
Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome
 , , and
, , and 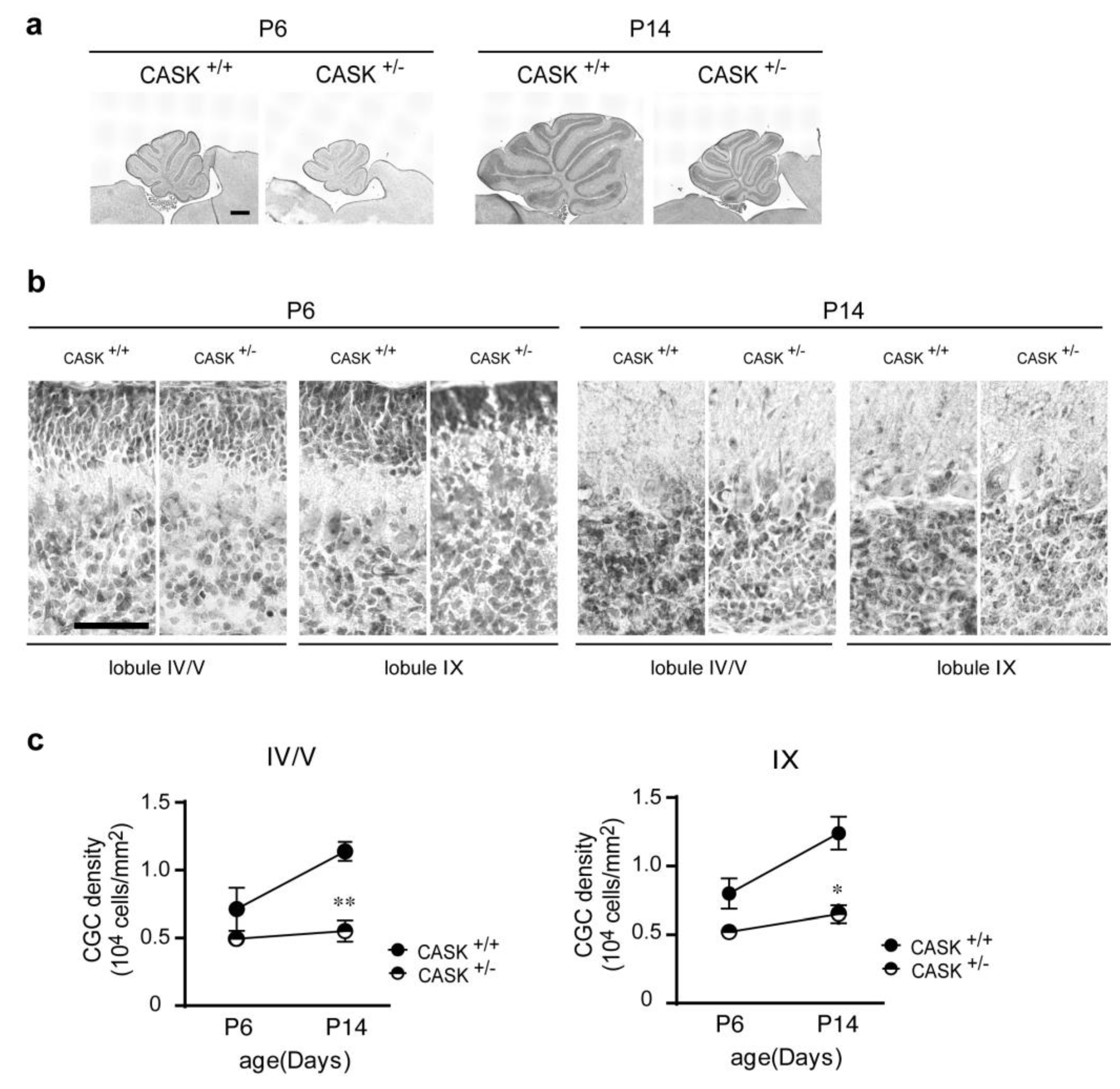{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Housing Conditions
2.2. Genotyping
2.3. Plasmid Construction
2.4. Cell Lines
2.5. Lentiviral Preparation
2.6. Cell Culture and Immunocytochemistry
2.7. TUNEL Assay
2.8. Immunoblotting
2.9. Coimmunoprecipitation
2.10. RT-qPCR
2.11. Hematoxylin & Eosin Staining
2.12. Intracellular Localization of CASK Mutants
2.13. Prediction of CASK Protein Structure and Structural Analysis
2.14. Sample Size and Statistical Analysis
3. Results
3.1. Density of CG Cells Is Decreased in CASK+/− Mice
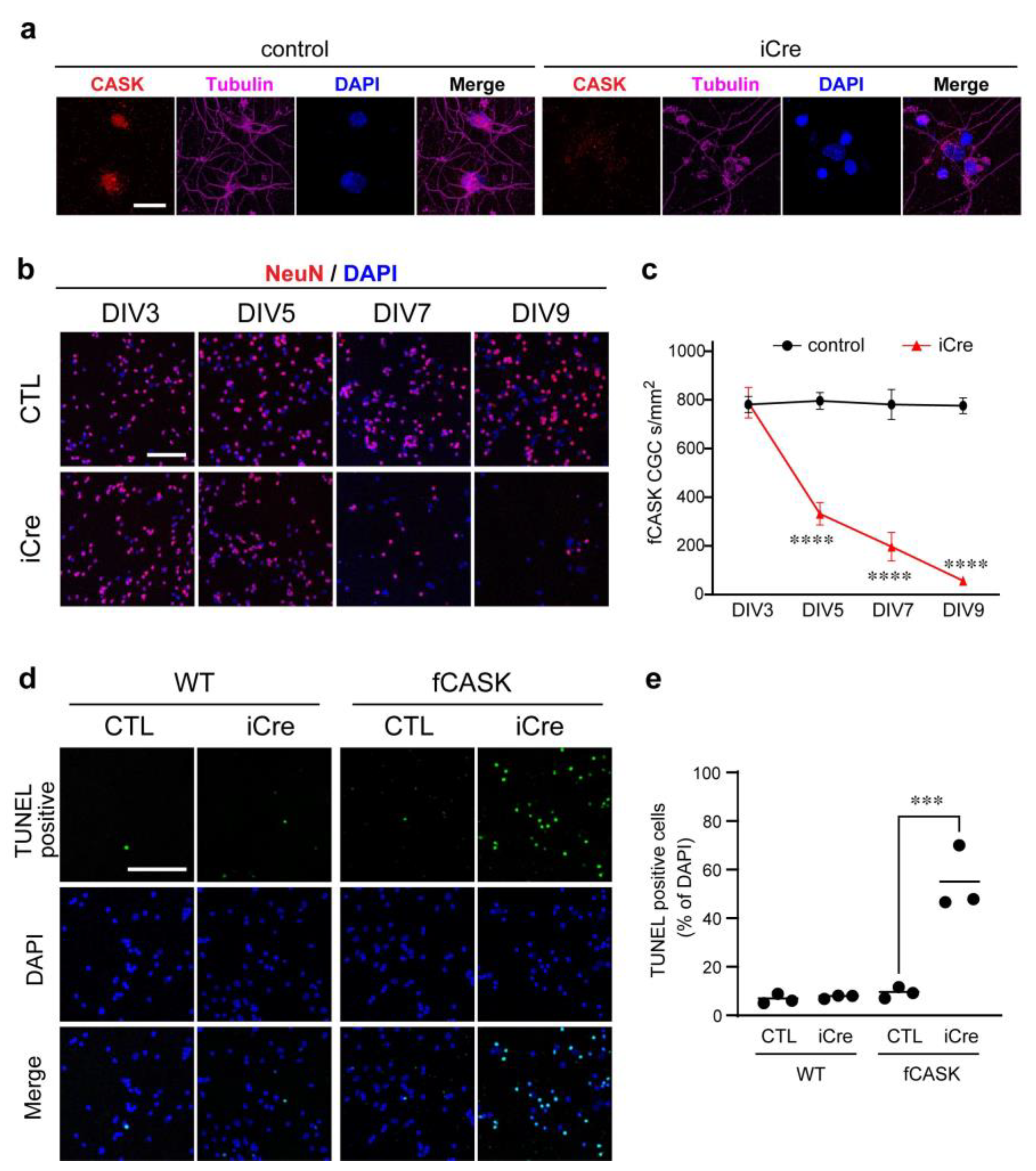
3.2. Apoptosis Occurs in CASK KO CG Cells
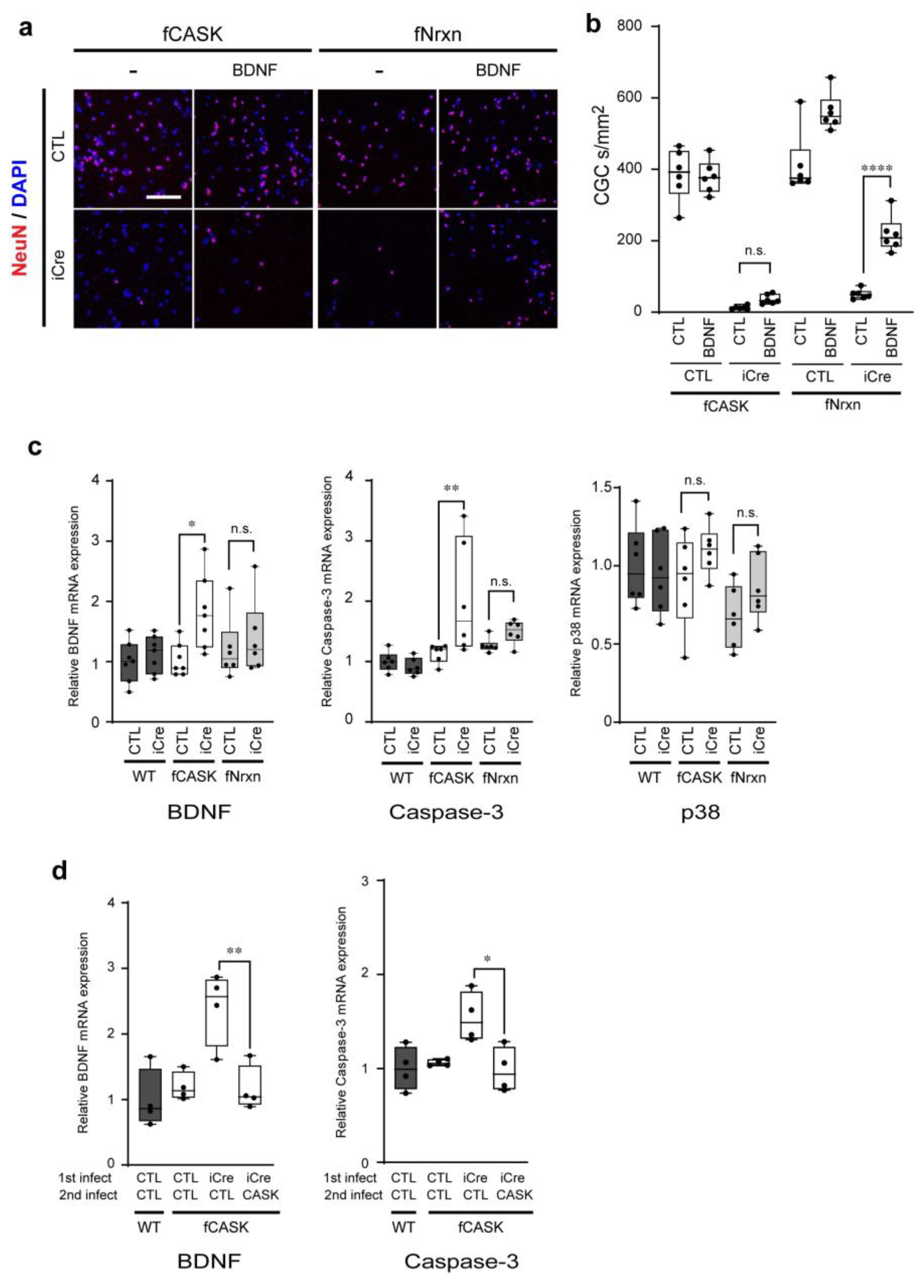
3.3. CG Cell Death in CASK KO Mice Is Independent of BDNF Secretion Mediated by Neurexins
3.4. CaMK, PDZ, and SH3 Domains of CASK Are Required for the Survival of CG Cells
3.5. Missense Mutations Identified from Patients with Cerebellar Hypoplasia Affect CG Cell Survival
3.6. R106P, R255C, and Y268H Mutations Reside the Binding Interface between CASK-CaMK Domain and Liprin-α2
3.7. Machine Learning-Based Analysis Indicates R106P, R255C, and Y268H Mutations Disrupt Structure of Binding Interface between CASK-CaMK Domain and Liprin-α2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hata, Y.; Butz, S.; Sudhof, T.C. CASK: A novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 1996, 16, 2488–2494. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Yang, F.C.; Kharazia, V.; Naisbitt, S.; Cohen, A.R.; Weinberg, R.J.; Sheng, M. Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J. Cell Biol. 1998, 142, 139–151. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Wang, T.F.; Yang, F.C.; Sheng, M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 2000, 404, 298–302. [Google Scholar] [CrossRef]
- Biederer, T.; Sara, Y.; Mozhayeva, M.; Atasoy, D.; Liu, X.; Kavalali, E.T.; Südhof, T.C. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 2002, 297, 1525–1531. [Google Scholar] [CrossRef]
- Hsueh, Y.P. Calcium/calmodulin-dependent serine protein kinase and mental retardation. Ann. Neurol. 2009, 66, 438–443. [Google Scholar] [CrossRef]
- Butz, S.; Okamoto, M.; Sudhof, T.C. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 1998, 94, 773–782. [Google Scholar] [CrossRef]
- Tabuchi, K.; Biederer, T.; Butz, S.; Sudhof, T.C. CASK participates in alternative tripartite complexes in which Mint 1 competes for binding with caskin 1, a novel CASK-binding protein. J. Neurosci. 2002, 22, 4264–4273. [Google Scholar] [CrossRef]
- Olsen, O.; Moore, K.A.; Fukata, M.; Kazuta, T.; Trinidad, J.C.; Kauer, F.W.; Streuli, M.; Misawa, H.; Burlingame, A.L.; Nicoll, R.A.; et al. Neurotransmitter release regulated by a MALS–liprin-α presynaptic complex. J. Cell Biol. 2005, 170, 1127–1134. [Google Scholar] [CrossRef]
- Stafford, R.L.; Ear, J.; Knight, M.J.; Bowie, J.U. The molecular basis of the Caskin1 and Mint1 interaction with CASK. J. Mol. Biol. 2011, 412, 3–13. [Google Scholar] [CrossRef]
- Wei, Z.; Zheng, S.; Spangler, S.A.; Yu, C.; Hoogenraad, C.C.; Zhang, M. Liprin-mediated large signaling complex organization revealed by the liprin-alpha/CASK and liprin-alpha/liprin-beta complex structures. Mol. Cell 2011, 43, 586–598. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Q.; Chen, Y.; Zhu, S.; Mi, J.; Wang, J.; Zhang, M. Structural Basis for the High-Affinity Interaction between CASK and Mint1. Structure 2020, 28, 664–673.e3. [Google Scholar] [CrossRef]
- LaConte, L.E.; Chavan, V.; Liang, C.; Willis, J.; Schonhense, E.M.; Schoch, S.; Mukherjee, K. CASK stabilizes neurexin and links it to liprin-alpha in a neuronal activity-dependent manner. Cell. Mol. Life Sci. 2016, 73, 3599–3621. [Google Scholar] [CrossRef]
- Mukherjee, K.; Sharma, M.; Urlaub, H.; Bourenkov, G.P.; Jahn, R.; Sudhof, T.C.; Wahl, M.C. CASK Functions as a Mg2+-independent neurexin kinase. Cell 2008, 133, 328–339. [Google Scholar] [CrossRef]
- Wang, G.S.; Hong, C.J.; Yen, T.Y.; Huang, H.Y.; Ou, Y.; Huang, T.N.; Jung, W.G.; Kuo, T.Y.; Sheng, M.; Wang, T.F.; et al. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 2004, 42, 113–128. [Google Scholar] [CrossRef]
- Wang, T.F.; Ding, C.N.; Wang, G.S.; Luo, S.C.; Lin, Y.L.; Ruan, Y.; Hevner, R.; Rubenstein, J.L.; Hsueh, Y.P. Identification of Tbr-1/CASK complex target genes in neurons. J. Neurochem. 2004, 91, 1483–1492. [Google Scholar] [CrossRef]
- Atasoy, D.; Schoch, S.; Ho, A.; Nadasy, K.A.; Liu, X.; Zhang, W.; Mukherjee, K.; Nosyreva, E.D.; Fernandez-Chacon, R.; Missler, M.; et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. USA 2007, 104, 2525–2530. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Hackett, A.; Tarpey, P.S.; Licata, A.; Cox, J.; Whibley, A.; Boyle, J.; Rogers, C.; Grigg, J.; Partington, M.; Stevenson, R.E.; et al. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur. J. Hum. Genet. 2010, 18, 544–552. [Google Scholar] [CrossRef]
- Seto, T.; Hamazaki, T.; Nishigaki, S.; Kudo, S.; Shintaku, H.; Ondo, Y.; Shimojima, K.; Yamamoto, T. A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly. Intractable Rare Dis. Res. 2017, 6, 177–182. [Google Scholar] [CrossRef]
- Burglen, L.; Chantot-Bastaraud, S.; Garel, C.; Milh, M.; Touraine, R.; Zanni, G.; Petit, F.; Afenjar, A.; Goizet, C.; Barresi, S.; et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: Confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J. Rare Dis. 2012, 7, 18. [Google Scholar] [CrossRef]
- Dimitratos, S.D.; Stathakis, D.G.; Nelson, C.A.; Woods, D.F.; Bryant, P.J. The location of human CASK at Xp11.4 identifies this gene as a candidate for X-linked optic atrophy. Genomics 1998, 51, 308–309. [Google Scholar] [CrossRef]
- Piluso, G.; D’Amico, F.; Saccone, V.; Bismuto, E.; Rotundo, I.L.; Di Domenico, M.; Aurino, S.; Schwartz, C.E.; Neri, G.; Nigro, V. A missense mutation in CASK causes FG syndrome in an Italian family. Am. J. Hum. Genet. 2009, 84, 162–177. [Google Scholar] [CrossRef]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.A.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef]
- Moog, U.; Kutsche, K.; Kortum, F.; Chilian, B.; Bierhals, T.; Apeshiotis, N.; Balg, S.; Chassaing, N.; Coubes, C.; Das, S.; et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J. Med. Genet. 2011, 48, 741–751. [Google Scholar] [CrossRef]
- Mukherjee, K.; LaConte, L.E.W.; Srivastava, S. The Non-Linear Path from Gene Dysfunction to Genetic Disease: Lessons from the MICPCH Mouse Model. Cells 2022, 11, 1131. [Google Scholar] [CrossRef]
- Hayashi, S.; Uehara, D.T.; Tanimoto, K.; Mizuno, S.; Chinen, Y.; Fukumura, S.; Takanashi, J.I.; Osaka, H.; Okamoto, N.; Inazawa, J. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS ONE 2017, 12, e0181791. [Google Scholar] [CrossRef]
- Moog, U.; Bierhals, T.; Brand, K.; Bautsch, J.; Biskup, S.; Brune, T.; Denecke, J.; de Die-Smulders, C.E.; Evers, C.; Hempel, M.; et al. Phenotypic and molecular insights into CASK-related disorders in males. Orphanet J. Rare Dis. 2015, 10, 44. [Google Scholar] [CrossRef]
- Mori, T.; Kasem, E.A.; Suzuki-Kouyama, E.; Cao, X.; Li, X.; Kurihara, T.; Uemura, T.; Yanagawa, T.; Tabuchi, K. Deficiency of calcium/calmodulin-dependent serine protein kinase disrupts the excitatory-inhibitory balance of synapses by down-regulating GluN2B. Mol. Psychiatry 2019, 24, 1079–1092. [Google Scholar] [CrossRef]
- Srivastava, S.; McMillan, R.; Willis, J.; Clark, H.; Chavan, V.; Liang, C.; Zhang, H.; Hulver, M.; Mukherjee, K. X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol. Commun. 2016, 4, 30. [Google Scholar] [CrossRef]
- Patel, P.A.; Hegert, J.V.; Cristian, I.; Kerr, A.; LaConte, L.E.W.; Fox, M.A.; Srivastava, S.; Mukherjee, K. Complete loss of the X-linked gene CASK causes severe cerebellar degeneration. J. Med. Genet. 2022, 59, 1044–1057. [Google Scholar] [CrossRef]
- Tibbe, D.; Ferle, P.; Krisp, C.; Nampoothiri, S.; Mirzaa, G.; Assaf, M.; Parikh, S.; Kutsche, K.; Kreienkamp, H.J. Regulation of Liprin-alpha phase separation by CASK is disrupted by a mutation in its CaM kinase domain. Life Sci. Alliance 2022, 5, e202201512. [Google Scholar] [CrossRef]
- Mehta, A.; Shirai, Y.; Kouyama-Suzuki, E.; Zhou, M.; Yoshizawa, T.; Yanagawa, T.; Mori, T.; Tabuchi, K. IQSEC2 Deficiency Results in Abnormal Social Behaviors Relevant to Autism by Affecting Functions of Neural Circuits in the Medial Prefrontal Cortex. Cells 2021, 10, 2724. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki-Kouyama, E.; Kawase, S.; Kurihara, T.; Yasumura, M.; Yoshida, T.; Fukai, S.; Yamazaki, M.; Fei, P.; Abe, M.; et al. Neurexins play a crucial role in cerebellar granule cell survival by organizing autocrine machinery for neurotrophins. Cell Rep. 2022, 39, 110624. [Google Scholar] [CrossRef]
- Cull-Candy, S.G.; Howe, J.R.; Ogden, D.C. Noise and single channels activated by excitatory amino acids in rat cerebellar granule neurones. J. Physiol. 1988, 400, 189–222. [Google Scholar] [CrossRef]
- Tabuchi, K.; Sudhof, T.C. Structure and evolution of neurexin genes: Insight into the mechanism of alternative splicing. Genomics 2002, 79, 849–859. [Google Scholar] [CrossRef]
- Kasem, E.; Kurihara, T.; Tabuchi, K. Neurexins and neuropsychiatric disorders. Neurosci. Res. 2018, 127, 53–60. [Google Scholar] [CrossRef]
- Cao, X.; Tabuchi, K. Functions of synapse adhesion molecules neurexin/neuroligins and neurodevelopmental disorders. Neurosci. Res. 2017, 116, 3–9. [Google Scholar] [CrossRef]
- Treutlein, B.; Gokce, O.; Quake, S.R.; Sudhof, T.C. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E1291–E1299. [Google Scholar] [CrossRef]
- Zhang, Y.; Nie, Y.; Mu, Y.; Zheng, J.; Xu, X.; Zhang, F.; Shu, J.; Liu, Y. A de novo variant in CASK gene causing intellectual disability and brain hypoplasia: A case report and literature review. Ital. J. Pediatr. 2022, 48, 73. [Google Scholar] [CrossRef]
- LaConte, L.E.W.; Chavan, V.; DeLuca, S.; Rubin, K.; Malc, J.; Berry, S.; Gail Summers, C.; Mukherjee, K. An N-terminal heterozygous missense CASK mutation is associated with microcephaly and bilateral retinal dystrophy plus optic nerve atrophy. Am. J. Med. Genet. 2019, 179, 94–103. [Google Scholar] [CrossRef]
- Cristofoli, F.; Devriendt, K.; Davis, E.E.; Van Esch, H.; Vermeesch, J.R. Novel CASK mutations in cases with syndromic microcephaly. Hum. Mutat. 2018, 39, 993–1001. [Google Scholar] [CrossRef]
- LaConte, L.E.W.; Chavan, V.; Elias, A.F.; Hudson, C.; Schwanke, C.; Styren, K.; Shoof, J.; Kok, F.; Srivastava, S.; Mukherjee, K. Two microcephaly-associated novel missense mutations in CASK specifically disrupt the CASK-neurexin interaction. Hum. Genet. 2018, 137, 231–246. [Google Scholar] [CrossRef]
- Pan, Y.E.; Tibbe, D.; Harms, F.L.; Reissner, C.; Becker, K.; Dingmann, B.; Mirzaa, G.; Kattentidt-Mouravieva, A.A.; Shoukier, M.; Aggarwal, S.; et al. Missense mutations in CASK, coding for the calcium-/calmodulin-dependent serine protein kinase, interfere with neurexin binding and neurexin-induced oligomerization. J. Neurochem. 2021, 157, 1331–1350. [Google Scholar] [CrossRef]
- Takanashi, J.; Okamoto, N.; Yamamoto, Y.; Hayashi, S.; Arai, H.; Takahashi, Y.; Maruyama, K.; Mizuno, S.; Shimakawa, S.; Ono, H.; et al. Clinical and radiological features of Japanese patients with a severe phenotype due to CASK mutations. Am. J. Med. Genet. 2012, 158A, 3112–3118. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Sheng, M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J. Neurosci. 1999, 19, 7415–7425. [Google Scholar] [CrossRef]
- Huang, T.N.; Hsueh, Y.P. Calcium/calmodulin-dependent serine protein kinase (CASK), a protein implicated in mental retardation and autism-spectrum disorders, interacts with T-Brain-1 (TBR1) to control extinction of associative memory in male mice. J. Psychiatry Neurosci. 2017, 42, 37–47. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Kouyama-Suzuki, E.; Shirai, Y.; Cao, X.; Yanagawa, T.; Mori, T.; Tabuchi, K. Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome. Cells 2023, 12, 1177. https://doi.org/10.3390/cells12081177
Guo Q, Kouyama-Suzuki E, Shirai Y, Cao X, Yanagawa T, Mori T, Tabuchi K. Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome. Cells. 2023; 12(8):1177. https://doi.org/10.3390/cells12081177
Chicago/Turabian StyleGuo, Qi, Emi Kouyama-Suzuki, Yoshinori Shirai, Xueshan Cao, Toru Yanagawa, Takuma Mori, and Katsuhiko Tabuchi. 2023. "Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome" Cells 12, no. 8: 1177. https://doi.org/10.3390/cells12081177