
FGF9, a Potent Mitogen, Is a New Ligand for Integrin αvβ3, and the FGF9 Mutant Defective in Integrin Binding Acts as an Antagonist
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of FGF9
2.3. Docking Simulation
2.4. Binding of Soluble Integrin αvβ3 to FGF9
2.5. FGF9 Binding to the FGFR1 D2D3 Fragment and Heparin
2.6. Binding of R108E to Heparin
2.7. Cell Migration Assay
2.8. Invasion Assay
2.9. BrdU Incorporation Assay
2.10. Statistical Analysis
3. Results
3.1. FGF9 Directly Binds to Integrin αvβ3
3.2. Generation of Integrin-Binding Defective FGF9 Mutants
3.3. R108E Is Defective in Inducing Migration and Invasion of Colon Cancer Cells
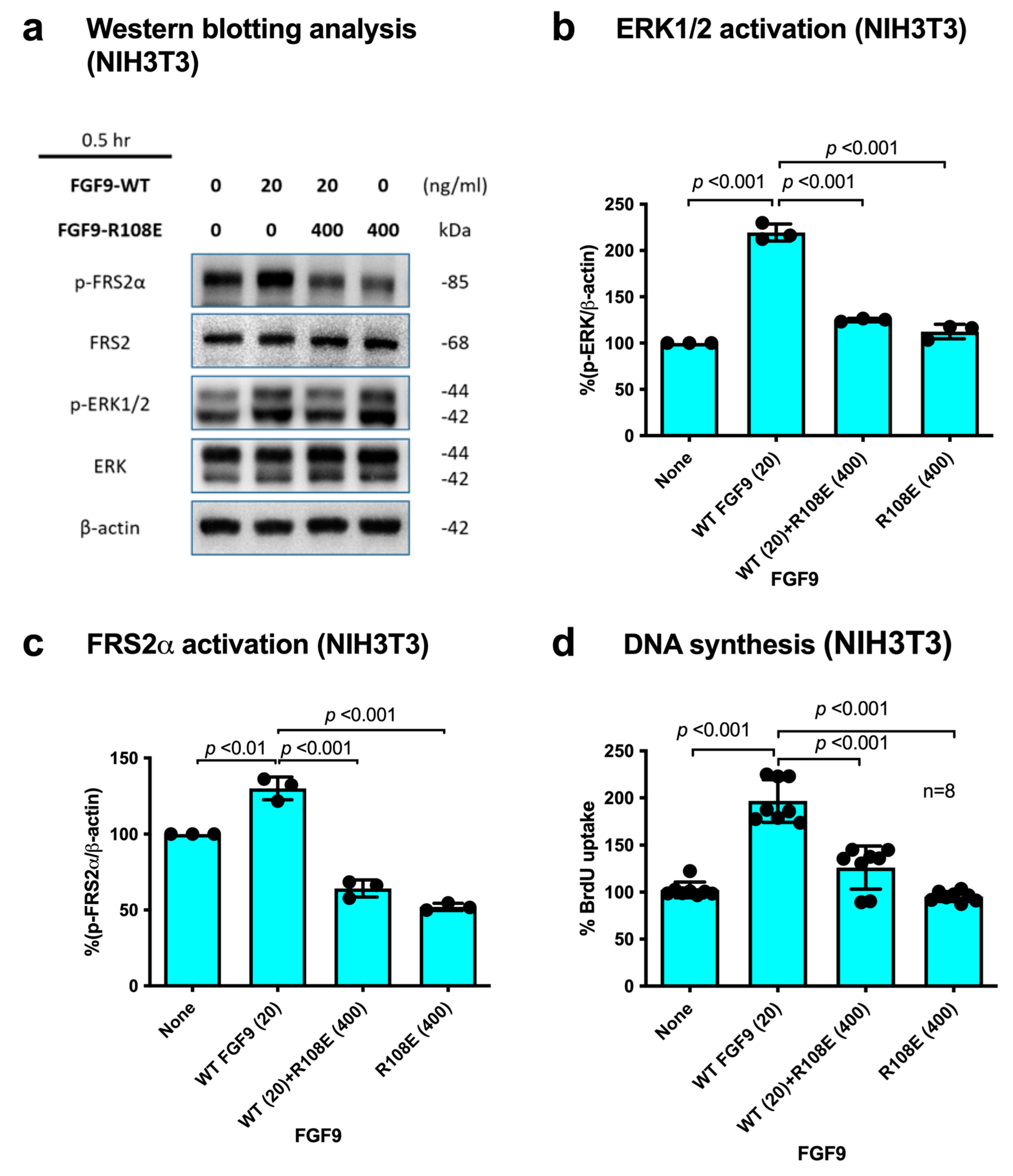
3.4. R108E Suppresses Activation of FRS2 and ERK1/2 and DNA Synthesis Induced by WT FGF9 (Dominant-Negative Effect) in NIH3T3 Cells
4. Discussion
R108E (FGF9 Antagonist) Has Potential as Therapeutic in Cancer
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P. Integrin and Growth Factor Receptor Crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Naruo, K.; Seko, C.; Kuroshima, K.; Matsutani, E.; Sasada, R.; Kondo, T.; Kurokawa, T. Novel secretory heparin-binding factors from human glioma cells (glia-activating factors) involved in glial cell growth. Purification and biological properties. J. Biol. Chem. 1993, 268, 2857–2864. [Google Scholar] [CrossRef]
- Klint, P.; Claesson-Welsh, L. Signal transduction by fibroblast growth factor receptors. Front. Biosci. 1999, 4, d165-77. [Google Scholar] [CrossRef]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef]
- Colvin, J.S.; White, A.C.; Pratt, S.J.; Ornitz, D.M. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 2001, 128, 2095–2106. [Google Scholar] [CrossRef]
- Colvin, J.S.; Green, R.P.; Schmahl, J.; Capel, B.; Ornitz, D.M. Male-to-Female Sex Reversal in Mice Lacking Fibroblast Growth Factor 9. Cell 2001, 104, 875–889. [Google Scholar] [CrossRef]
- DiNapoli, L.; Batchvarov, J.; Capel, B. FGF9 promotes survival of germ cells in the fetal testis. Development 2006, 133, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.H.; Yu, K.; Lavine, K.J.; Ornitz, D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev. Biol. 2007, 307, 300–313. [Google Scholar] [CrossRef]
- Lavine, K.J.; Yu, K.; White, A.C.; Zhang, X.; Smith, C.; Partanen, J.; Ornitz, D.M. Endocardial and Epicardial Derived FGF Signals Regulate Myocardial Proliferation and Differentiation In Vivo. Dev. Cell 2005, 8, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Pirvola, U.; Zhang, X.; Mantela, J.; Ornitz, D.M.; Ylikoski, J. Fgf9 signaling regulates inner ear morphogenesis through epithelial–mesenchymal interactions. Dev. Biol. 2004, 273, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-M.; Weng, H.-Y.; Lai, M.-S.; Wang, L.-W.; Yang, S.-H.; Wu, C.-C.; Wang, C.-Y.; Huang, B.-M. Original FGF9 promotes cell proliferation and tumorigenesis in TM3 mouse Leydig progenitor cells. Am. J. Cancer Res. 2022, 12, 5613–5630. [Google Scholar] [PubMed]
- Chang, M.; Lai, M.; Hong, S.; Pan, B.; Huang, H.; Yang, S.; Wu, C.; Sun, H.S.; Chuang, J.; Wang, C.; et al. FGF9/FGFR2 increase cell proliferation by activating ERK1/2, Rb/E2F1, and cell cycle pathways in mouse Leydig tumor cells. Cancer Sci. 2018, 109, 3503–3518. [Google Scholar] [CrossRef]
- Arai, D.; E. Hegab, A.; Soejima, K.; Kuroda, A.; Ishioka, K.; Yasuda, H.; Naoki, K.; Kagawa, S.; Hamamoto, J.; Yin, Y.; et al. Characterization of the cell of origin and propagation potential of the fibroblast growth factor 9-induced mouse model of lung adenocarcinoma. J. Pathol. 2015, 235, 593–605. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, C.; Hamana, T.; Liu, J.; Wang, C.; An, L.; McKeehan, W.L.; Wang, F. Overexpression of FGF9 in Prostate Epithelial Cells Augments Reactive Stroma Formation and Promotes Prostate Cancer Progression. Int. J. Biol. Sci. 2015, 11, 948–960. [Google Scholar] [CrossRef]
- Chen, T.-M.; Shih, Y.-H.; Tseng, J.T.; Lai, M.-C.; Wu, C.-H.; Li, Y.-H.; Tsai, S.-J.; Sun, H.S. Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation. Nucleic Acids Res. 2014, 42, 2932–2944. [Google Scholar] [CrossRef]
- Ren, C.; Chen, H.; Han, C.; Fu, D.; Wang, F.; Wang, D.; Ma, L.; Zhou, L.; Han, D. The anti-apoptotic and prognostic value of fibroblast growth factor 9 in gastric cancer. Oncotarget 2016, 7, 36655–36665. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Chaudhuri, S.R.; Roy, S.S. FGF9-induced ovarian cancer cell invasion involves VEGF-A/VEGFR2 augmentation by virtue of ETS1 upregulation and metabolic reprogramming. J. Cell. Biochem. 2018, 119, 8174–8189. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, F.; Ornitz, D.M. Mesothelial- and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development 2011, 138, 3169–3177. [Google Scholar] [CrossRef]
- Teishima, J.; Shoji, K.; Hayashi, T.; Miyamoto, K.; Ohara, S.; Matsubara, A. Relationship between the localization of fibroblast growth factor 9 in prostate cancer cells and postoperative recurrence. Prostate Cancer Prostatic Dis. 2012, 15, 8–14. [Google Scholar] [CrossRef]
- Chang, M.-M.; Wu, S.-Z.; Yang, S.-H.; Wu, C.-C.; Wang, C.-Y.; Huang, B.-M. FGF9/FGFR1 promotes cell proliferation, epithelial-mesenchymal transition, M2 macrophage infiltration and liver metastasis of lung cancer. Transl. Oncol. 2021, 14, 101208. [Google Scholar] [CrossRef] [PubMed]
- Ohgino, K.; Soejima, K.; Yasuda, H.; Hayashi, Y.; Hamamoto, J.; Naoki, K.; Arai, D.; Ishioka, K.; Sato, T.; Terai, H.; et al. Expression of fibroblast growth factor 9 is associated with poor prognosis in patients with resected non-small cell lung cancer. Lung Cancer 2014, 83, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Wu, C.-Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; et al. Direct Binding of Integrin αvβ3 to FGF1 Plays a Role in FGF1 Signaling. J. Biol. Chem. 2008, 283, 18066–18075. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Tran, V.; Nishikawa, K.; Kaneda, T.; Hamada, Y.; Kawaguchi, N.; Fujita, M.; Takada, Y.K.; Matsuura, N.; Zhao, M.; et al. A Dominant-Negative FGF1 Mutant (the R50E Mutant) Suppresses Tumorigenesis and Angiogenesis. PLoS ONE 2013, 8, e57927. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Hatori, N.; Kawaguchi, N.; Hamada, Y.; Shih, T.-C.; Wu, C.-Y.; Lam, K.S.; Matsuura, N.; Yamamoto, H.; Takada, Y.K.; et al. The integrin-binding defective FGF2 mutants potently suppress FGF2 signalling and angiogenesis. Biosci. Rep. 2017, 37, BSR20170173. [Google Scholar] [CrossRef]
- Wang, W.; Malcolm, B.A. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 1999, 26, 680–682. [Google Scholar] [CrossRef]
- Saegusa, J.; Akakura, N.; Wu, C.-Y.; Hoogland, C.; Ma, Z.; Lam, K.S.; Liu, F.-T.; Takada, Y.K.; Takada, Y. Pro-inflammatory Secretory Phospholipase A2 Type IIA Binds to Integrins αvβ3 and α4β1 and Induces Proliferation of Monocytic Cells in an Integrin-dependent Manner. J. Biol. Chem. 2008, 283, 26107–26115. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Takada, Y.K.; Takada, Y. Integrins αvβ3 and α4β1 Act as Coreceptors for Fractalkine, and the Integrin-Binding Defective Mutant of Fractalkine Is an Antagonist of CX3CR1. J. Immunol. 2012, 189, 5809–5819. [Google Scholar] [CrossRef]
- Wang, S.; Lin, H.; Zhao, T.; Huang, S.; Fernig, D.G.; Xu, N.; Wu, F.; Zhou, M.; Jiang, C.; Tian, H. Expression and purification of an FGF9 fusion protein in E. coli, and the effects of the FGF9 subfamily on human hepatocellular carcinoma cell proliferation and migration. Appl. Microbiol. Biotechnol. 2017, 101, 7823–7835. [Google Scholar] [CrossRef]
- Sun, C.; Fukui, H.; Hara, K.; Zhang, X.; Kitayama, Y.; Eda, H.; Tomita, T.; Oshima, T.; Kikuchi, S.; Watari, J.; et al. FGF9 from cancer-associated fibroblasts is a possible mediator of invasion and anti-apoptosis of gastric cancer cells. BMC Cancer 2015, 15, 333. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Ieguchi, K.; Cedano-Prieto, D.M.; Fong, A.; Wilkerson, C.; Chen, J.Q.; Wu, M.; Lo, S.-H.; Cheung, A.T.W.; Wilson, M.D.; et al. An Integrin Binding-defective Mutant of Insulin-like Growth Factor-1 (R36E/R37E IGF1) Acts as a Dominant-negative Antagonist of the IGF1 Receptor (IGF1R) and Suppresses Tumorigenesis but Still Binds to IGF1R. J. Biol. Chem. 2013, 288, 19593–19603. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.K.; Yu, J.; Shimoda, M.; Takada, Y. Integrin Binding to the Trimeric Interface of CD40L Plays a Critical Role in CD40/CD40L Signaling. J. Immunol. 2019, 203, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-S.; Taniguchi, Y.; Yunoki, Y.; Masai, S.; Nogi, M.; Doi, H.; Sekiguchi, K.; Nakagawa, M. Simultaneous binding of bFGF to both FGFR and integrin maintains properties of primed human induced pluripotent stem cells. Regen. Therapy 2024, 25, 113e127. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FGF9 | β3 | αv |
|---|---|---|
| Thr52, Asp53, Asp55, His56, Leu57, Pro78, Asn79, Gly80, Thr81, Arg108, Val110, Asp111, Ser112, Gly113, Leu114, Tyr115, Asn119, Glu120, Tyr125, Gly126, Ser127, Glu128, Lys129, Leu130, Thr131, Gln132, Glu133, Cys134, Asn151, Leu152, Lys154, Asp203, Leu204, Leu205, Ser206, Glu207, Ser208 | Ser121, Tyr122, Ser123, Met124, Lys125, Asp126, Asp127, Leu128, Trp129, Tyr166, Cys177, Tyr178, Asp179. Met180, Lys181, Yhr182, Arg214, Asn215, Arg216, Asp217, Ala218, Pro219, Glu220, Asp251, Thr311, Glu312, Asn313, Leu333, Ser334, Met335, Asp336, Ser337 | Asp146, Asp148, Ala149, Asp150, Tyr178, Trp179, Gln182, Asn205, Asn206, Gln207, Leu208, Ala209, Arg211, Thr212, Ala213, Gln214, Ala215, Phe217, Asp218, Asn260, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-C.; Takada, Y.K.; Cheng, C.-W.; Maekawa, Y.; Mori, S.; Takada, Y. FGF9, a Potent Mitogen, Is a New Ligand for Integrin αvβ3, and the FGF9 Mutant Defective in Integrin Binding Acts as an Antagonist. Cells 2024, 13, 307. https://doi.org/10.3390/cells13040307
Chang C-C, Takada YK, Cheng C-W, Maekawa Y, Mori S, Takada Y. FGF9, a Potent Mitogen, Is a New Ligand for Integrin αvβ3, and the FGF9 Mutant Defective in Integrin Binding Acts as an Antagonist. Cells. 2024; 13(4):307. https://doi.org/10.3390/cells13040307
Chicago/Turabian StyleChang, Chih-Chieh, Yoko K. Takada, Chao-Wen Cheng, Yukina Maekawa, Seiji Mori, and Yoshikazu Takada. 2024. "FGF9, a Potent Mitogen, Is a New Ligand for Integrin αvβ3, and the FGF9 Mutant Defective in Integrin Binding Acts as an Antagonist" Cells 13, no. 4: 307. https://doi.org/10.3390/cells13040307