
Understanding Developmental Cell Death Using Drosophila as a Model System
by
 , , ,
, , ,
Ruchi Umargamwala
1,*,
Jantina Manning
1,
Loretta Dorstyn
1,
Donna Denton
1 and
Sharad Kumar
1,2,* 1
Centre for Cancer Biology, University of South Australia and SA Pathology, Adelaide, SA 5001, Australia
2
Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5005, Australia
*
Authors to whom correspondence should be addressed.
Cells 2024, 13(4), 347; https://doi.org/10.3390/cells13040347
Submission received: 23 January 2024
/
Revised: 9 February 2024
/
Accepted: 13 February 2024
/
Published: 16 February 2024
(This article belongs to the Special Issue Drosophila Models in Autophagy and Aging)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cell death plays an essential function in organismal development, wellbeing, and ageing. Many types of cell deaths have been described in the past 30 years. Among these, apoptosis remains the most conserved type of cell death in metazoans and the most common mechanism for deleting unwanted cells. Other types of cell deaths that often play roles in specific contexts or upon pathological insults can be classed under variant forms of cell death and programmed necrosis. Studies in Drosophila have contributed significantly to the understanding and regulation of apoptosis pathways. In addition to this, Drosophila has also served as an essential model to study the genetic basis of autophagy-dependent cell death (ADCD) and other relatively rare types of context-dependent cell deaths. Here, we summarise what is known about apoptosis, ADCD, and other context-specific variant cell death pathways in Drosophila, with a focus on developmental cell death.
1. Introduction
Maintaining the correct cell numbers via cell division and eliminating harmful or redundant cells is crucial for the fitness of all metazoans [1]. Cells can die from different mechanisms such as accidental cell death (ACD) that can induce necrosis, or through genetically controlled methods of regulated cell death (RCD) [2]. More specifically, programmed cell death (PCD) is a specific subset of RCD that occurs exclusively under physiological conditions, with apoptosis being the most frequently used and conserved cell death pathway [3]. However, with the emergence of context-specific, non-apoptotic modes of cell deletion such as autophagy-dependent cell death (ADCD), Drosophila has prevailed as an excellent model system to better understand the genetic, molecular, and regulatory mechanisms underlying the unconventional forms of developmental cell death. Herein, we review our knowledge of the conserved apoptotic machinery in Drosophila and highlight tissue-specific modes of cell death, such as ADCD and other variant cell deaths, which support development and organismal homeostasis.
2. Evolutionarily Conserved Apoptotic Cell Death
The genetic machinery of apoptosis was first discovered in the nematode, Caenorhabditis elegans, where 131 of the 1090 cells generated during the development of the hermaphrodite worm died by this process, even though the extra cells had no effect on the viability of the worm [4,5]. Pioneering morphological and genetic studies in C. elegans initiated by Sulston, Horvitz, and Brenner lead to the discovery of the genetics of PCD execution that underpins the core apoptosis machinery in metazoans [4,6]. The primary apoptosis machinery in C. elegans comprises four cell death genes/proteins. Cell death abnormal (CED)-9 is crucial for cell survival, whereas the other three, EGL-1, CED-4, and CED-3, are required for executing cell death. The BH3-only protein, EGL-1, initiates cell death by sequestering CED-9, allowing the adaptor CED-4 to activate CED-3, a cysteine protease that cleaves target proteins, resulting in apoptosis and the clearance of the cell corpse [7,8,9,10]. This elegant pathway, while far more complex, is essentially conserved in mammals, where BH3-only, EGL-1-like proteins initiate apoptosis by blocking the survival function of CED-9-like BCL-2 family members to initiate APAF-1 (CED-4-like)-dependent caspase-9 (CED-3 like) activation (Figure 1) [11]. Although apoptosis signalling in mammals can also be initiated by extrinsic mechanisms mediated by the tumour necrosis factor receptor (TNFR) family, all apoptotic cell deaths in mammals involve the caspase, cysteine protease (CED-3) family of enzymes which are initially activated using an activating platform (apoptosome or DISC) that is functionally analogous to the CED-4 complex (the CED-4 apoptosome) (Figure 1) [11,12].
3. The Apoptotic Machinery in Drosophila
Drosophila melanogaster was first adopted as a model organism more than 110 years ago by Thomas Hunt Morgan to study the generational inheritance of genetic information [13]. Since then, Drosophila has played a critical role in understanding the genetics of many complex biological pathways which are evolutionarily conserved across phylogeny. The well-defined developmental stages in Drosophila comprising embryo, larva, pupa, and adult have allowed genetic studies to explore molecular pathways that drive spatiotemporal regulation of development, including developmental cell death.
The main apoptotic machinery in Drosophila includes the conserved family of caspases, although the upstream mechanisms that regulate the activation of caspases are divergent from C. elegans and mammals (Figure 1). The three pro-apoptotic genes, grim, reaper and head involution defective (hid) (also known as RHG genes that are transcribed on the H99 locus) act as initiators of apoptosis, with the loss of any individual gene resulting in suppression of cell death [14,15,16]. Antagonising RHG are the Drosophila inhibitor of apoptosis proteins (IAPs), Diap1 and Diap2. Diap1 acts as the major cell death inhibitor and blocks caspase activation by utilising its E3 ligase function to catalyse transfer of ubiquitin moieties to Dronc, thus resulting in its proteasomal degradation [17,18]. On the other hand, the binding of RHG proteins to Diap1 results in Diap1 autoubiquitination and degradation, which triggers rapid and spontaneous caspase activation and apoptosis [17,19].
There are seven caspases in Drosophila of which Dronc (Drosophila Nedd2-like caspase) is the sole apoptosis initiator caspase that has the most sequence homology to mammalian caspase-2 but is most functionally similar to CED-3 in C. elegans and caspase-9 in mammals [20,21]. The two effector caspases, Drice and Dcp-1, require activation by active Dronc and are functionally similar to mammalian caspase-3 and caspase-7 [22]. The caspase Dredd is partially similar to mammalian caspase-8 and caspase-10; however, its function is mainly attributed to mediating innate immune responses [23,24]. The functions of the other three caspases, Strica, Decay and Damm, are mostly apoptosis independent. Strica has a long amino-terminal Ser/Thr-rich region which remains functionally undefined, whereas Decay and Damm are similar to caspase-3 and caspase-7 but their deletion has no effect on cell death, suggesting a level of redundancy in the Drosophila cell death machinery [25,26,27,28].
Analogous to CED-4-mediated activation of CED-3 in C. elegans and APAF-1-dependent caspase-9 activation in mammals, Dronc activation requires the Drosophila APAF-1-related killer (Dark) apoptosome [29]. Although sharing functional similarities, the octameric CED-4 and Dark apoptosomes, as well as the heptameric APAF-1 apoptosome, assemble and activate caspases using different molecular mechanisms (discussed in detail in [30]). Also, unlike in mammals, where cytochrome c release from the mitochondria promotes the formation of the apoptosome and activates caspase-9, CED-4 and Dark apoptosomes have no such requirement [30]. In all cases, procaspase recruitment to the apoptosome occurs via homotypic interactions between conserved caspase activation and recruitment domains (CARDs) in these proteins; however, the stoichiometry is vastly different between species [31]. For example, the CED-4 apoptosome binds two molecules of CED-3 and facilitates CED-3 autocatalytic activity [32]. Similarly, the structure of the Dark apoptosome is also octameric, however eight molecules of Dronc are recruited in Dark [33]. Conversely, the heptameric assembly of APAF-1 binds three to four molecules of procaspase-9 [30,34]. Nonetheless, the principal role of this complex is to facilitate proximity-induced autoactivation of procaspase (zymogen) molecules [35].
Drosophila has two BCL-2-like proteins, Debcl and Buffy, both of which are more similar to the mammalian BAX-like protein, BOK, than other members of the prosurvival BCL-2 family proteins [36]. In overexpression and RNAi experiments, Debcl appears to serve a proapoptotic function, whereas Buffy suppresses cell death [37,38,39]. However, neither of these proteins appear essential for apoptosis or cell survival, as single or combined (double) mutants of Debcl and Buffy are viable and do not modulate PCD [40]. Recently, a BH3-like protein has also been described based on sequence homology to the conserved BH3 motif [41]. This protein, Sayonara, can complex with Buffy and Debcl and induce apoptosis when overexpressed; however, synr mutants are viable without adverse developmental or cell death phenotypes [41]. Thus, like Buffy and Debcl, the physiological function of Sayonara in the core apoptosis machinery remains ambiguous. It is possible that in Drosophila, BCL-2 family members have evolved to have context-dependent function(s) outside the main apoptosis pathway. However, Drosophila, unlike C. elegans and mammals, relies on RHG proteins to initiate apoptosis and Diap1 to inhibit caspase activation. The mammalian BCL-2 family members (including proapoptotic BH3-only proteins, prosurvival BCL-2 proteins, and pore forming tBID, BAX, and BAK) are mainly involved in controlling the release of cytochrome c from the mitochondria that is required for APAF-1 apoptosome formation and caspase-9 activation [42]. As cytochrome c is not required for apoptosome formation and caspase activation in Drosophila, it is likely that this specific apoptosis regulatory function for BCL-2 proteins evolved much later.
4. Autophagy Machinery in Drosophila
Originally established as a cell degradation and recycling process, autophagy is also associated with context-dependent cell death processes. Autophagy is an evolutionarily conserved response to adverse cellular events such as starvation, hypoxia, and microbial infection, protecting cells from stress-induced death [43]. This occurs via sequestration of redundant or detrimental substrates (including damaged organelles) into double-membraned vesicular structures known as autophagosomes, followed by their degradation via lysosomal-mediated pathways [44]. The recycling of substrates through autophagy releases energy and monomers of complex biomolecules (fatty acids and amino acids) into the intracellular environment, which can be utilised by cells to ensure cellular homeostasis when compromised [45]. While autophagy operates constitutively under basal conditions to maintain cellular health, elevated levels occur when cells face adversities [46].
The core machinery of autophagy was first discovered through genetic screens performed in Saccharomyces cerevisiae (yeast), unveiling 41 genes encoding important autophagy regulatory components [47,48]. These genes are highly conserved in mammals and Drosophila [48,49,50].
In Drosophila, autophagy is activated upon the formation of the Atg1 initiation complex, comprising Atg1 (a serine/threonine kinase, homologous to mammalian ULK1), Atg13, Atg17 (FIP200 in mammals) and Atg101 [49,51] (Figure 2). The Atg1 complex translocates to the isolation membrane of the developing autophagosome (also known as the phagophore), where it recruits Atg9 as well as the class III phosphatidylinositol 3-kinase (PtdIns3K) complex [52]. The PtdIns3K complex, consisting of vacuolar protein sorting (VPS), VPS34 and VPS15, Atg6 (Beclin 1 in mammals) and Atg14, phosphorylates phosphatidylinositol to phosphatidylinositol-3-phosphate (PI(3)P), which then promotes the nucleation of the autophagosomal membrane (Figure 2) [53,54].
The expansion of the autophagosome is dependent on two ubiquitin-like conjugation systems (Figure 2). Firstly, the E1- and E2-like enzymes, Atg7 and Atg10, respectively, coordinate attachment of Atg16 to Atg5-Atg12, forming the E3-like complex, Atg5-Atg12-Ag16 [55,56]. The second ubiquitin-like system involves the tethering of Atg8a (similar to mammalian LC3 and GABARAP family members) to the autophagosomal membrane [57]. Atg8a is cleaved by the cysteine protease, Atg4, and becomes lipidated through the attachment of phosphatidylethanolamine (PE) to form Atg8a-PE by Atg7 (E1-like enzyme) and Atg3 (E2-like enzyme), the latter of which is recruited by the Atg5-Atg12-Atg16 complex to the developing autophagosome [57,58].
Finally, fusion of autophagosomes to lysosomes degrades biomaterials and is coordinated by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) in combination with tethering factors targeting the autophagosomal Atg8a proteins and lysosomal RAB7 proteins [59].
Autophagy is tightly controlled by the conserved protein kinase, target of rapamycin (TOR), which forms the catalytic subunit of two distinct multiprotein complexes, mTORC1 and mTORC2 (mechanistic target of rapamycin) [60]. These complexes are distinguishable by their additional components—raptor, lst8, lobe (mammalian PRAS40) in mTORC1, and rict-1 (mammalian Rictor), sinh-1 (mammalian mSin1) and Lst8 in mTORC2 [61]. mTORC1 is the primary sensor of nutrient and amino acid availability, supporting cellular metabolism, homeostasis, and autophagy [62]. Although the roles of mTORC2 are yet to be fully elucidated, some functions include the regulation of metabolism and cell proliferation [62,63,64]. In nutrient-rich conditions, autophagy is downregulated by mTORC1-mediated hyperphosphorylation of Atg13 which prevents its association with Atg1. This is attenuated under starvation conditions and mTORC1 inhibition by rapamycin treatment, where dephosphorylation of Atg13 promotes the formation of the Atg1 complex [65,66]. Interestingly, an increased expression of Atg13 can promote Atg1 phosphorylation by TOR and subsequently inhibit autophagy in Drosophila, demonstrating dual roles for Atg13 in autophagy suppression and activation [51].
5. Autophagy-Dependent Cell Death (ADCD)
The primary role of autophagy is to promote cell survival under basal and adverse conditions. Challenging this dogma are several context-specific physiological processes that reveal the cytotoxic nature of autophagy.
Following the stress signalling that accompanies cell death, autophagy is often observed in cells undergoing apoptosis. However, a distinction must be made between autophagy that accompanies cell death, and autophagy that promotes cell killing. For example, in Parkinson’s disease and Danon disease, the accumulation of autophagic structures was initially regarded as a feature of cell death by autophagy, when in actuality, the increase in autophagic vacuoles was as a result of defective autophagy and not increased cell death [67,68,69].
To overcome such confusion, the Nomenclature Committee on Cell Death (NCCD) proposed the following criteria to define ADCD: (1) inhibition of autophagy must prevent cell death, (2) the process must be functionally dependent on two or more autophagy-related genes/proteins participating in ADCD, (3) and ADCD must occur independently of other cell death processes, such as apoptosis and necrosis [3,70].
6. Apoptosis and ADCD in Drosophila Development
The development of Drosophila is marked by drastic morphological changes from an early embryo to three larval stages, followed by the pupation and formation of the adult fly. This requires tight regulation and coordination of both apoptotic and autophagic machinery, with specific tissues relying on one or both these processes to undergo remodelling.
6.1. Embryogenesis
Drosophila embryogenesis is a highly dynamic process featuring the rapid transformation of numerous tissue structures at specific developmental stages. The earliest instance of apoptotic cell death in Drosophila was found to occur at stage 11 of embryogenesis [71]. Apoptosis begins in the dorsal area of the head region and leads to a retraction of the germ band, allowing dying cells along the dorsal ridge and ventral midline to facilitate closure of the dorsal tissue. Widespread cell death during neurodevelopmental stages 15 and 16 causes head involution, condensation of the ventral cord and the restructuring of brain lobes [71]. Whilst caspases are the primary drivers of these early events, late embryogenesis events, including degradation of the amnioserosa (AS) or extraembryonic tissue, rely on induction of autophagy to coordinate caspase-dependent cell death [72]. However, a subsequent study by Cormier et al. [73] demonstrated that Atg1 mutants did not significantly attenuate caspase-dependent AS extrusion, suggesting that there is no strict requirement for autophagy induction in this process.
6.2. Midgut
The Drosophila larval midgut (anterior and posterior midgut) comprises a single layer of epithelial cells [74,75]. The proventriculus, also known as the cardia, is located anteriorly to the midgut midbody and controls passaging of food into the midgut [76]. The four arm-like structures, collectively known as the gastric caeca, branch off from the proventriculus [77].
The larval midgut undergoes drastic morphological changes during metamorphosis to give rise to the adult midgut. Despite the majority of Drosophila tissues dying via apoptosis, the larval midgut degrades strictly via ADCD. Beginning at −4 h relative to puparium formation (RPF), a dramatic increase in autophagy coinciding with the transcriptional upregulation of autophagy genes promotes contraction of gastric caeca, removal of the proventriculus, and condensation of the larval midgut, giving rise to the adult midgut by +12 h RPF [78,79]. Midgut histolysis remains unaffected by caspase inhibition; however, ablation of the autophagy-related genes, Atg1, Atg2 and Atg18, significantly delays gastric caeca and midgut retraction. However, as midgut degradation is not completely blocked, this suggests potential involvement of other catabolic pathways to facilitate complete midgut degradation [78]. Regardless, the midgut remains the best example of ADCD in vivo.
6.3. Salivary Glands
The salivary glands are large tubular structures extending from the larval mouth which undergoes abrupt degradation during metamorphosis. Following midgut histolysis, salivary gland cell death is initiated at +10 h RPF and is degraded by +15 h RPF [80,81]. Increased nuclear permeability of salivary gland cells to acridine orange observed at +14 h RPF confirmed the involvement of apoptotic machinery in salivary gland degeneration; however, only a partial block in histolysis was achieved upon expression of the apoptosis inhibitor, baculovirus p35 [81]. Subsequent studies showed that intact apoptotic and autophagic machinery was required to effectively induce salivary gland cell death, with the loss of either pathway delaying tissue removal [82]. This suggests that the histolysis of the salivary glands is dependent on both autophagy and apoptosis.
6.4. Fat Body
The fat body, analogous to vertebrate adipose tissue, is an essential component of Drosophila which serves an important role in meeting metabolic demands during larval and adult stages of life [83]. Prior to metamorphosis, wandering larvae stop feeding in order to undergo pupation. In response to starvation, robust levels of autophagy promote fat body shrinkage to provide energy for survival [84]. Additionally, Drosophila must rely on stored energy to fuel pupal-to-adult transition [85]. Individualisation of fat body cells during metamorphosis causes their redistribution within the pupa, and these are progressively eliminated at the early stages of life [83]. In a study by Scott et al. [86], it was shown that increasing autophagy levels by overexpressing Atg1 resulted in caspase-dependent fat body cell death, as demonstrated by DNA fragmentation and disruption of the cytoskeleton. Thus, in the context of fat body remodelling, late larval development relies on autophagy-dependent energy production for survival, whereas during metamorphosis, autophagy precedes apoptosis as opposed to directly coordinating cellular demise.
6.5. Myogenesis
The formation of Drosophila musculature occurs in two waves: the first wave of myogenesis during embryogenesis gives rise to mesodermal-derived somatic muscles that support larval motility, whereas the second myogenic wave initiates the formation of adult muscles responsible for flight, walking, and mating behaviours [87]. During metamorphosis, whilst most larval muscles undergo histolysis, some are retained into adulthood to facilitate proper body patterning prior to undergoing PCD [88,89]. Specifically, within the abdomen, the dorsal external oblique muscles (DEOM) are degraded during early metamorphosis at 12 h RPF, whereas the dorsal internal oblique muscles (DIOM) persist until eclosion and are removed within 24 h of adulthood [90,91]. Although increased levels of autophagy have been reported in degenerating DEOMs, knockdown of Atg1, Atg5 and Atg18 did not affect DEOM removal [92]. In contrast, overexpression of p35 caused retention of DEOMs, signifying that while the role of autophagy in DEOM PCD is redundant, this process is largely dependent on apoptosis [92,93].
6.6. Neurogenesis
The formation of the Drosophila central nervous system (CNS) begins during embryogenesis, where neuroblasts (NBs) undergo asymmetric divisions to replenish the stem cell pool and produce ganglion mother cells (GMCs), the latter which further divide to create neurons and glial cells [94]. Whilst a majority of NBs cease to proliferate at +30 h RFP during pupal development, NBs within a region of the brain known as the mushroom body (MB) continue dividing up to 10 h prior to eclosion of the adult fly, following which they undergo cell death [95]. Elimination of MB NBs is required to complete neurogenesis, and, in a similar manner to the salivary glands, this is dependent on both autophagy and apoptosis. Downregulation of autophagy or RNAi-mediated inhibition of RHG genes results in persistence of MB NBs for up to 3 days and 7 days post-eclosion, respectively. Conversely, MB NBs survive well into adulthood upon ablation of both pathways [96].
6.7. Oogenesis
Drosophila oogenesis is a highly dynamic process involving cell-to-cell communication and maintenance of stem cell niches to support egg production [97]. Cytoblasts undergo multiple rounds of division to form an oogenic cyst consisting of 16 germ cells, of which only one cell proceeds to become an oocyte and the remaining cells develop into supporting nurse cells [98]. During the later stages of oogenesis, nurse cells expel cytoplasm-containing maternal mRNAs, organelles, and proteins into the maturing oocyte, leading to PCD of nurse cells while causing no detrimental effects to the attached oocyte [99,100]. Though there is an established apoptotic role in the removal of egg chambers during germarium and mid-oogenesis stages, recent findings suggest that this process is also triggered by nutrient deprivation [101,102]. Under starvation conditions, knockdown of Atg1 and Atg7, as well as Dcp-1 effector caspase mutants, fail to activate autophagy, causing a persistence of egg chambers [101]. Conversely, mutants of the Diap1/2 protein, dBruce, enhance egg chamber degradation, suggesting that while Dcp-1 can promote autophagy, dBruce operates antagonistically in this process [101,103].
7. Signalling Mechanisms Regulating ADCD in Drosophila
7.1. Ecdysone Signalling
The transition of the Drosophila larvae to an adult fly is defined by stages of larval molting, followed by metamorphosis during which cell death results in a structural reorganisation of obsolete larval tissues and the formation of adult tissues [104]. This is tightly regulated by the steroid hormone, 20-hydroxyecdysone (20E), in a spatiotemporal manner. Produced in the prothoracic glands from dietary cholesterol, the precursor of 20E, ecdysone, is secreted into the haemolymph and is oxidised into active 20E within the target tissues [105]. Significant pulses of ecdysone titre release occur at the late third instar, prepupal, and pupal stage, although other smaller waves of ecdysone release facilitate larval molting during the first and second instar stages [106,107,108]. Ecdysone release at the late larval stage initiates prepupa formation and degradation of the larval midgut at −4 h RPF [109]. A subsequent ecdysone titre pulse +10–12 h RPF signifies pupa development and salivary gland degradation, the latter which is complete by +15–16 h RPF. A final pulse at 30 h RPF coordinates adult development [110].
Downstream signalling through ecdysone is mediated by the heterodimeric receptor complex, ecdysone receptor/ultraspiracle (EcR/USP), which can activate a transcription of ecdysone-responsive transcription factor genes such as Broad-Complex (BrC), E74, E75 and E93 [111,112] (Figure 3). E93, in particular, was previously implicated in the degradation of salivary glands through autophagy and apoptosis [112]. However, a study by Duncan, et al. [113] showed that the allelic mutations performed by Lee and Baehrecke [111] were of the β subunit of the mitochondrial enzyme, isocitrate dehydrogenase-3 (Idh3b), not the three “type” alleles of the E93 family, E931, E932 and E933. Consequently, Duncan, Kiefel and Duncan [113] demonstrate that mutations in Idh3b alleles prevent autophagy initiation during salivary gland degradation. Although E93 has also been shown to activate autophagy through the downregulation of growth signalling in the removal of MB NBs [96], recent studies confirm that E93 does not regulate expression of cell death genes in the salivary glands [114].
Larval fat bodies also rely on ecdysone receptors to regulate growth signalling and induce autophagy. Whilst the loss of ecdysone receptors prevents autophagy, inhibition of growth signalling can restore autophagy levels [115,116]. Additionally, the knockdown of EcR has been shown to block larval midgut degradation by reducing the transcription of autophagy-related genes, Atg1, Atg13, Atg17 and Atg8a, amongst multiple others, thereby confirming its necessity in Drosophila ADCD [117].
7.2. Growth Signalling
Early Drosophila larval development is driven by growth signalling pathways which converge on mTORC1 (Figure 3) [118]. Insulin-like peptides (ILPs) bind to the Drosophila insulin receptor (dInR), causing downstream phosphorylation of the insulin receptor substrate protein, CHICO [119]. Subsequently, phosphorylation of the active subunit of Drosophila PI3K, Dp110, catalyses the conversion of phosphatidylinositol 4,5-biphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3) [120,121]. Antagonising the function of Dp110 is Drosophila PTEN (dPTEN) which converts PIP3 into PIP2 [122]. Acting as a secondary messenger, PIP3 recruits PDK1 and phosphorylates AKT at the plasma membrane, enabling AKT-dependent phosphorylation of Drosophila TSC2 (dTSC2) and subsequently maintaining the activity of mTORC1 [121,123].
As inhibition of growth signalling is required for autophagy induction, mTORC1 downregulation is essential for midgut removal by ADCD [124]. As expected, inhibition of PI3K can similarly promote midgut degradation, as does the expression of negative regulators of the PI3K pathway, dPTEN and TSC1/2 [125].
Salivary glands also demonstrate the requirement for growth arrest prior to autophagy induction and histolysis [126]. The expression of Dp110 and AKT results in persistent enlarged salivary glands, which undergo apoptosis- and autophagy-dependent histolysis upon removal of PIP3 and AKT from the outer membrane of salivary gland cells [82]. A similar morphological phenotype is observed when a positive regulator of PI3K signalling, Ras, is expressed in the salivary glands [82]. Another pathway that regulates growth signalling and ADCD involves the Warts (wts) family of genes, Wts, Hippo (Hpo), Mob-as-tumour suppressor (Mats), Salvador (Sav), Merlin (Mer) and Expanded (Ex), which are important for growth control [127]. Wts facilitates autophagy in salivary glands whereas homozygous mutation of wts prevents the removal of AKT from the salivary gland cell cortex during puparium formation, in turn inhibiting salivary gland degradation. This defect was shown to be rescued by ectopic expression of Atg1, reaffirming that growth arrest precedes autophagy-dependent salivary gland histolysis [128]. Interestingly, the knockdown of Wts family members does not impede midgut degradation, suggesting that signals regulating growth and cell death occur in tissue-specific manners [125].
7.3. Decapentaplegic Signalling
The bone morphogenetic protein/transforming growth factor β (BMP/TGF-β) ligand, decapentaplegic (Dpp), has been known to regulate cell proliferation, cell fate, and body patterning during embryogenesis and larval development [129,130]. Dpp exists in a heterodimeric complex consisting of the type I receptor, Thickveins (Tkv), and the type II receptor, Punt (Put). Phosphorylation of Tkv by Put triggers Tkv kinase activity, causing the phosphorylation of downstream regulators, Mothers against Dpp (Mad) and Medea (Med) [131]. Acting as a complex, Mad and Med can deregulate or activate target genes such as Brinker (Brk), a negative regulator of Dpp-dependent genes, or Schnurri (Shn), responsible for facilitating the Dpp-mediated repression of Brk [131,132,133].
Recent studies have uncovered that Dpp signalling is required for retention of the Drosophila larval midgut until it begins to undergo histolysis during the third instar stage [117]. In the presence of Dpp signalling through the expression of Dpp or Tkv, ecdysone production within prothoracic glands and transcriptional upregulation of ecdysone-responsive genes in the midgut is repressed, thereby inhibiting ADCD (Figure 3). Conversely, knockdown of Mad and Med, or blocking Dpp signalling through expression of its inhibitory SMAD protein, Dad, restored autophagy levels and accelerated midgut removal. Hence, there is convergence of multiple signalling pathways in the initiation and regulation of ADCD (summarised in Figure 3).
8. Alternative Cell Death Pathways in Drosophila
In addition to apoptosis and ADCD, non-canonical forms of cell deaths have also been reported in Drosophila, as summarised below.
8.1. Parthanatos
Parthanatos, related to the Greek term for the personification of death, “thanatos”, describes cell death involving a family of DNA repair enzymes collectively known as poly(ADP-ribose) polymerases (PARPs) [134]. PARP-1 in particular, has a variety of nuclear roles including base excision repair (BER) to single-stranded DNA break repair [135]. Whilst PARP-1 instigates a cell-protective response to acute DNA aberrations, an overaccumulation of DNA damage results in the hyperactivation of PARP-1, leading to the exhaustion of cellular energy due to a depletion of NAD+ and consequent translocation of an apoptosis-inducing factor (AIF)-DNase complex from mitochondria into the nucleus. Subsequently, AIF can trigger condensation of nuclear chromatin, disruption of mitochondrial membrane potential, externalisation of phosphatidylserine on the cell surface and cell death [136,137].
In a study by Tarayrah-Ibraheim et al. [138], it was shown that parthanatos was responsible for the elimination of primordial germ cells (PGCs) during Drosophila embryogenesis. Permeabilisation of lysosomal membranes releases cathepsin B, triggering mobilisation of AIF from mitochondria. Another lysosomal hydrolase, DNase II, is translocated into the nucleus by AIF and disrupts chromatin architecture, causing hyperactivation of PARP-1. A positive feedback loop between PARP-1 and AIF results in PGC death. Despite lysosomal leakage that suggests the involvement of autophagy, genetic inhibition of autophagy genes did not attenuate cell death, inferring that autophagy is not critical for this process.
8.2. HtrA2/Omi in Caspase-Independent Cell Death
HtrA2/Omi is a serine protease released from mitochondria upon induction of apoptosis. In a similar manner to mammalian Smac/DIABLO and Drosophila RHG proteins, HtrA2 can bind to and prevent IAPs from suppressing caspase-3 activity [139,140,141].
Recently, an intriguing role for HtrA2 has been unveiled in Drosophila germ cell death (GCD). During Drosophila spermatogenesis, germ cells undergo multiple rounds of synchronous divisions but remain connected by cytoplasmic bridges due to incomplete cytokinesis. Up to a quarter of differentiated spermatocytes undergo spontaneous cell death, and the rate of GCD can increase during starvation conditions to support survival of spermatogonial stem cell populations [142,143]. Drosophila GCD has been shown to occur independently of caspase activation [144]. Instead, the catalytic activity of HtrA2, but not its IAP-inhibiting function, controls lysosomal and mitochondrial-driven cell death by interacting with Pink1, an important modulator of mitochondrial homeostasis [144]. Furthermore, Endonuclease G (EndoG), a proapoptotic mitochondrial enzyme, is released from mitochondria to induce cell death by altering chromatin dynamics in parallel to HtrA2-mediated cell death [144,145].
9. Conclusions
Drosophila has provided the cell death community a highly accessible tool to investigate and understand tissue homeostasis and physiological processes of apoptotic and non-apoptotic forms of PCD. Combined molecular and genetic studies in Drosophila have provided a significant insight into the diversity of cell death pathways, including the evolutionary divergence of apoptotic machinery and the manner by which caspase activation is triggered (e.g., removal of Diap1, rather than the involvement of BH3-proteins, BCL-2 and Bax/Bak-dependent mitochondrial membrane permeabilisation). Investigations involving Drosophila also played a vital role in establishing autophagy as a regulator of cell survival and cell death. Whilst genetic evidence and the rationale for death by autophagy (ADCD) has been uncovered, exactly how cells die by autophagy requires further investigation. Additionally, Drosophila models have provided much benefit to in vivo studies into alternate forms of cell death involving non-canonical roles of apoptotic machinery, enabling clear distinctions between the context- and tissue-specific roles of cell death players in diverse physiological settings. Finally, as dysregulated apoptosis and autophagy have been implicated in various diseases (see [146,147,148]), Drosophila should continue to contribute knowledge relevant to both the fundamentals of cellular homeostasis in organismal development and healthy ageing.
Author Contributions
R.U. and S.K. conceptualised this review; R.U. wrote the first version of the article and drafted figures. S.K., D.D., J.M. and L.D. edited various versions of this review and figures. All authors have read and agreed to the published version of the manuscript.
Funding
The recent cell death work in our laboratory has been supported by the following funding: National Health & Medical Research Council (NHMRC) of Australia Project Grant (1124490), Australian Research Council Discovery Project Grant (DP210100665), a University of South Australia Support Package, NHMRC Senior Principal Research Fellowship (GNT1103006) and NHMRC L3 Investigator Grant (2007739) to S.K., R.U. is a supported by an Australian Government Research Training Program Scholarship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Horvitz, H.R. Genetic Control of Programmed Cell Death in the Nematode Caenorhabditis Elegans. In Apoptosis; Mihich, E., Schimke, R.T., Eds.; Springer US: Boston, MA, USA, 1994; pp. 1–13. [Google Scholar]
- Hengartner, M.O. Genetic Control of Programmed Cell Death and Aging in the Nematode Caenorhabditis Elegans. Exp. Gerontol. 1997, 32, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The Embryonic Cell Lineage of the Nematode Caenorhabditis Elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef] [PubMed]
- Conradt, B.; Horvitz, H.R. The C. elegans Protein Egl-1 Is Required for Programmed Cell Death and Interacts with the Bcl-2–Like Protein Ced-9. Cell 1998, 93, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.S.; Desnoyers, S.; Hoeppner, D.J.; Hengartner, M.O. Interaction between the C. elegans Cell-Death Regulators Ced-9 and Ced-4. Nature 1997, 385, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chai, J.; Lee, E.S.; Gu, L.; Liu, Q.; He, J.; Wu, J.W.; Kokel, D.; Li, H.; Hao, Q.; et al. Structure of the Ced-4-Ced-9 Complex Provides Insights into Programmed Cell Death in Caenorhabditis Elegans. Nature 2005, 437, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Miller, L.K. Caenorhabditis Elegans Ced-4 Stimulates Ced-3 Processing and Ced-3-Induced Apoptosis. Curr. Biol. 1997, 7, 455–460. [Google Scholar] [CrossRef]
- Kumar, S.; Dorstyn, L.; Lim, Y. The Role of Caspases as Executioners of Apoptosis. Biochem. Soc. Trans. 2021, 50, 33–45. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of Bcl-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.H. Sex Limited Inheritance in Drosophila. Science 1910, 32, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Nordstrom, W.; Gish, B.; Abrams, J.M. Grim, a Novel Cell Death Gene in Drosophila. Genes Dev. 1996, 10, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Tahaoglu, E.; Steller, H. Cell Killing by the Drosophila Gene Reaper. Science 1996, 271, 805–807. [Google Scholar] [CrossRef]
- Haining, W.N.; Carboy-Newcomb, C.; Wei, C.L.; Steller, H. The Proapoptotic Function of Drosophila Hid Is Conserved in Mammalian Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 4936–4941. [Google Scholar] [CrossRef]
- Wang, S.L.; Hawkins, C.J.; Yoo, S.J.; Müller, H.-A.J.; Hay, B.A. The Drosophila Caspase Inhibitor Diap1 Is Essential for Cell Survival and Is Negatively Regulated by Hid. Cell 1999, 98, 453–463. [Google Scholar] [CrossRef]
- Wilson, R.; Goyal, L.; Ditzel, M.; Zachariou, A.; Baker, D.A.; Agapite, J.; Steller, H.; Meier, P. The Diap1 Ring Finger Mediates Ubiquitination of Dronc and Is Indispensable for Regulating Apoptosis. Nat. Cell Biol. 2002, 4, 445–450. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Bergmann, A.; Gonen, H.; Ciechanover, A.; Steller, H. Regulation of Drosophila Iap1 Degradation and Apoptosis by Reaper and Ubcd1. Nat. Cell Biol. 2002, 4, 432–438. [Google Scholar] [CrossRef]
- Daish, T.J.; Mills, K.; Kumar, S. Drosophila Caspase Dronc Is Required for Specific Developmental Cell Death Pathways and Stress-Induced Apoptosis. Dev. Cell 2004, 7, 909–915. [Google Scholar] [CrossRef]
- Dorstyn, L.; Colussi, P.A.; Quinn, L.M.; Richardson, H.; Kumar, S. Dronc, an Ecdysone-Inducible Drosophila Caspase. Proc. Natl. Acad. Sci. USA 1999, 96, 4307–4312. [Google Scholar] [CrossRef]
- Denton, D.; Aung-Htut, M.T.; Kumar, S. Developmentally Programmed Cell Death in Drosophila. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3499–3506. [Google Scholar] [CrossRef]
- Chen, P.; Rodriguez, A.; Erskine, R.; Thach, T.; Abrams, J.M. Dredd, a Novel Effector of the Apoptosis Activators Reaper, Grim, and Hid in Drosophila. Dev. Biol. 1998, 201, 202–216. [Google Scholar] [CrossRef]
- Leulier, F.; Rodriguez, A.; Khush, R.S.; Abrams, J.M.; Lemaitre, B. The Drosophila Caspase Dredd Is Required to Resist Gram-Negative Bacterial Infection. EMBO Rep. 2000, 1, 353–358. [Google Scholar] [CrossRef]
- Doumanis, J.; Quinn, L.; Richardson, H.; Kumar, S. Strica, a Novel Drosophila Melanogaster Caspase with an Unusual Serine/Threonine-Rich Prodomain, Interacts with Diap1 and Diap2. Cell Death Differ. 2001, 8, 387–394. [Google Scholar] [CrossRef]
- Dorstyn, L.; Read, S.H.; Quinn, L.M.; Richardson, H.; Kumar, S. Decay, a Novel Drosophila Caspase Related to Mammalian Caspase-3 and Caspase-7. J. Biol. Chem. 1999, 274, 30778–30783. [Google Scholar] [CrossRef]
- Harvey, N.L.; Daish, T.; Mills, K.; Dorstyn, L.; Quinn, L.M.; Read, S.H.; Richardson, H.; Kumar, S. Characterization of the Drosophila Caspase, Damm. J. Biol. Chem. 2001, 276, 25342–25350. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Woodfield, S.E.; Lee, T.V.; Fan, Y.; Antonio, C.; Bergmann, A. Genetic Control of Programmed Cell Death (Apoptosis) in Drosophila. Fly 2009, 3, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Kumar, S. A Biochemical Analysis of the Activation of the Drosophila Caspase Dronc. Cell Death Differ. 2008, 15, 461–470. [Google Scholar] [CrossRef]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New Insights into Apoptosome Structure and Function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H. Caspase Recruitment Domains for Protein interactions in Cellular Signaling (Review). Int. J. Mol. Med. 2019, 43, 1119–1127. [Google Scholar] [CrossRef]
- Qi, S.; Pang, Y.; Hu, Q.; Liu, Q.; Li, H.; Zhou, Y.; He, T.; Liang, Q.; Liu, Y.; Yuan, X. Crystal Structure of the Caenorhabditis Elegans Apoptosome Reveals an Octameric Assembly of Ced-4. Cell 2010, 141, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yu, X.; Topf, M.; Dorstyn, L.; Kumar, S.; Ludtke, S.J.; Akey, C.W. Structure of the Drosophila Apoptosome at 6.9 å Resolution. Structure 2011, 19, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-Dimensional Structure of the Apoptosome: Implications for Assembly, Procaspase-9 Binding, and Activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Shi, Y. Apoptosome: A Platform for the Activation of Initiator Caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Doumanis, J.; Dorstyn, L.; Kumar, S. Molecular Determinants of the Subcellular Localization of the Drosophila Bcl-2 Homologues Debcl and Buffy. Cell Death Differ. 2007, 14, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Colussi, P.A.; Quinn, L.M.; Huang, D.C.; Coombe, M.; Read, S.H.; Richardson, H.; Kumar, S. Debcl, a Proapoptotic Bcl-2 Homologue, Is a Component of the Drosophila Melanogaster Cell Death Machinery. J. Cell Biol. 2000, 148, 703–714. [Google Scholar] [CrossRef]
- Quinn, L.; Coombe, M.; Mills, K.; Daish, T.; Colussi, P.; Kumar, S.; Richardson, H. Buffy, a Drosophila Bcl-2 Protein, Has Anti-Apoptotic and Cell Cycle Inhibitory Functions. EMBO J. 2003, 22, 3568–3579. [Google Scholar] [CrossRef]
- Igaki, T.; Kanuka, H.; Inohara, N.; Sawamoto, K.; Núñez, G.; Okano, H.; Miura, M. Drob-1, a Drosophila Member of the Bcl-2/Ced-9 Family That Promotes Cell Death. Proc. Natl. Acad. Sci. USA 2000, 97, 662–667. [Google Scholar] [CrossRef]
- Sevrioukov, E.A.; Burr, J.; Huang, E.W.; Assi, H.H.; Monserrate, J.P.; Purves, D.C.; Wu, J.N.; Song, E.J.; Brachmann, C.B. Drosophila Bcl-2 Proteins Participate in Stress-Induced Apoptosis, but Are Not Required for Normal Development. Genesis 2007, 45, 184–193. [Google Scholar] [CrossRef]
- Ikegawa, Y.; Combet, C.; Groussin, M.; Navratil, V.; Safar-Remali, S.; Shiota, T.; Aouacheria, A.; Yoo, S.K. Evidence for Existence of an Apoptosis-Inducing BH3-Only Protein, Sayonara, in Drosophila. EMBO J. 2023, 42, e110454. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The Bcl-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less Than a Decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. An Overview of Macroautophagy in Yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef]
- Manent, J.; Banerjee, S.; de Matos Simoes, R.; Zoranovic, T.; Mitsiades, C.; Penninger, J.; Simpson, K.; Humbert, P.; Richardson, H. Autophagy Suppresses Ras-Driven Epithelial Tumourigenesis by Limiting the Accumulation of Reactive Oxygen Species. Oncogene 2017, 36, 5576–5592. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Y.; Neufeld, T.P. An Atg1/Atg13 Complex with Multiple Roles in Tor-Mediated Autophagy Regulation. Mol. Biol. Cell 2009, 20, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging Roles of Atg Proteins and Membrane Lipids in Autophagosome Formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-Phosphate in the Regulation of Autophagy Membrane Dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p Is Required for the Function of the Apg12p-Apg5p Conjugate in the Yeast Autophagy Pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klionsky, D.J. The Regulation of Autophagy—Unanswered Questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Demir, E.; Kacew, S. Drosophila as a Robust Model System for Assessing Autophagy: A Review. Toxics 2023, 11, 682. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-Like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Frappaolo, A.; Giansanti, M.G. Using Drosophila Melanogaster to Dissect the Roles of the Mtor Signaling Pathway in Cell Growth. Cells 2023, 12, 2622. [Google Scholar] [CrossRef] [PubMed]
- Takahara, T.; Maeda, T. Evolutionarily Conserved Regulation of Tor Signalling. J. Biochem. 2013, 154, 1–10. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. Mtor Signaling Pathway and Mtor Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/Pkb by the Rictor-Mtor Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Bian, Y.H.; Xu, J.; Zhao, W.Y.; Zhang, Z.Z.; Tu, L.; Cao, H.; Zhang, Z.G. Targeting Mtorc2 Component Rictor Inhibits Cell Proliferation and Promotes Apoptosis in Gastric Cancer. Am. J. Transl. Res. 2017, 9, 4317–4330. [Google Scholar]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-Mediated Induction of Autophagy Via an Apg1 Protein Kinase Complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-Dependent Mtorc1 Association with the Ulk1-Atg13-Fip200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic Cell Death: The Story of a Misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.-L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.; Blanz, J.; Von Figura, K.; Saftig, P. Accumulation of Autophagic Vacuoles and Cardiomyopathy in Lamp-2-Deficient Mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Abrams, J.M.; White, K.; Fessler, L.I.; Steller, H. Programmed Cell Death During Drosophila Embryogenesis. Development 1993, 117, 29–43. [Google Scholar] [CrossRef]
- Mohseni, N.; McMillan, S.C.; Chaudhary, R.; Mok, J.; Reed, B.H. Autophagy Promotes Caspase-Dependent Cell Death During Drosophila Development. Autophagy 2009, 5, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Cormier, O.; Mohseni, N.; Voytyuk, I.; Reed, B.H. Autophagy Can Promote but Is Not Required for Epithelial Cell Extrusion in the Amnioserosa of the Drosophila Embryo. Autophagy 2012, 8, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Apidianakis, Y.; Rahme, L.G. Drosophila Melanogaster as a Model for Human Intestinal Infection and Pathology. Dis. Models Mech. 2011, 4, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Younossi-Hartenstein, A.; Ortiz, P.A.; Hartenstein, V. A Novel Tissue in an Established Model System: The Drosophila Pupal Midgut. Dev. Genes Evol. 2011, 221, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Zeng, X.; Zheng, Z.; Hou, S.X. The Adult Drosophila Gastric and Stomach Organs Are Maintained by a Multipotent Stem Cell Pool at the Foregut/Midgut Junction in the Cardia (Proventriculus). Cell Cycle 2011, 10, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- LaJeunesse, D.R.; Johnson, B.; Presnell, J.S.; Catignas, K.K.; Zapotoczny, G. Peristalsis in the Junction Region of the Drosophila Larval Midgut Is Modulated by Dh31 Expressing Enteroendocrine Cells. BMC Physiol. 2010, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Shravage, B.; Simin, R.; Mills, K.; Berry, D.L.; Baehrecke, E.H.; Kumar, S. Autophagy, Not Apoptosis, Is Essential for Midgut Cell Death in Drosophila. Curr. Biol. 2009, 19, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Yin, V.P.; Thummel, C.S. Mechanisms of Steroid-Triggered Programmed Cell Death in Drosophila. Proc. Semin. Cell Dev. Biol. 2005, 16, 237–243. [Google Scholar] [CrossRef]
- Jiang, C.; Baehrecke, E.H.; Thummel, C.S. Steroid Regulated Programmed Cell Death During Drosophila Metamorphosis. Development 1997, 124, 4673–4683. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Steroid Regulation of Programmed Cell Death During Drosophila Development. Cell Death Differ. 2000, 7, 1057–1062. [Google Scholar] [CrossRef]
- Berry, D.L.; Baehrecke, E.H. Growth Arrest and Autophagy Are Required for Salivary Gland Cell Degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, X.; Xi, Y. Fat Body Remodeling and Homeostasis Control in Drosophila. Life Sci. 2016, 167, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and Regulation of Starvation-Induced Autophagy in the Drosophila Fat Body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Aguila, J.R.; Suszko, J.; Gibbs, A.G.; Hoshizaki, D.K. The Role of Larval Fat Cells in Adult Drosophila Melanogaster. J. Exp. Biol. 2007, 210, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.C.; Juhász, G.; Neufeld, T.P. Direct Induction of Autophagy by Atg1 Inhibits Cell Growth and Induces Apoptotic Cell Death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Poovathumkadavil, P.; Jagla, K. Genetic Control of Muscle Diversification and Homeostasis: Insights from Drosophila. Cells 2020, 9, 1543. [Google Scholar] [CrossRef]
- Kimura, K.; Truman, J. Postmetamorphic Cell Death in the Nervous and Muscular Systems of Drosophila Melanogaster. J. Neurosci. 1990, 10, 403–411. [Google Scholar] [CrossRef]
- Roy, S.; VijayRaghavan, K. Patterning Muscles Using Organizers: Larval Muscle Templates and Adult Myoblasts Actively Interact to Pattern the Dorsal Longitudinal Flight Muscles of Drosophila. J. Cell Biol. 1998, 141, 1135–1145. [Google Scholar] [CrossRef]
- Kuleesha, Y.; Puah, W.C.; Wasser, M. Live Imaging of Muscle Histolysis in Drosophila Metamorphosis. BMC Dev. Biol. 2016, 16, 12. [Google Scholar] [CrossRef]
- Wasser, M.; Bte Osman, Z.; Chia, W. East and Chromator Control the Destruction and Remodeling of Muscles During Drosophila Metamorphosis. Dev. Biol. 2007, 307, 380–393. [Google Scholar] [CrossRef]
- Zirin, J.; Cheng, D.; Dhanyasi, N.; Cho, J.; Dura, J.-M.; VijayRaghavan, K.; Perrimon, N. Ecdysone Signaling at Metamorphosis Triggers Apoptosis of Drosophila Abdominal Muscles. Dev. Biol. 2013, 383, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Vishal, K.; Bawa, S.; Brooks, D.; Bauman, K.; Geisbrecht, E.R. Thin Is Required for Cell Death in the Drosophila Abdominal Muscles by Targeting Diap1. Cell Death Dis. 2018, 9, 740. [Google Scholar] [CrossRef] [PubMed]
- Homem, C.C.F.; Knoblich, J.A. Drosophila Neuroblasts: A Model for Stem Cell Biology. Development 2012, 139, 4297–4310. [Google Scholar] [CrossRef] [PubMed]
- Lin, S. The Making of the Drosophila Mushroom Body. Front. Physiol. 2023, 14, 1091248. [Google Scholar] [CrossRef] [PubMed]
- Pahl, M.C.; Doyle, S.E.; Siegrist, S.E. E93 Integrates Neuroblast Intrinsic State with Developmental Time to Terminate Mb Neurogenesis Via Autophagy. Curr. Biol. 2019, 29, 750–762.e753. [Google Scholar] [CrossRef]
- Hudson, A.M.; Cooley, L. Methods for Studying Oogenesis. Methods 2014, 68, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Lehmann, R. Drosophila Oogenesis: Versatile Spn Doctors. Curr. Biol. 1999, 9, R55–R58. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stravopodis, D.J.; Papassideri, I.; Robert-Nicoud, M.; Margaritis, L.H. Stage-Specific Apoptotic Patterns During Drosophila Oogenesis. Eur. J. Cell Biol. 2000, 79, 610–620. [Google Scholar] [CrossRef]
- Peterson, J.S.; Barkett, M.; McCall, K. Stage-Specific Regulation of Caspase Activity in Drosophila Oogenesis. Dev. Biol. 2003, 260, 113–123. [Google Scholar] [CrossRef]
- Hou, Y.-C.C.; Chittaranjan, S.; Barbosa, S.G.; McCall, K.; Gorski, S.M. Effector Caspase Dcp-1 and Iap Protein Bruce Regulate Starvation-Induced Autophagy During Drosophila Melanogaster Oogenesis. J. Cell Biol. 2008, 182, 1127–1139. [Google Scholar] [CrossRef]
- Laundrie, B.; Peterson, J.S.; Baum, J.S.; Chang, J.C.; Fileppo, D.; Thompson, S.R.; McCall, K. Germline Cell Death Is Inhibited by P-Element Insertions Disrupting the Dcp-1/Pita Nested Gene Pair in Drosophila. Genetics 2003, 165, 1881–1888. [Google Scholar] [CrossRef]
- Nezis, I.P.; Shravage, B.V.; Sagona, A.P.; Johansen, T.; Baehrecke, E.H.; Stenmark, H. Autophagy as a Trigger for Cell Death: Autophagic Degradation of Inhibitor of Apoptosis dBruce Controls DNA Fragmentation During Late Oogenesis in Drosophila. Autophagy 2010, 6, 1214–1215. [Google Scholar] [CrossRef]
- Riddiford, L.M. Hormones and Drosophila Development. Dev. Drosoph. Melanogaster 1993, 2, 899–939. [Google Scholar]
- Nakagawa, Y.; Sonobe, H. Subchapter 98a—20-Hydroxyecdysone. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 560–563. [Google Scholar]
- Berreur, P.; Porcheron, P.; Moriniere, M.; Berreur-Bonnenfant, J.; Belinski-Deutsch, S.; Busson, D.; Lamour-Audit, C. Ecdysteroids During the Third Larval Instar in 1(3)Ecd-1ts, a Temperature-Sensitive Mutant of Drosophila Melanogaster. Gen. Comp. Endocrinol. 1984, 54, 76–84. [Google Scholar] [CrossRef]
- Nicolson, S.; Denton, D.; Kumar, S. Ecdysone-Mediated Programmed Cell Death in Drosophila. The Int J Dev Biol. 2015, 59, 23–32. [Google Scholar] [CrossRef]
- Kannangara, J.R.; Mirth, C.K.; Warr, C.G. Regulation of Ecdysone Production in Drosophila by Neuropeptides and Peptide Hormones. Open Biol. 2021, 11, 200373. [Google Scholar] [CrossRef] [PubMed]
- Thummel, C.S. Molecular Mechanisms of Developmental Timing in C. elegans and Drosophila. Dev. Cell 2001, 1, 453–465. [Google Scholar] [CrossRef]
- Xu, T.; Jiang, X.; Denton, D.; Kumar, S. Ecdysone Controlled Cell and Tissue Deletion. Cell Death Differ. 2020, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Baehrecke, E.H. Steroid Regulation of Autophagic Programmed Cell Death During Development. Development 2001, 128, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Wendel, D.P.; Reid, P.; Lam, G.; Thummel, C.S.; Baehrecke, E.H. E93 Directs Steroid-Triggered Programmed Cell Death in Drosophila. Mol. Cell 2000, 6, 433–443. [Google Scholar] [CrossRef]
- Duncan, D.M.; Kiefel, P.; Duncan, I. Mutants for Drosophila Isocitrate Dehydrogenase 3b Are Defective in Mitochondrial Function and Larval Cell Death. G3 Genes Genomes Genet. 2017, 7, 789–799. [Google Scholar] [CrossRef]
- Lam, G.; Nam, H.J.; Velentzas, P.D.; Baehrecke, E.H.; Thummel, C.S. Drosophila E93 Promotes Adult Development and Suppresses Larval Responses to Ecdysone During Metamorphosis. Dev. Biol. 2022, 481, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Lindmo, K.; Juhász, G.; Sass, M.; Seglen, P.O.; Brech, A.; Stenmark, H. Programmed Autophagy in the Drosophila Fat Body Is Induced by Ecdysone through Regulation of the Pi3k Pathway. Dev. Cell 2004, 7, 179–192. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Li, S. E93 Predominantly Transduces 20-Hydroxyecdysone Signaling to Induce Autophagy and Caspase Activity in Drosophila Fat Body. Insect Biochem. Mol. Biol. 2014, 45, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Xu, T.; Dayan, S.; Nicolson, S.; Kumar, S. Dpp Regulates Autophagy-Dependent Midgut Removal and Signals to Block Ecdysone Production. Cell Death Differ. 2019, 26, 763–778. [Google Scholar] [CrossRef]
- Tracy, K.; Baehrecke, E.H. The Role of Autophagy in Drosophila Metamorphosis. Curr. Top. Dev. Biol. 2013, 103, 101–125. [Google Scholar] [PubMed]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of Life-Span by Loss of Chico, a Drosophila Insulin Receptor Substrate Protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef]
- Böhni, R.; Riesgo-Escovar, J.; Oldham, S.; Brogiolo, W.; Stocker, H.; Andruss, B.F.; Beckingham, K.; Hafen, E. Autonomous Control of Cell and Organ Size by Chico, a Drosophila Homolog of Vertebrate Irs1-4. Cell 1999, 97, 865–875. [Google Scholar] [CrossRef]
- Kozma, S.C.; Thomas, G. Regulation of Cell Size in Growth, Development and Human Disease: Pi3k, Pkb and S6k. BioEssays 2002, 24, 65–71. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and Function of 3-Phosphorylated Inositol Lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The Two Torcs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Nicolson, S.; Denton, D.; Kumar, S. Distinct Requirements of Autophagy-Related Genes in Programmed Cell Death. Cell Death Differ. 2015, 22, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Chang, T.K.; Nicolson, S.; Shravage, B.; Simin, R.; Baehrecke, E.H.; Kumar, S. Relationship between Growth Arrest and Autophagy in Midgut Programmed Cell Death in Drosophila. Cell Death Differ. 2012, 19, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Baehrecke, E.H. Caspases Function in Autophagic Programmed Cell Death in Drosophila. Development 2004, 131, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.V.G.; Irvine, K.D. The Fat and Warts Signaling Pathways: New Insights into Their Regulation, Mechanism and Conservation. Development 2008, 135, 2827–2838. [Google Scholar] [CrossRef]
- Dutta, S.; Baehrecke, E.H. Warts Is Required for Pi3k-Regulated Growth Arrest, Autophagy, and Autophagic Cell Death in Drosophila. Curr. Biol. 2008, 18, 1466–1475. [Google Scholar] [CrossRef]
- Ferguson, E.L.; Anderson, K.V. Decapentaplegic Acts as a Morphogen to Organize Dorsal-Ventral Pattern in the Drosophila Embryo. Cell 1992, 71, 451–461. [Google Scholar] [CrossRef]
- Akiyama, T.; Gibson, M.C. Decapentaplegic and Growth Control in the Developing Drosophila Wing. Nature 2015, 527, 375–378. [Google Scholar] [CrossRef]
- Affolter, M.; Marty, T.; Vigano, M.A.; Jaźwińska, A. Nuclear Interpretation of Dpp Signaling in Drosophila. EMBO J. 2001, 20, 3298–3305. [Google Scholar] [CrossRef]
- Marty, T.; Müller, B.; Basler, K.; Affolter, M. Schnurri Mediates Dpp-Dependent Repression of Brinker Transcription. Nat. Cell Biol. 2000, 2, 745–749. [Google Scholar] [CrossRef]
- Minami, M.; Kinoshita, N.; Kamoshida, Y.; Tanimoto, H.; Tabata, T. Brinker Is a Target of Dpp in Drosophila That Negatively Regulates Dpp-Dependent Genes. Nature 1999, 398, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. Parp-1 Cleavage Fragments: Signatures of Cell-Death Proteases in Neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular Characterization of Mitochondrial Apoptosis-Inducing Factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(Adp-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Tarayrah-Ibraheim, L.; Maurice, E.C.; Hadary, G.; Ben-Hur, S.; Kolpakova, A.; Braun, T.; Peleg, Y.; Yacobi-Sharon, K.; Arama, E. Dnase Ii Mediates a Parthanatos-Like Developmental Cell Death Pathway in Drosophila Primordial Germ Cells. Nat. Commun. 2021, 12, 2285. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. Htra2 Promotes Cell Death through Its Serine Protease Activity and Its Ability to Antagonize Inhibitor of Apoptosis Proteins. J. Biol. Chem. 2002, 277, 445–454. [Google Scholar] [CrossRef]
- van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The Serine Protease Omi/Htra2 Is Released from Mitochondria During Apoptosis. Omi Interacts with Caspase-Inhibitor Xiap and Induces Enhanced Caspase Activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef]
- Martins, L.M.; Iaccarino, I.; Tenev, T.; Gschmeissner, S.; Totty, N.F.; Lemoine, N.R.; Savopoulos, J.; Gray, C.W.; Creasy, C.L.; Dingwall, C.; et al. The Serine Protease Omi/Htra2 Regulates Apoptosis by Binding Xiap through a Reaper-Like Motif. J. Biol. Chem. 2002, 277, 439–444. [Google Scholar] [CrossRef]
- Zohar-Fux, M.; Ben-Hamo-Arad, A.; Arad, T.; Volin, M.; Shklyar, B.; Hakim-Mishnaevski, K.; Porat-Kuperstein, L.; Kurant, E.; Toledano, H. The Phagocytic Cyst Cells in Drosophila Testis Eliminate Germ Cell Progenitors Via Phagoptosis. Sci. Adv. 2022, 8, eabm4937. [Google Scholar] [CrossRef]
- Yang, H.; Yamashita, Y.M. The Regulated Elimination of Transit-Amplifying Cells Preserves Tissue Homeostasis During Protein Starvation in Drosophila Testis. Development 2015, 142, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Yacobi-Sharon, K.; Namdar, Y.; Arama, E. Alternative Germ Cell Death Pathway in Drosophila Involves Htra2/Omi, Lysosomes, and a Caspase-9 Counterpart. Dev. Cell 2013, 25, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.L.; Yamashita, Y.M. Germ Cell Connectivity Enhances Cell Death in Response to DNA Damage in the Drosophila Testis. Elife 2017, 6, e27960. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Denton, D.; O’Keefe, L.; Kumar, S. Drosophila as a Model to Understand Autophagy Deregulation in Human Disorders. Prog. Mol. Biol. Transl. Sci. 2020, 172, 375–409. [Google Scholar]
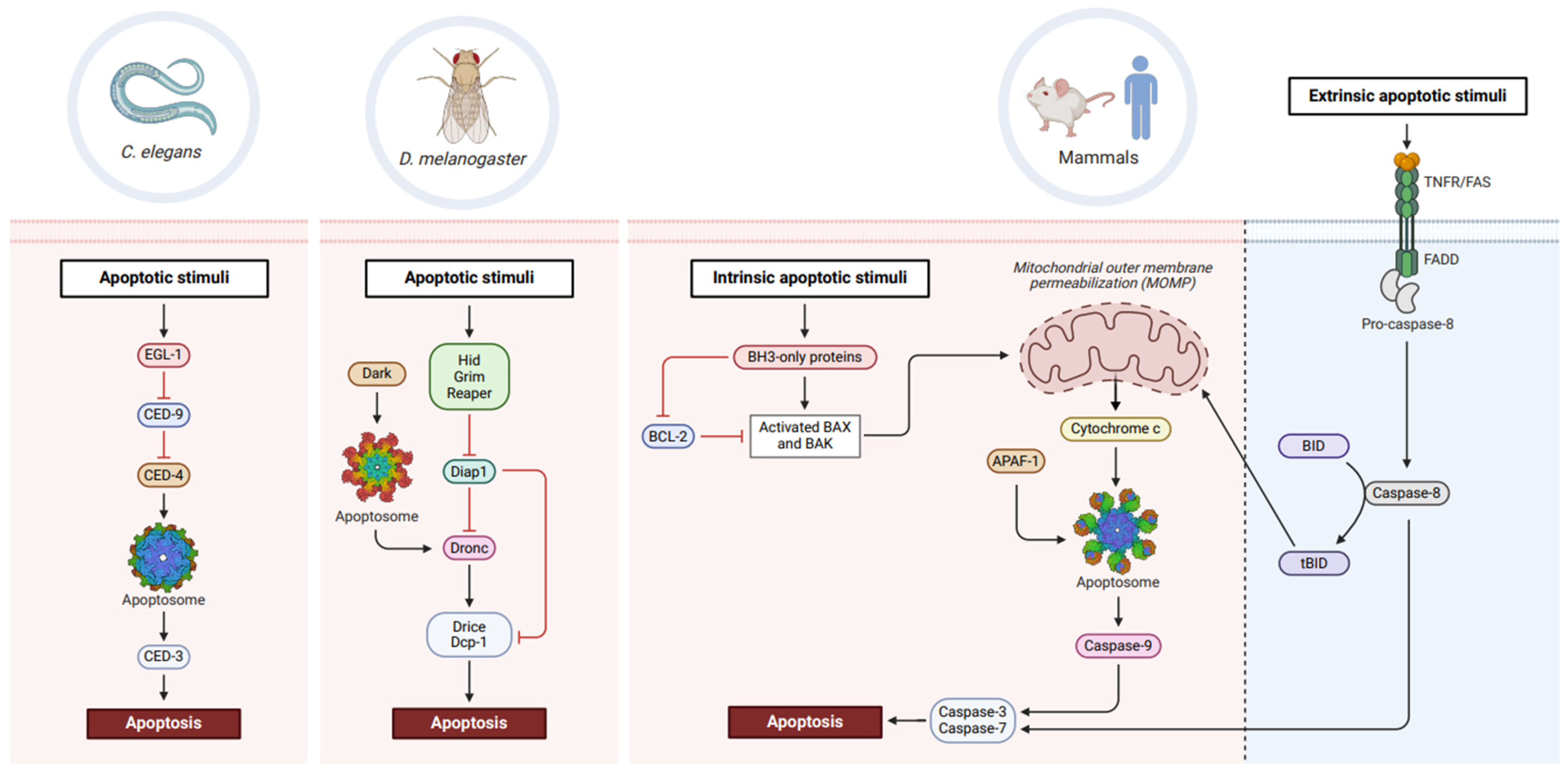
Figure 1.
Conservation of apoptotic machinery across phylogeny. In C. elegans, the BH3-only protein, EGL-1, inhibits the prosurvival factor, CED-9. This releases CED-4 which forms the apoptosome and promotes autocatalytic cleavage of CED-3 into an active caspase, thereby eventuating in cell death. Drosophila apoptosis proceeds with RHG proteins inhibiting Diap1, leading to Dark apoptosome-mediated activation of Dronc. Activation of downstream effector caspases, Drice and Dcp-1, signals cellular demise. In mammals, BH3-only proteins inhibit BCL-2 prosurvival activity, allowing activated BAX/BAK proteins to trigger mitochondrial outer membrane permeabilisation (MOMP). Cytochrome c release coordinates APAF-1 apoptosome formation and enables caspase-9 activation, subsequently promoting cell death in a caspase-3 and -7-dependent manner. Extrinsic lethal stimuli recognised by death receptors (TNFR, FAS) activate caspase-8, leading to caspase-3 and -7 activation and cell death. Truncation of BID to tBID by caspase-8 can also trigger MOMP and feed into intrinsic apoptotic pathways. Created with BioRender.com.
Figure 1.
Conservation of apoptotic machinery across phylogeny. In C. elegans, the BH3-only protein, EGL-1, inhibits the prosurvival factor, CED-9. This releases CED-4 which forms the apoptosome and promotes autocatalytic cleavage of CED-3 into an active caspase, thereby eventuating in cell death. Drosophila apoptosis proceeds with RHG proteins inhibiting Diap1, leading to Dark apoptosome-mediated activation of Dronc. Activation of downstream effector caspases, Drice and Dcp-1, signals cellular demise. In mammals, BH3-only proteins inhibit BCL-2 prosurvival activity, allowing activated BAX/BAK proteins to trigger mitochondrial outer membrane permeabilisation (MOMP). Cytochrome c release coordinates APAF-1 apoptosome formation and enables caspase-9 activation, subsequently promoting cell death in a caspase-3 and -7-dependent manner. Extrinsic lethal stimuli recognised by death receptors (TNFR, FAS) activate caspase-8, leading to caspase-3 and -7 activation and cell death. Truncation of BID to tBID by caspase-8 can also trigger MOMP and feed into intrinsic apoptotic pathways. Created with BioRender.com.

Figure 2.
The molecular machinery of autophagy. Autophagy is an evolutionarily conserved process comprising (1) intiation, (2) nucleation, (3) expansion, and (4) fusion and degradation. Under basal or optimal cell conditions, mTORC1 remains active and phosphorylates Atg13, thereby preventing recruitment of Atg1. Cellular stressors including nutrient deprivation or energy depletion inactive mTORC1, thereby enabling formation of the Atg1 initiation complex (Atg1, Atg13, Atg17, Atg101). Nucleation occurs upon Atg1-dependent phoshophorylation and recruitment of Atg9, as well as the PtdIns3K complex (VPS15, VPS34, Atg6, Atg14). Expansion of the autophagosome relies on two ubiquitin-like conjugation systems—Atg12 and Atg8a. Conjugation of Atg16 to Atg5-Atg12 forms an E3-like complex that recruits the E2-like Atg3 to facilitate attachment of PE to Atg8a (Atg8a-PE) at the autophagosomal membrane. Fusion of autophagosomes to lysosomes relies on tethering proteins (SNAREs) and lysosomal membrane proteins (RAB7), forming an autolysosome within which lysosomal acid hydrolases degrade sequestered proteins and organelles. Created with BioRender.com.
Figure 2.
The molecular machinery of autophagy. Autophagy is an evolutionarily conserved process comprising (1) intiation, (2) nucleation, (3) expansion, and (4) fusion and degradation. Under basal or optimal cell conditions, mTORC1 remains active and phosphorylates Atg13, thereby preventing recruitment of Atg1. Cellular stressors including nutrient deprivation or energy depletion inactive mTORC1, thereby enabling formation of the Atg1 initiation complex (Atg1, Atg13, Atg17, Atg101). Nucleation occurs upon Atg1-dependent phoshophorylation and recruitment of Atg9, as well as the PtdIns3K complex (VPS15, VPS34, Atg6, Atg14). Expansion of the autophagosome relies on two ubiquitin-like conjugation systems—Atg12 and Atg8a. Conjugation of Atg16 to Atg5-Atg12 forms an E3-like complex that recruits the E2-like Atg3 to facilitate attachment of PE to Atg8a (Atg8a-PE) at the autophagosomal membrane. Fusion of autophagosomes to lysosomes relies on tethering proteins (SNAREs) and lysosomal membrane proteins (RAB7), forming an autolysosome within which lysosomal acid hydrolases degrade sequestered proteins and organelles. Created with BioRender.com.

Figure 3.
Regulatory mechanisms underlying developmental PCD in Drosophila. Ecdysone, produced in the prothoracic glands from dietary cholesterol, circulates in haemolymph and transcriptionally upregulates ecdysone-responsive genes (BrC, E74, E75) in target tissues. Subsequent transcription of autophagy and apoptotic genes coordinates tissue deletion at multiple stages of Drosophila development. Downregulation of PI3K signalling inhibits mTORC1, thereby activating autophagy. Dpp signalling blocks ecdysone production and transcription of ecdysone-response genes, and must therefore be downregulated to trigger ADCD. Created with BioRender.com.
Figure 3.
Regulatory mechanisms underlying developmental PCD in Drosophila. Ecdysone, produced in the prothoracic glands from dietary cholesterol, circulates in haemolymph and transcriptionally upregulates ecdysone-responsive genes (BrC, E74, E75) in target tissues. Subsequent transcription of autophagy and apoptotic genes coordinates tissue deletion at multiple stages of Drosophila development. Downregulation of PI3K signalling inhibits mTORC1, thereby activating autophagy. Dpp signalling blocks ecdysone production and transcription of ecdysone-response genes, and must therefore be downregulated to trigger ADCD. Created with BioRender.com.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Umargamwala, R.; Manning, J.; Dorstyn, L.; Denton, D.; Kumar, S. Understanding Developmental Cell Death Using Drosophila as a Model System. Cells 2024, 13, 347. https://doi.org/10.3390/cells13040347
AMA Style
Umargamwala R, Manning J, Dorstyn L, Denton D, Kumar S. Understanding Developmental Cell Death Using Drosophila as a Model System. Cells. 2024; 13(4):347. https://doi.org/10.3390/cells13040347
Chicago/Turabian StyleUmargamwala, Ruchi, Jantina Manning, Loretta Dorstyn, Donna Denton, and Sharad Kumar. 2024. "Understanding Developmental Cell Death Using Drosophila as a Model System" Cells 13, no. 4: 347. https://doi.org/10.3390/cells13040347
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.