q.; Nguyen, Q.; Dzierlega, K.; Huang, Y.; Yokota, T. CRISPR-Generated Animal Models of Duchenne Muscular Dystrophy. Genes 2020, 11, 342. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Gersbach, C.A. Genome-Editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the Next-Generation of CAR T-Cells with CRISPR-Cas9 Gene Editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.Gov/show/NCT05397184 (accessed on 15 December 2022).
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Food and Drug Administratio. Available online: https://www.fda.gov/Vaccines-Blood-Biologics/Cellular-Gene-Therapy-Products/Approved-Cellular-and-Gene-Therapy-Products (accessed on 15 December 2022).
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy with TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.; Pulsipher, M.A.; Moskop, A.; Hu, Z.-H.; Phillips, C.L.; Hall, E.M.; Margossian, S.P.; Nikiforow, S.; Martin, P.L.; Oshrine, B.; et al. Real-World Outcomes for Pediatric and Young Adult Patients with Relapsed or Refractory (R/R) B-Cell Acute Lymphoblastic Leukemia (ALL) Treated with Tisagenlecleucel: Update from the Center for International Blood and Marrow Transplant Research (CIBMTR) Registry. Blood 2021, 138 (Suppl. S1), 428. [Google Scholar] [CrossRef]
- Jacobson, C.; Locke, F.L.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Hill, B.T.; Timmerman, J.M.; Deol, A.; et al. Long-Term (≥4 Year and ≥5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL). Blood 2021, 138 (Suppl. S1), 1764. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T Immunotherapy: Overcoming the Challenges of T Cell Exhaustion. eBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T Cell Malignancies Using CAR-Based Immunotherapy: Challenges and Potential Solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Maus, M.V. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 Blockade Modulates Chimeric Antigen Receptor (CAR)–Modified T Cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04417764 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03081715 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT02793856 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03044743 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03545815 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03747965 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03399448 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://Clinicaltrials.gov/show/NCT04637763 (accessed on 15 December 2022).
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I Study of CAR-T Cells with PD-1 and TCR Disruption in Mesothelin-Positive Solid Tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–Specific TCR–Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-Cell Receptor: Long-Term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal Checkpoint Regulates T Cell Neoantigen Reactivity and Susceptibility to PD1 Blockade. Med 2022, 3, 682–704.e8. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05566223 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04426669 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04767308 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04037566 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04976218 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05066165 (accessed on 15 December 2022).
- Qasim, W. Allogeneic CAR T Cell Therapies for Leukemia. Am. J. Hematol. 2019, 94 (Suppl. S1), S50–S54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 Clinical Trial of CRISPR-Engineered CAR19 Universal T Cells for Treatment of Children with Refractory B Cell Leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03166878 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03398967 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04557436 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04502446 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04438083 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04035434 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04244656 (accessed on 15 December 2022).
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, S.; Nastoupil, L.J.; Essell, J.; Dsouza, L.; Hart, D.; Matsuda, E.; Satterfield, T.; Nesheiwat, T.; Hammad, A.; Davi, F.; et al. A First-in-Human Phase 1, Multicenter, Open-Label Study of CB-010, a Next-Generation CRISPR-Edited Allogeneic Anti-CD19 CAR-T Cell Therapy with a PD-1 Knockout, in Patients with Relapsed/Refractory B Cell Non-Hodgkin Lymphoma (ANTLER Study). Blood 2022, 140 (Suppl. S1), 9457–9458. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05309733 (accessed on 15 December 2022).
- von Kalle, C.; Deichmann, A.; Schmidt, M. Vector Integration and Tumorigenesis. Hum. Gene Ther. 2014, 25, 475–481. [Google Scholar] [CrossRef]
- Dai, Z.; Mu, W.; Zhao, Y.; Jia, X.; Liu, J.; Wei, Q.; Tan, T.; Zhou, J. The Rational Development of CD5-Targeting Biepitopic CARs with Fully Human Heavy-Chain-Only Antigen Recognition Domains. Mol. Ther. 2021, 29, 2707–2722. [Google Scholar] [CrossRef]
- Iyer, S.P.; Sica, R.A.; Ho, P.J.; Hu, B.; Zain, J.; Prica, A.; Weng, W.-K.; Kim, Y.H.; Khodadoust, M.S.; Palomba, M.L.; et al. S262: The Cobalt-Lym Study of Ctx130: A Phase 1 Dose Escalation Study of Cd70-Targeted Allogeneic Crispr-Cas9–Engineered Car T Cells in Patients with Relapsed/Refractory (R/R) T-Cell Malignancies. HemaSphere 2022, 6, 163–164. [Google Scholar] [CrossRef]
- Si, J.; Shi, X.; Sun, S.; Zou, B.; Li, Y.; An, D.; Lin, X.; Gao, Y.; Long, F.; Pang, B.; et al. Hematopoietic Progenitor Kinase1 (HPK1) Mediates T Cell Dysfunction and Is a Druggable Target for T Cell-Based Immunotherapies. Cancer Cell 2020, 38, 551–566.e11. [Google Scholar] [CrossRef]
- Zhang, N.; Si, J.; Li, G.; Wang, Y.; Long, F.; Wang, T.; Song, Y.; Liao, X.; Gao, G.; Dimitrov, D.S. Decreasing HPK1 Expression in CD19 CAR-T Cells: A Novel Strategy to Overcome Challenges of Cell Therapy for Adult (r/r) B-ALL. J. Clin. Oncol. 2022, 40 (Suppl. 16), 7041. [Google Scholar] [CrossRef]
- Cossette, D.; Aiyer, S.; Kimball, C.; Luby, C.; Zarate, J.; Eng, J.; Doshi, V.; Cole, R.; Kolluri, N.; Jaligama, S.; et al. Clinical-Scale Production and Characterization of Ntla-5001—A Novel Approach to Manufacturing CRISPR/Cas9 Engineered T Cell Therapies. Blood 2021, 138 (Suppl. S1), 3881. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03655678 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05477563 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03745287 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05356195 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05329649 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04208529 (accessed on 15 December 2022).
- Khosravi, M.A.; Abbasalipour, M.; Concordet, J.-P.; Berg, J.V.; Zeinali, S.; Arashkia, A.; Azadmanesh, K.; Buch, T.; Karimipoor, M. Targeted Deletion of BCL11A Gene by CRISPR-Cas9 System for Fetal Hemoglobin Reactivation: A Promising Approach for Gene Therapy of Beta Thalassemia Disease. Eur. J. Pharmacol. 2019, 854, 398–405. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Bhatia, M.; Mapara, M.Y.; Molinari, L.; Sharma, A.; Lobitz, S.; de Montalembert, M.; Rondelli, D.; Steinberg, M.; et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Severe Sickle Cell Disease. Blood 2022, 140 (Suppl. S1), 29–31. [Google Scholar] [CrossRef]
- Locatelli, F.; Lang, P.; Li, A.; Corbacioglu, S.; de la Fuente, J.; Wall, D.A.; Liem, R.; Meisel, R.; Mapara, M.Y.; Shah, A.J.; et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. Blood 2022, 140 (Suppl. S1), 4899–4901. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04925206 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04390971 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05577312 (accessed on 15 December 2022).
- Shi, J.; Fang, R.; Gao, Z.; Shi, Z.; Kuang, Z.; Zhang, Y.; Zhang, L.; Yang, H.; Zhang, Y.; Zou, D.; et al. Preliminary Safety and Efficacy Results of EDI001: An Investigator Initiated Trial on CRISPR/Cas9-Modified Autologous CD34+ Hematopoietic Stem and Progenitor Cells for Patients with Transfusion Dependent β-Thalassemia. Blood 2022, 140 (Suppl. S1), 10652–10653. [Google Scholar] [CrossRef]
- Fu, B.; Liao, J.; Chen, S.; Li, W.; Wang, Q.; Hu, J.; Yang, F.; Hsiao, S.; Jiang, Y.; Wang, L.; et al. CRISPR-Cas9-Mediated Gene Editing of the BCL11A Enhancer for Pediatric Β0/Β0 Transfusion-Dependent β-Thalassemia. Nat. Med. 2022, 28, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04819841 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04774536 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03728322 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04211480 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05444894 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04853576 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05210530 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03872479 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05120830 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05514249 (accessed on 15 December 2022).
- De Dreuzy, E.; Heath, J.; Zuris, J.A.; Sousa, P.; Viswanathan, R.; Scott, S.; Da Silva, J.; Ta, T.; Capehart, S.; Wang, T.; et al. EDIT-301: An Experimental Autologous Cell Therapy Comprising Cas12a-RNP Modified MPB-CD34+ Cells for the Potential Treatment of SCD. Blood 2019, 134 (Suppl. S1), 4636. [Google Scholar] [CrossRef]
- Sousa, P.; Janoudi, T.; deDreuzy, E.; Shearman, M.S.; Zhang, K.; Chang, K.-H. Preclinical Development of EDIT301, an Autologous Cell Therapy Comprising AsCas12a-RNP Modified Mobilized Peripheral Blood-CD34+ Cells for the Potential Treatment of Transfusion Dependent Beta Thalassemia. Blood 2021, 138 (Suppl. S1), 1858. [Google Scholar] [CrossRef]
- Editas Medicine. Editas Medicine Announces Positive Safety and Efficacy Data from the First Two Patients Treated in the RUBY Trial of EDIT-301 for the Treatment of Severe Sickle Cell Disease. Press Release. Available online: https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-positive-safety-and-efficacy-data (accessed on 6 December 2022).
- Kanter, J.; Di Persio, J.F.; Leavey, P.; Shyr, D.C.; Thompson, A.A.; Porteus, M.H.; Intondi, A.; Lahiri, P.; Dever, D.P.; Petrusich, A.; et al. Trial In Progress: A First-in-Human Phase 1/2 Study of the Correction of a Single Nucleotide Mutation in Autologous HSCs (GPH101) to Convert HbS to HbA for Treating Severe SCD. In 2021 ASH Annual Meeting and Exposition; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Graphite Bio. Graphite Bio Doses First Patient with Investigational Gene Editing Therapy GPH101 for Sickle Cell Disease [Press Release]. Available online: https://ir.graphitebio.com/press-releases/detail/76/graphite-bio-doses-first-patient-with-investigational-gene (accessed on 11 August 2022).
- Graphite Bio. Graphite Bio Announces Voluntary Pause of Phase 1/2 CEDAR Study of Nulabeglogene Autogedtemcel (Nula-Cel) for Sickle Cell Disease. [Press Release]. Available online: https://ir.graphitebio.com/press-releases/detail/84/graphite-bio-announces-voluntary-pause-of-phase-12-cedar (accessed on 5 January 2023).
- Ramzy, A.; Thompson, D.M.; Ward-Hartstonge, K.A.; Ivison, S.; Cook, L.; Garcia, R.V.; Loyal, J.; Kim, P.T.W.; Warnock, G.L.; Levings, M.K.; et al. Implanted Pluripotent Stem-Cell-Derived Pancreatic Endoderm Cells Secrete Glucose-Responsive C-Peptide in Patients with Type 1 Diabetes. Cell Stem Cell 2021, 28, 2047–2061.e5. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Thompson, D.; Donner, T.W.; Bellin, M.D.; Hsueh, W.; Pettus, J.; Wilensky, J.; Daniels, M.; Wang, R.M.; Brandon, E.P.; et al. Insulin Expression and C-Peptide in Type 1 Diabetes Subjects Implanted with Stem Cell-Derived Pancreatic Endoderm Cells in an Encapsulation Device. Cell Rep. Med. 2021, 2, 100466. [Google Scholar] [CrossRef]
- CRISPR Theraputics. CRISPR Therapeutics and ViaCyte, Inc. Announce First Patient Dosed in Phase 1 Clinical Trial of Novel Gene-Edited Cell Replacement Therapy for Treatment of Type 1 Diabetes (T1D). [Press Release]. Available online: https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-and-viacyte-inc-announce-first-patient-dosed-in-phase-1-clinical-trial-of-novel-gene-edited-cell-replacement-therapy-for-treatment-of-type-1-diabetes-t1d (accessed on 2 February 2023).
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.-L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 Mutations in Leber Congenital Amaurosis and Delineation of the Associated Phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a Gene-Editing Approach to Restore Vision Loss in Leber Congenital Amaurosis Type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Editas Medicine. Press Release: Editas Medicine Announces Clinical Data Demonstrating Proof of Concept of EDIT-101 from Phase 1/2 BRILLIANCE Trial. 2022. Available online: https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-clinical-data-demonstrating-proof (accessed on 15 December 2022).
- Longhurst, H. In Vivo CRISPR/Cas9 Editing of KLKB1 in Patients with Hereditary Angioedema: A First-in Human Study. In American College of Allergy, Asthma, and Immunology 2002 Annual Scientific Meeting; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Intellia Theraputics. Press Release: Intellia Therapeutics Presents New Interim Data from First-in-Human Study of NTLA-2002 for the Treatment of Hereditary Angioedema (HAE) at the American College of Allergy, Asthma & Immunology 2022 Annual Scientific Meeting. 2022. Available online: https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-new-interim-data-first-human (accessed on 15 December 2022).
- Emery, A.E. Population Frequencies of Inherited Neuromuscular Diseases--a World Survey. Neuromuscul. Disord. NMD 1991, 1, 19–29. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05144386 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05143307 (accessed on 15 December 2022).
- Mancuso, P.; Chen, C.; Kaminski, R.; Gordon, J.; Liao, S.; Robinson, J.A.; Smith, M.D.; Liu, H.; Sariyer, I.K.; Sariyer, R.; et al. CRISPR Based Editing of SIV Proviral DNA in ART Treated Non-Human Primates. Nat. Commun. 2020, 11, 6065. [Google Scholar] [CrossRef]
- Excision BioTheraputics. Press Release: Excision BioTherapeutics Doses First Participant in EBT-101 Phase 1/2 Trial Evaluating EBT-101 as a Potential Cure for HIV, 2022. [Press Release]. Available online: https://www.globenewswire.com/news-release/2022/09/15/2516733/0/en/Excision-BioTherapeutics-Doses-First-Participant-in-EBT-101-Phase-1-2-Trial-Evaluating-EBT-101-as-a-Potential-Cure-for-HIV.html (accessed on 15 September 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03164135 (accessed on 15 December 2022).
- Blanpain, C.; Libert, F.; Vassart, G.; Parmentier, M. CCR5 and HIV Infection. Recept. Channels 2002, 8, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03057912 (accessed on 15 December 2022).
- Tian, R.; Liu, J.; Fan, W.; Li, R.; Cui, Z.; Jin, Z.; Huang, Z.; Xie, H.; Li, L.; Huang, Z.; et al. Gene Knock-out Chain Reaction Enables High Disruption Efficiency of HPV18 E6/E7 Genes in Cervical Cancer Cells. Mol. Ther. Oncolytics 2022, 24, 171–179. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, H.; Wang, T.; He, D.; Tian, R.; Cui, Z.; Tian, X.; Gao, Q.; Ma, X.; Yang, J.; et al. In Vitro and in Vivo Growth Inhibition of Human Cervical Cancer Cells via Human Papillomavirus E6/E7 MRNAs’ Cleavage by CRISPR/Cas13a System. Antivir. Res. 2020, 178, 104794. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04560790 (accessed on 15 December 2022).
- Yin, D.; Ling, S.; Wang, D.; Dai, Y.; Jiang, H.; Zhou, X.; Paludan, S.R.; Hong, J.; Cai, Y. Targeting Herpes Simplex Virus with CRISPR-Cas9 Cures Herpetic Stromal Keratitis in Mice. Nat. Biotechnol. 2021, 39, 567–577. [Google Scholar] [CrossRef]
- BDGene Theraputics. BD111 of BDgene Passed the FDA Orphan Drug Application. [151 Press Release]. Available online: https://www.bdgenetherapeutics.com/en/news/77.html (accessed on 24 June 2022).
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05398029 (accessed on 15 December 2022).
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Rao, A.S.; Lindholm, D.; Rivas, M.A.; Knowles, J.W.; Montgomery, S.B.; Ingelsson, E. Large-Scale Phenome-Wide Association Study of PCSK9 Variants Demonstrates Protection Against Ischemic Stroke. Circ. Genom. Precis. Med. 2018, 11, e002162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base Editing Therapy Targeting PCSK9 in Non-Human Primate and Mouse Models. Circulation 2022, 147, 242–253. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In Vivo Adenine Base Editing of PCSK9 in Macaques Reduces LDL Cholesterol Levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Chiesa, R.; Georgiadis, C.; Ottaviano, G.; Syed, F.; Braybrook, T.; Etuk, A.; Zhan, H.; Gkazi, S.A.; Preece, R.; Adams, S.; et al. Tvt CAR7: Phase 1 Clinical Trial of Base-Edited “Universal” CAR7 T Cells for Paediatric Relapsed/Refractory T-ALL. Blood. 2022, 140 (Suppl. 1), 4579–4580. [Google Scholar] [CrossRef]
- Georgiadis, C.; Rasaiyaah, J.; Gkazi, S.A.; Preece, R.; Etuk, A.; Christi, A.; Qasim, W. Base-Edited CAR T Cells for Combinational Therapy against T Cell Malignancies. Leukemia 2021, 35, 3466–3481. [Google Scholar] [CrossRef] [PubMed]
- UCL. World-First Use of Base-Edited CAR T-Cells to Treat Resistant Leukaemia; UCL Great Ormond Street Institute of Child Health. Available online: https://www.ucl.ac.uk/child-health/news/2022/dec/world-first-use-base-edited-car-t-cells-treat-resistant-leukaemia (accessed on 21 December 2022).
- Bain, B.J.; Bates, I.; Laffan, M.A. Dacie and Lewis Practical Haematology, 12th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Harbottle, J.A. Immunotherapy to Get on Point with Base Editing. Drug Discov. Today 2021, 26, 2350–2357. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, J.F.; Friedman, J.R.; Meyer, W.K.; Vissing, H.; Thiesen, H.J.; Rauscher, F.J. 3rd. Krüppel-Associated Boxes Are Potent Transcriptional Repression Domains. Proc. Natl. Acad. Sci. USA 1994, 91, 4509–4513. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-Guided Activation of Endogenous Human Genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR Technologies for Precise Epigenome Editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-Based System for Inducing Site-Specific DNA Methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient Targeted DNA Methylation with Chimeric DCas9-Dnmt3a-Dnmt3L Methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly Specific Epigenome Editing by CRISPR-Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [Green Version]
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing. Gene Ther. 2022, 29, 81–93. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal Degeneration Mutants in the Mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and Mutation Independent Therapy via CRISPR-Cas9 Mediated Cellular Reprogramming in Rod Photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, M.M.; Rancourt, D.; Farrar, G.J.; Kenna, P.; Hazel, M.; Bush, R.A.; Sieving, P.A.; Sheils, D.M.; McNally, N.; Creighton, P.; et al. Retinopathy Induced in Mice by Targeted Disruption of the Rhodopsin Gene. Nat. Genet. 1997, 15, 216–219. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The Molecular and Cellular Basis of Rhodopsin Retinitis Pigmentosa Reveals Potential Strategies for Therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Larhammar, D.; Nordström, K.; Larsson, T.A. Evolution of Vertebrate Rod and Cone Phototransduction Genes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 2867–2880. [Google Scholar] [CrossRef] [Green Version]
- Deenen, J.C.W.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; van Engelen, B.G.M. Population-Based Incidence and Prevalence of Facioscapulohumeral Dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD Associated DNA Rearrangements Are Due to Deletions of Integral Copies of a 3.2 Kb Tandemly Repeated Unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA Rearrangements Associated with Facioscapulohumeral Muscular Dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. CRISPR/DCas9-Mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-Fl in FSH Muscular Dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.P.; Livingston, D.M. Mechanisms of BRCA1 Tumor Suppression. Cancer Discov. 2012, 2, 679–684. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Hayward, N.K. The CDKN2A (P16) Gene and Human Cancer. Mol. Med. Camb. Mass 1997, 3, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Hesson, L.B.; Cooper, W.N.; Latif, F. The Role of RASSF1A Methylation in Cancer. Dis. Markers 2007, 23, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Ullah, U.; Andrabi, S.B.A.; Tripathi, S.K.; Dirasantha, O.; Kanduri, K.; Rautio, S.; Gross, C.C.; Lehtimäki, S.; Bala, K.; Tuomisto, J.; et al. Transcriptional Repressor HIC1 Contributes to Suppressive Function of Human Induced Regulatory T Cells. Cell Rep. 2018, 22, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Arechavaleta-Velasco, F.; Perez-Juarez, C.E.; Gerton, G.L.; Diaz-Cueto, L. Progranulin and Its Biological Effects in Cancer. Med. Oncol. 2017, 34, 194. [Google Scholar] [CrossRef]
- Kleiber, K.; Strebhardt, K.; Martin, B.T. The Biological Relevance of FHL2 in Tumour Cells and Its Role as a Putative Cancer Target. Anticancer Res. 2007, 27, 55–61. [Google Scholar] [PubMed]
- Therrien, M.; Wong, A.M.; Kwan, E.; Rubin, G.M. Functional Analysis of CNK in RAS Signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 13259–13263. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA Methylation Inhibitors in Cancer: Recent and Future Approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.-H.; Lee, H.S.; Kim, W.H. Promoter Methylation and Silencing of PTEN in Gastric Carcinoma. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 285–291. [Google Scholar] [CrossRef] [Green Version]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter Methylation of the PTEN Gene Is a Common Molecular Change in Breast Cancer. Genes. Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. PTEN Methylation Is Associated with Advanced Stage and Microsatellite Instability in Endometrial Carcinoma. Int. J. Cancer 2001, 91, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/DCas9 System. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.D.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-Run Epigenetic Editing Prevents Senescence Entry in Primary Breast Cells from Healthy Donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.J.; Stampfer, M.R.; Aldaz, C.M. Increased P16 Expression with First Senescence Arrest in Human Mammary Epithelial Cells and Extended Growth Capacity with P16 Inactivation. Oncogene 1998, 17, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Allach El Khattabi, L.; Backer, S.; Pinard, A.; Dieudonné, M.-N.; Tsatsaris, V.; Vaiman, D.; Dandolo, L.; Bloch-Gallego, E.; Jammes, H.; Barbaux, S. A Genome-Wide Search for New Imprinted Genes in the Human Placenta Identifies DSCAM as the First Imprinted Gene on Chromosome 21. Eur. J. Hum. Genet. EJHG 2019, 27, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Eggermann, T.; Perez de Nanclares, G.; Maher, E.R.; Temple, I.K.; Tümer, Z.; Monk, D.; Mackay, D.J.G.; Grønskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting Disorders: A Group of Congenital Disorders with Overlapping Patterns of Molecular Changes Affecting Imprinted Loci. Clin. Epigenetics 2015, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Barlow, D.P. Genomic Imprinting: A Mammalian Epigenetic Discovery Model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman Syndrome: Insights into Genomic Imprinting and Neurodevelopmental Phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagli, A.I.; Mathews, J.; Williams, C.A. Angelman Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 Gene Therapy for Angelman Syndrome Traps Ube3a-ATS Long Non-Coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef] [Green Version]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, L.C.; Bomfim, L.M.; Dittz, U.V.T.; Velozo, C.d.A.; da Cunha, R.D.; Tanuri, A. Repression of HIV-1 Reactivation Mediated by CRISPR/DCas9-KRAB in Lymphoid and Myeloid Cell Models. Retrovirology 2022, 19, 12. [Google Scholar] [CrossRef]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-Wide Binding of the CRISPR Endonuclease Cas9 in Mammalian Cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-Wide Analysis Reveals Characteristics of off-Target Sites Bound by the Cas9 Endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-Mediated Direct Mutation of Cancer Genes in the Mouse Liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Tollefsbol, T.O. Targeting Cancer Epigenetics with CRISPR-DCAS9: Principles and Prospects. Methods San Diego Calif. 2021, 187, 77–91. [Google Scholar] [CrossRef]
- Cebrian-Serrano, A.; Davies, B. CRISPR-Cas Orthologues and Variants: Optimizing the Repertoire, Specificity and Delivery of Genome Engineering Tools. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2017, 28, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther.-Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A Review of the Challenges and Approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in Cancer Biology and Therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutškov, K.; Jouhilahti, E.-M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human Pluripotent Reprogramming with CRISPR Activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Catchpole, R.J.; Terns, M.P. New Type III CRISPR Variant and Programmable RNA Targeting Tool: Oh, Thank Heaven for Cas7-11. Mol. Cell 2021, 81, 4354–4356. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An Unwinding Activity That Covalently Modifies Its Double-Stranded RNA Substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Greeve, J.; Navaratnam, N.; Scott, J. Characterization of the Apolipoprotein B MRNA Editing Enzyme: No Similarity to the Proposed Mechanism of RNA Editing in Kinetoplastid Protozoa. Nucleic Acids Res. 1991, 19, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR Protein Family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinshteyn, B.; Nishikura, K. Adenosine-to-Inosine RNA Editing. WIREs Syst. Biol. Med. 2009, 1, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Woolf, T.; Chase, J.; Stinchcomb, D. Toward the Therapeutic Editing of Mutated RNA Sequences. Proc Natl Acad Sci USA 1995, 92, 8298–8302. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.M. The Genome Editing Revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Palaz, F.; Kalkan, A.K.; Can, Ö.; Demir, A.N.; Tozluyurt, A.; Özcan, A.; Ozsoz, M. CRISPR-Cas13 System as a Promising and Versatile Tool for Cancer Diagnosis, Therapy, and Research. ACS Synth. Biol. 2021, 10, 1245–1267. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A Cytosine Deaminase for Programmable Single-Base RNA Editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Bot, J.F.; van der Oost, J.; Geijsen, N. The Double Life of CRISPR–Cas13. Curr. Opin. Biotechnol. 2022, 78, 102789. [Google Scholar] [CrossRef] [PubMed]
- van Beljouw, S.P.B.; Haagsma, A.C.; Rodríguez-Molina, A.; van den Berg, D.F.; Vink, J.N.A.; Brouns, S.J.J. The GRAMP CRISPR-Cas Effector Is an RNA Endonuclease Complexed with a Caspase-like Peptidase. Science 2021, 373, 1349–1353. [Google Scholar] [CrossRef]
- Kato, K.; Zhou, W.; Okazaki, S.; Isayama, Y.; Nishizawa, T.; Gootenberg, J.S.; Abudayyeh, O.O.; Nishimasu, H. Structure and Engineering of the Type III-E CRISPR-Cas7-11 Effector Complex. Cell 2022, 185, 2324–2337.e16. [Google Scholar] [CrossRef]
- Zang, H.; Zhang, Q.; Li, X. Non-Coding RNA Networks in Pulmonary Hypertension. Front. Genet. 2021, 12, 703860. [Google Scholar] [CrossRef]
- Kalpachidou, T.; Kummer, K.K.; Kress, M. Non-Coding RNAs in Neuropathic Pain. Neuronal Signal. 2020, 4, NS20190099. [Google Scholar] [CrossRef] [Green Version]
- Dinger, M.E.; Mercer, T.R.; Mattick, J.S. RNAs as Extracellular Signaling Molecules. J. Mol. Endocrinol. 2008, 40, 151–159. [Google Scholar] [CrossRef]
- Lin, Y.; Li, J.; Li, C.; Tu, Z.; Li, S.; Li, X.-J.; Yan, S. Application of CRISPR/Cas9 System in Establishing Large Animal Models. Front. Cell Dev. Biol. 2022, 10, 919155. [Google Scholar] [CrossRef]
- Zhao, J.; Lai, L.; Ji, W.; Zhou, Q. Genome Editing in Large Animals: Current Status and Future Prospects. Natl. Sci. Rev. 2019, 6, 402–420. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Ouyang, H.; Yu, T.; Chen, X.; Pang, D.; Tang, X.; Chen, C. Preparation of a New Type 2 Diabetic Miniature Pig Model via the CRISPR/Cas9 System. Cell Death Dis. 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cui, Y.; Shen, B.; Niu, Y.; Zhao, X.; Wang, L.; Wang, J.; Li, W.; Zhou, Q.; Ji, W.; et al. Germline Acquisition of Cas9/RNA-Mediated Gene Modifications in Monkeys. Cell Res. 2015, 25, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional Disruption of the Dystrophin Gene in Rhesus Monkey Using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Li, S.; Li, X.-J. A CRISPR Monkey Model Unravels a Unique Function of PINK1 in Primate Brains. Mol. Neurodegener. 2019, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Tu, Z.; Xiao, C.; Yan, S.; Ma, X.; Guo, X.; Chen, X.; Yin, P.; Yang, Z.; et al. CRISPR/Cas9-Mediated PINK1 Deletion Leads to Neurodegeneration in Rhesus Monkeys. Cell Res. 2019, 29, 334–336. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wu, S.; Ma, X.; Li, X.; Cheng, T.; Chen, Z.; Wu, J.; Lv, L.; Li, L.; Xu, L.; et al. Co-Editing PINK1 and DJ-1 Genes Via Adeno-Associated Virus-Delivered CRISPR/Cas9 System in Adult Monkey Brain Elicits Classical Parkinsonian Phenotype. Neurosci. Bull. 2021, 37, 1271–1288. [Google Scholar] [CrossRef]
- Sun, Z.; Ye, J.; Yuan, J. PINK1 Mediates Neuronal Survival in Monkey. Protein Cell 2022, 13, 4–5. [Google Scholar] [CrossRef]
- Yang, W.; Guo, X.; Tu, Z.; Chen, X.; Han, R.; Liu, Y.; Yan, S.; Wang, Q.; Wang, Z.; Zhao, X.; et al. PINK1 Kinase Dysfunction Triggers Neurodegeneration in the Primate Brain without Impacting Mitochondrial Homeostasis. Protein Cell 2022, 13, 26–46. [Google Scholar] [CrossRef]
- Kang, Y.; Zheng, B.; Shen, B.; Chen, Y.; Wang, L.; Wang, J.; Niu, Y.; Cui, Y.; Zhou, J.; Wang, H.; et al. CRISPR/Cas9-Mediated Dax1 Knockout in the Monkey Recapitulates Human AHC-HH. Hum. Mol. Genet. 2015, 24, 7255–7264. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Zhao, H.; Li, B.; Yan, S.; Wang, L.; Tang, Y.; Li, Z.; Bai, D.; Li, C.; Lin, Y.; et al. CRISPR/Cas9-Mediated Disruption of SHANK3 in Monkey Leads to Drug-Treatable Autism-like Symptoms. Hum. Mol. Genet. 2018, 28, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tu, Z.; Xu, H.; Yan, S.; Yan, H.; Zheng, Y.; Yang, W.; Zheng, J.; Li, Z.; Tian, R.; et al. Altered Neurogenesis and Disrupted Expression of Synaptic Proteins in Prefrontal Cortex of SHANK3-Deficient Non-Human Primate. Cell Res. 2017, 27, 1293–1297. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-Step Generation of Knockout Pigs by Zygote Injection of CRISPR/Cas System. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 System to Produce Genetically Engineered Pigs from In Vitro-Derived Oocytes and Embryos1. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xin, J.; Fan, N.; Zou, Q.; Huang, J.; Ouyang, Z.; Zhao, Y.; Zhao, B.; Liu, Z.; Lai, S.; et al. Generation of CRISPR/Cas9-Mediated Gene-Targeted Pigs via Somatic Cell Nuclear Transfer. Cell. Mol. Life Sci. 2015, 72, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, C.; Huang, J.; Yao, J.; Hai, T.; Zheng, Q.; Wang, X.; Zhang, H.; Qin, G.; Cheng, J.; et al. One-Step Generation of Triple Gene-Targeted Pigs Using CRISPR/Cas9 System. Sci. Rep. 2016, 6, 20620. [Google Scholar] [CrossRef] [Green Version]
- Koppes, E.A.; Redel, B.K.; Johnson, M.A.; Skvorak, K.J.; Ghaloul-Gonzalez, L.; Yates, M.E.; Lewis, D.W.; Gollin, S.M.; Wu, Y.L.; Christ, S.E.; et al. A Porcine Model of Phenylketonuria Generated by CRISPR/Cas9 Genome Editing. JCI Insight 2020, 5, e141523. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002.e13. [Google Scholar] [CrossRef] [Green Version]
- Isakson, S.H.; Rizzardi, A.E.; Coutts, A.W.; Carlson, D.F.; Kirstein, M.N.; Fisher, J.; Vitte, J.; Williams, K.B.; Pluhar, G.E.; Dahiya, S.; et al. Genetically Engineered Minipigs Model the Major Clinical Features of Human Neurofibromatosis Type 1. Commun. Biol. 2018, 1, 158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, C.; Zhang, Y.; Jiang, Y.; Qin, Y.; Pang, D.; Zhang, G.; Liu, H.; Xie, Z.; Yuan, H.; et al. A CRISPR-Engineered Swine Model of COL2A1 Deficiency Recapitulates Altered Early Skeletal Developmental Defects in Humans. Bone 2020, 137, 115450. [Google Scholar] [CrossRef]
- Gao, H.; Zhao, C.; Xiang, X.; Li, Y.; Zhao, Y.; Li, Z.; Pan, D.; Dai, Y.; Hara, H.; Cooper, D.K.C.; et al. Production of A1,3-Galactosyltransferase and Cytidine Monophosphate-N-Acetylneuraminic Acid Hydroxylase Gene Double-Deficient Pigs by CRISPR/Cas9 and Handmade Cloning. J. Reprod. Dev. 2017, 63, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.-K.; Chen, C.-H.; Huang, C.-L.; Su, Y.-H.; Peng, S.-H.; Lin, T.-Y.; Tai, H.-C.; Yang, T.-S.; Tu, C.-F. Generation of GGTA1 Mutant Pigs by Direct Pronuclear Microinjection of CRISPR/Cas9 Plasmid Vectors. Anim. Biotechnol. 2017, 28, 174–181. [Google Scholar] [CrossRef]
- Joanna, Z.; Magdalena, H.; Agnieszka, N.-T.; Jacek, J.; Ryszard, S.; Zdzisław, S.; Barbara, G.; Daniel, L. The Production of UL16-Binding Protein 1 Targeted Pigs Using CRISPR Technology. 3 Biotech 2018, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Güell, M.; Niu, D.; George, H.; Lesha, E.; Grishin, D.; Aach, J.; Shrock, E.; Xu, W.; Poci, J.; et al. Genome-Wide Inactivation of Porcine Endogenous Retroviruses (PERVs). Science 2015, 350, 1101–1104. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Xu, W.; Kan, Y.; Zhao, H.-Y.; Zhou, Y.; Song, X.; Wu, J.; Xiong, J.; Goswami, D.; Yang, M.; et al. Extensive Germline Genome Engineering in Pigs. Nat. Biomed. Eng. 2021, 5, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Porrett, P.M.; Orandi, B.J.; Kumar, V.; Houp, J.; Anderson, D.; Cozette Killian, A.; Hauptfeld-Dolejsek, V.; Martin, D.E.; Macedon, S.; Budd, N.; et al. First Clinical-Grade Porcine Kidney Xenotransplant Using a Human Decedent Model. Am. J. Transplant. 2022, 22, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Griffith, B.P.; Goerlich, C.E.; Singh, A.K.; Rothblatt, M.; Lau, C.L.; Shah, A.; Lorber, M.; Grazioli, A.; Saharia, K.K.; Hong, S.N.; et al. Genetically Modified Porcine-to-Human Cardiac Xenotransplantation. N. Engl. J. Med. 2022, 387, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Wettere, A.J.V.; Wang, Z.; Harris, A.; White, K.L.; et al. A Sheep Model of Cystic Fibrosis Generated by CRISPR/Cas9 Disruption of the CFTR Gene. JCI Insight 2018, 3, e123529. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.K.; Pinzón, C.; Huggins, S.; Pryor, J.H.; Falck, A.; Herman, F.; Oldeschulte, J.; Chavez, M.B.; Foster, B.L.; White, S.H.; et al. Genetic Engineering a Large Animal Model of Human Hypophosphatasia in Sheep. Sci. Rep. 2018, 8, 16945. [Google Scholar] [CrossRef] [Green Version]
- Menchaca, A.; dos Santos-Neto, P.C.; Souza-Neves, M.; Cuadro, F.; Mulet, A.P.; Tesson, L.; Chenouard, V.; Guiffès, A.; Heslan, J.M.; Gantier, M.; et al. Otoferlin Gene Editing in Sheep via CRISPR-Assisted SsODN-Mediated Homology Directed Repair. Sci. Rep. 2020, 10, 5995. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene Editing Restores Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Niu, Y.; Zhou, J.; Chen, Y.; Cheng, Y.; Li, S.; Ai, Z.; Chu, C.; Wang, H.; Zheng, B.; et al. Generation of a Precise Oct4-HrGFP Knockin Cynomolgus Monkey Model via CRISPR/Cas9-Assisted Homologous Recombination. Cell Res. 2018, 28, 383–386. [Google Scholar] [CrossRef]
- Wu, M.; Wei, C.; Lian, Z.; Liu, R.; Zhu, C.; Wang, H.; Cao, J.; Shen, Y.; Zhao, F.; Zhang, L.; et al. Rosa26-Targeted Sheep Gene Knock-in via CRISPR-Cas9 System. Sci. Rep. 2016, 6, 24360. [Google Scholar] [CrossRef] [Green Version]
- Mehravar, M.; Shirazi, A.; Nazari, M.; Banan, M. Mosaicism in CRISPR/Cas9-Mediated Genome Editing. Dev. Biol. 2019, 445, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Yang, W.; Yan, S.; Yin, A.; Gao, J.; Liu, X.; Zheng, Y.; Zheng, J.; Li, Z.; Yang, S.; et al. Promoting Cas9 Degradation Reduces Mosaic Mutations in Non-Human Primate Embryos. Sci. Rep. 2017, 7, 42081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Duan, X.; An, X.; Feng, T.; Li, P.; Li, L.; Liu, J.; Wu, P.; Pan, D.; Du, X.; et al. Efficient RNA-Guided Base Editing for Disease Modeling in Pigs. Cell Discov. 2018, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, W.; Yang, Q.; Kang, Y.; Fan, Y.; Wei, J.; Liu, Z.; Dai, S.; Li, H.; Li, Z.; et al. Generation of a Hutchinson–Gilford Progeria Syndrome Monkey Model by Base Editing. Protein Cell 2020, 11, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ge, W.; Li, N.; Liu, Q.; Chen, F.; Yang, X.; Huang, X.; Ouyang, Z.; Zhang, Q.; Zhao, Y.; et al. Efficient Base Editing for Multiple Genes and Loci in Pigs Using Base Editors. Nat. Commun. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Yu, T.; Wang, L.; Yang, L.; Zhang, Y.; Liu, H.; Li, M.; Tang, X.; Liu, Z.; Li, Z.; et al. Efficient Base Editing by RNA-Guided Cytidine Base Editors (CBEs) in Pigs. Cell. Mol. Life Sci. 2020, 77, 719–733. [Google Scholar] [CrossRef]
Figure 1.
Summary of the various CRISPR-Cas systems discussed in this review.

Figure 2.
A scheme of CRISPR-Cas9 engineering in autologous CAR-T therapy. T cells are extracted from cancer patients and engineered with CRISPR-Cas9 to edit select genes. Later, chimeric antigen receptor (CAR) is randomly inserted into the genome via lentivirus or precisely engineered into a target locus via the CRISPR-Cas9 system to create an engineered autologous CAR-T cell. Engineered CAR-T cells are later given back to the cancer patients.
Figure 2.
A scheme of CRISPR-Cas9 engineering in autologous CAR-T therapy. T cells are extracted from cancer patients and engineered with CRISPR-Cas9 to edit select genes. Later, chimeric antigen receptor (CAR) is randomly inserted into the genome via lentivirus or precisely engineered into a target locus via the CRISPR-Cas9 system to create an engineered autologous CAR-T cell. Engineered CAR-T cells are later given back to the cancer patients.

Figure 3.
A scheme of CRISPR-Cas9 engineering in the allogenic CAR-T therapy. T cells are extracted from healthy donors and engineered with CRISPR-Cas9 technology to edit select genes including genes for TCR, PD1, and MHC Class I and II molecules, among others. Later, CAR is randomly inserted into the genome via lentivirus or precisely inserted at a target locus via the CRISPR-Cas9 system to create an engineered allogenic off-the-shelf CAR-T cell. These donor-derived allogenic CAR-T cells are manufactured in scale and given back to multiple cancer patients.
Figure 3.
A scheme of CRISPR-Cas9 engineering in the allogenic CAR-T therapy. T cells are extracted from healthy donors and engineered with CRISPR-Cas9 technology to edit select genes including genes for TCR, PD1, and MHC Class I and II molecules, among others. Later, CAR is randomly inserted into the genome via lentivirus or precisely inserted at a target locus via the CRISPR-Cas9 system to create an engineered allogenic off-the-shelf CAR-T cell. These donor-derived allogenic CAR-T cells are manufactured in scale and given back to multiple cancer patients.

Figure 4.
Delivery systems for base editors. (A) Base editors can be delivered directly to patients in vivo, typically through adeno-associated viruses or lipid nanoparticles. The base editor then acts directly in patients (e.g., VERVE-101 edits PCSK9 in the liver). (B) Base editors can also be used to modify T cells or HSCs ex vivo before the edited cells are infused into patients.
Figure 4.
Delivery systems for base editors. (A) Base editors can be delivered directly to patients in vivo, typically through adeno-associated viruses or lipid nanoparticles. The base editor then acts directly in patients (e.g., VERVE-101 edits PCSK9 in the liver). (B) Base editors can also be used to modify T cells or HSCs ex vivo before the edited cells are infused into patients.

Figure 5.
Emerging therapeutic applications for RNA editing by Cas enzymes.
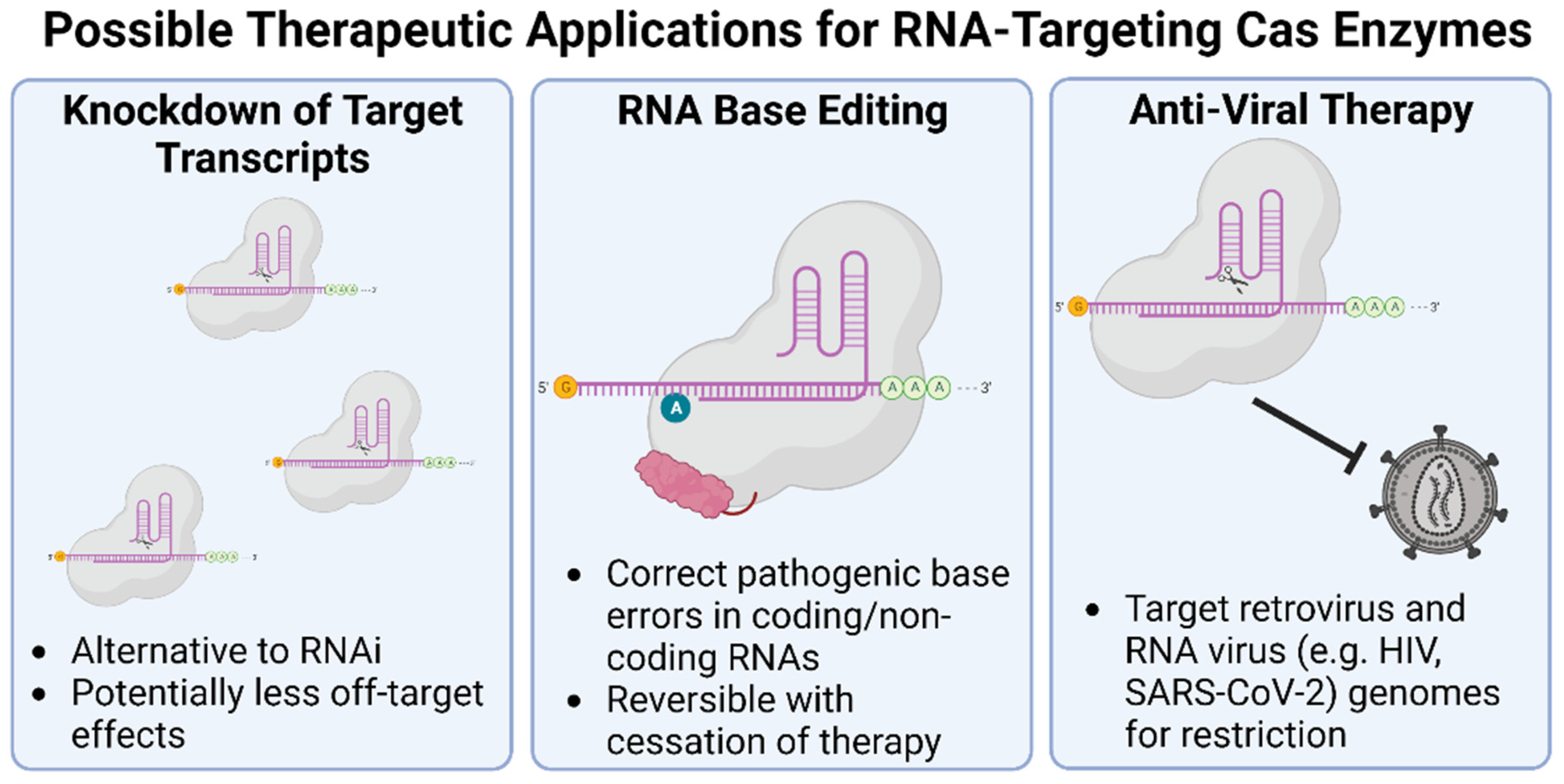
Figure 6.
Diagram showing the steps to generate large animal models. SCNT: somatic cell nuclear transfer.
Figure 6.
Diagram showing the steps to generate large animal models. SCNT: somatic cell nuclear transfer.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bhokisham, N.; Laudermilch, E.; Traeger, L.L.; Bonilla, T.D.; Ruiz-Estevez, M.; Becker, J.R. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells 2023, 12, 1103. https://doi.org/10.3390/cells12081103
AMA Style
Bhokisham N, Laudermilch E, Traeger LL, Bonilla TD, Ruiz-Estevez M, Becker JR. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells. 2023; 12(8):1103. https://doi.org/10.3390/cells12081103
Chicago/Turabian StyleBhokisham, Narendranath, Ethan Laudermilch, Lindsay L. Traeger, Tonya D. Bonilla, Mercedes Ruiz-Estevez, and Jordan R. Becker. 2023. "CRISPR-Cas System: The Current and Emerging Translational Landscape" Cells 12, no. 8: 1103. https://doi.org/10.3390/cells12081103
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.