
Cisplatin and Starvation Differently Sensitize Autophagy in Renal Carcinoma: A Potential Therapeutic Pathway to Target Variegated Drugs Resistant Cancerous Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
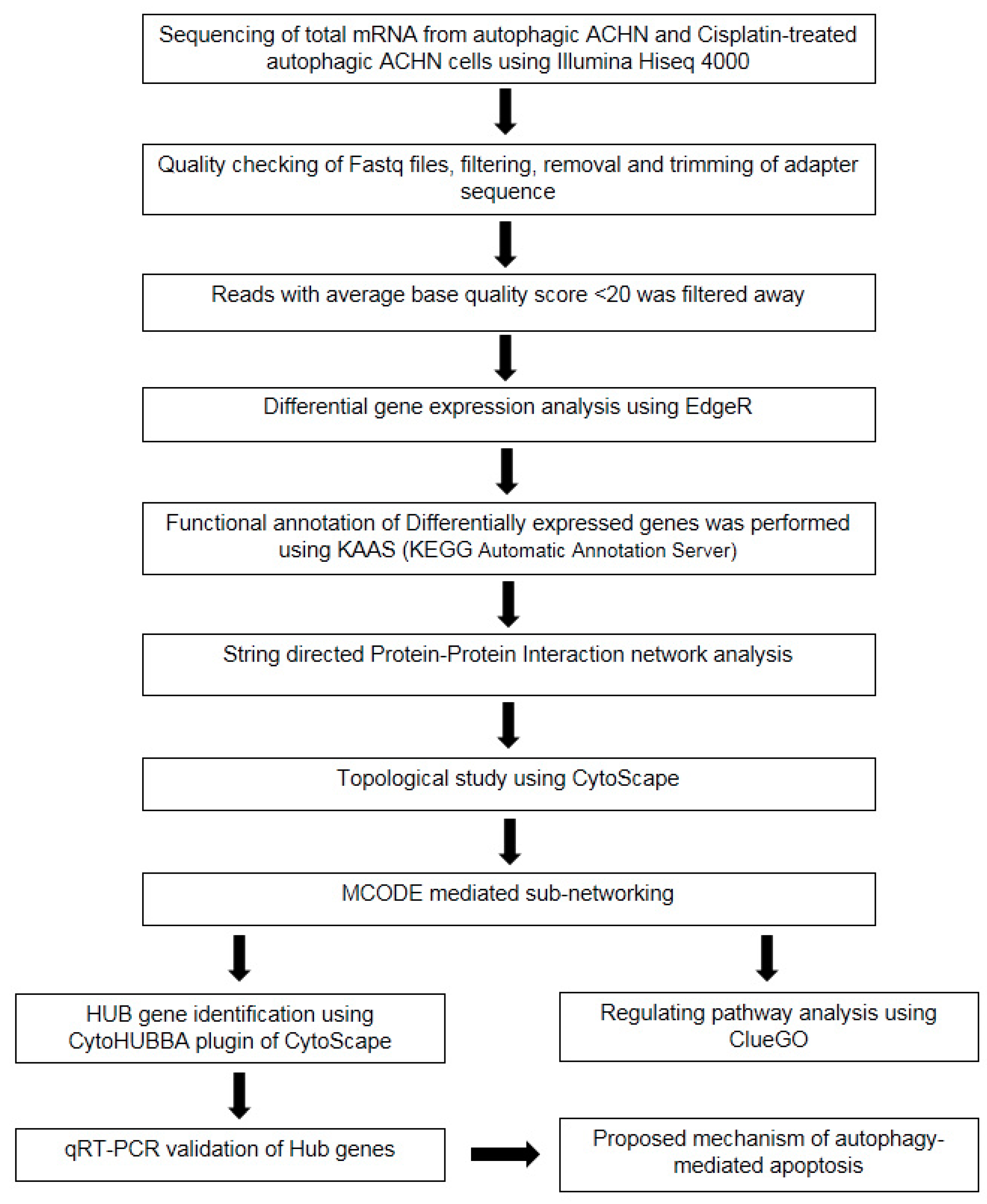
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies and Chemicals
2.3. Autophagy Detection after Complete Nutrient Deprivation via Qualitative and Quantitative Approaches
2.4. Cell Viability Determination by MTT Assay and Trypan Blue Exclusion Assay
2.5. Preparation of Cells for RNA Isolation and High-Throughput Illumina Sequencing
2.6. RNA Sequencing Data Pre-Processing, De Novo Transcriptome Assembly, and Analysis of Differential Gene Expression
2.7. Functional Enrichment Analysis of Identified Differentially Expressed Genes
2.8. Hub Gene Identification among the Upregulated DEGs
2.9. Cross-Validation of Identified Hub Genes by Quantitative Real-Time PCR
2.10. Apoptosis Assay by AnnexinV/FITC-PI Staining
2.11. Statistical Analysis
3. Results
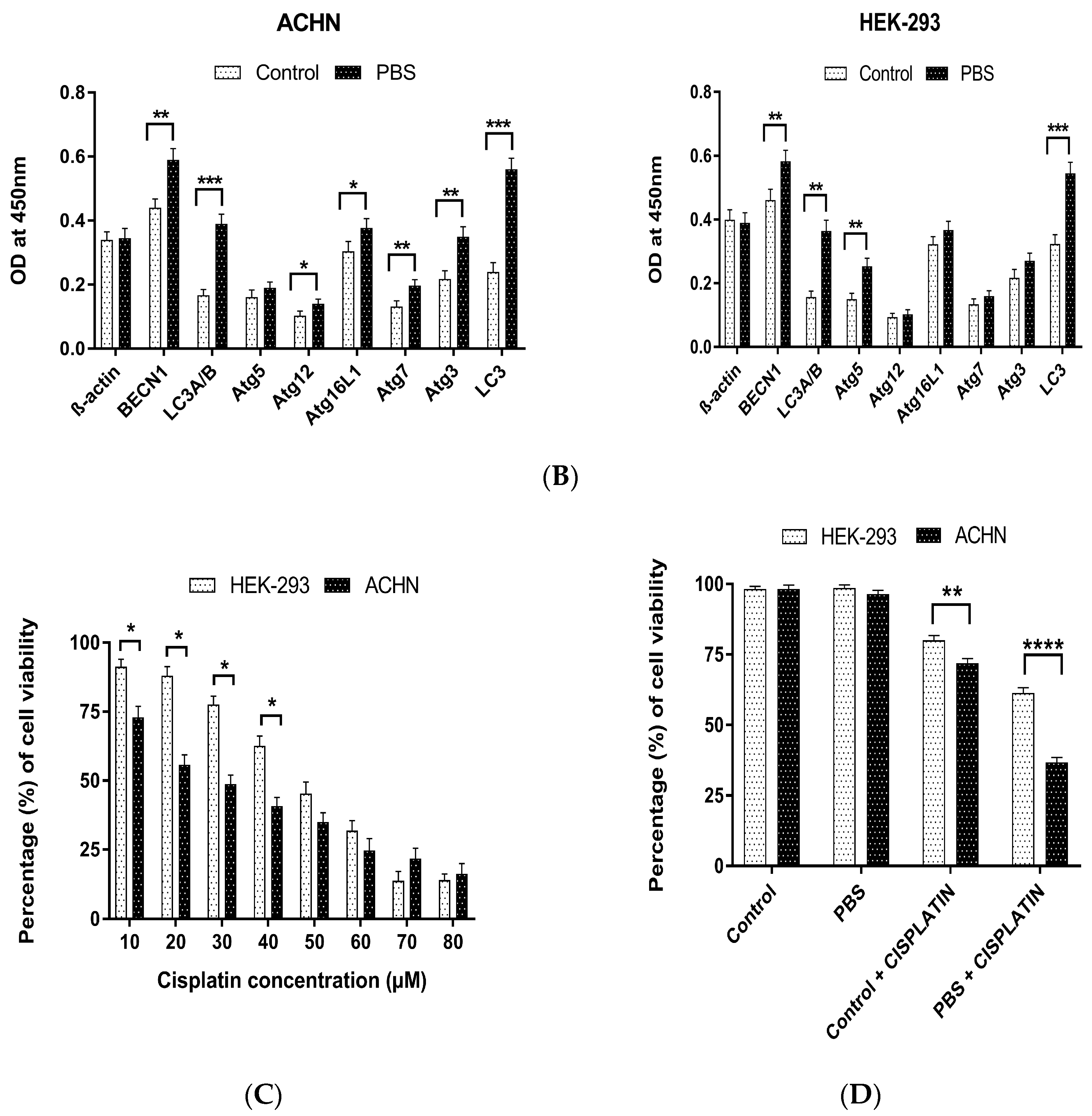
3.1. Effect of Cisplatin under Starvation-Induced Autophagic Environment
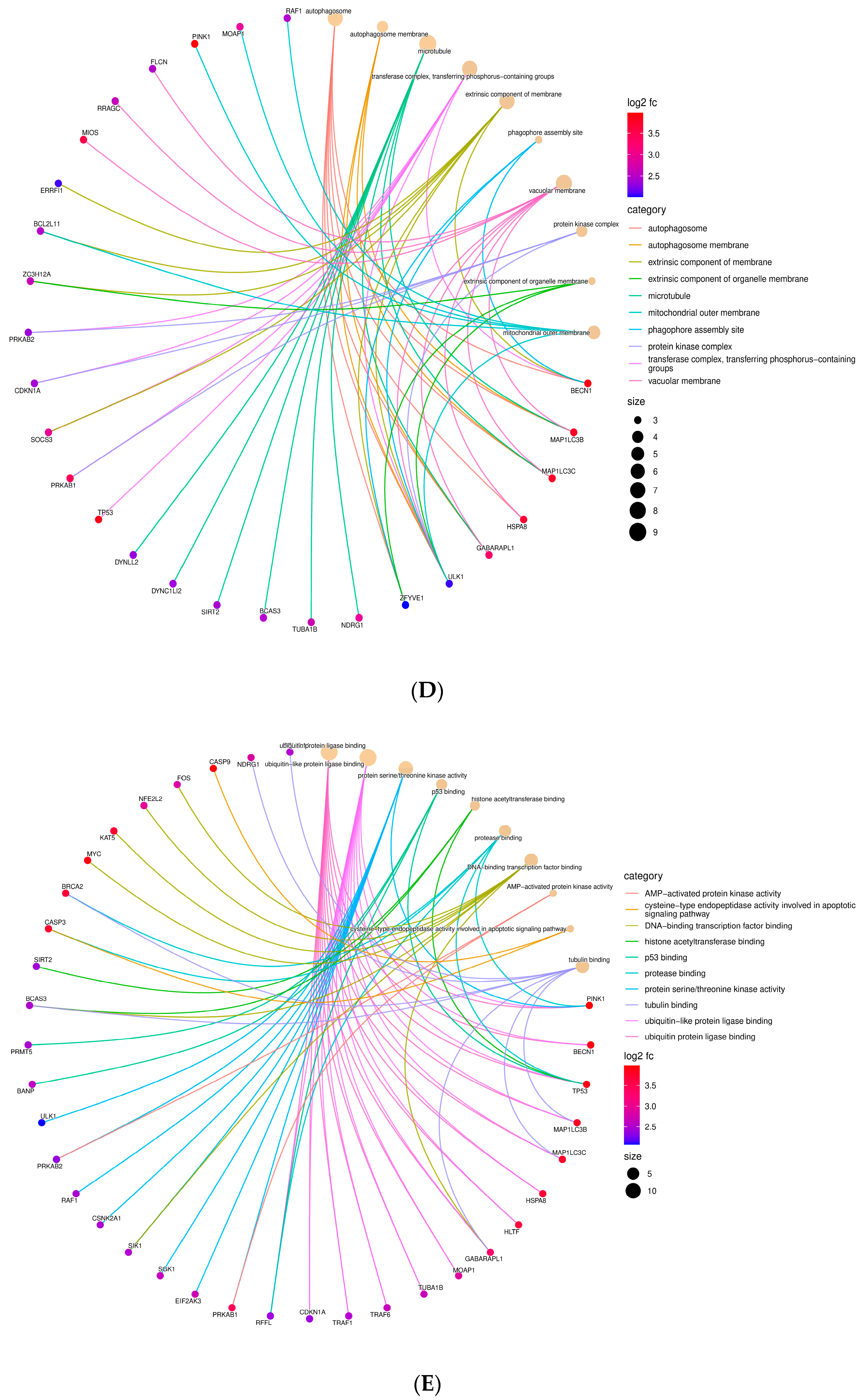
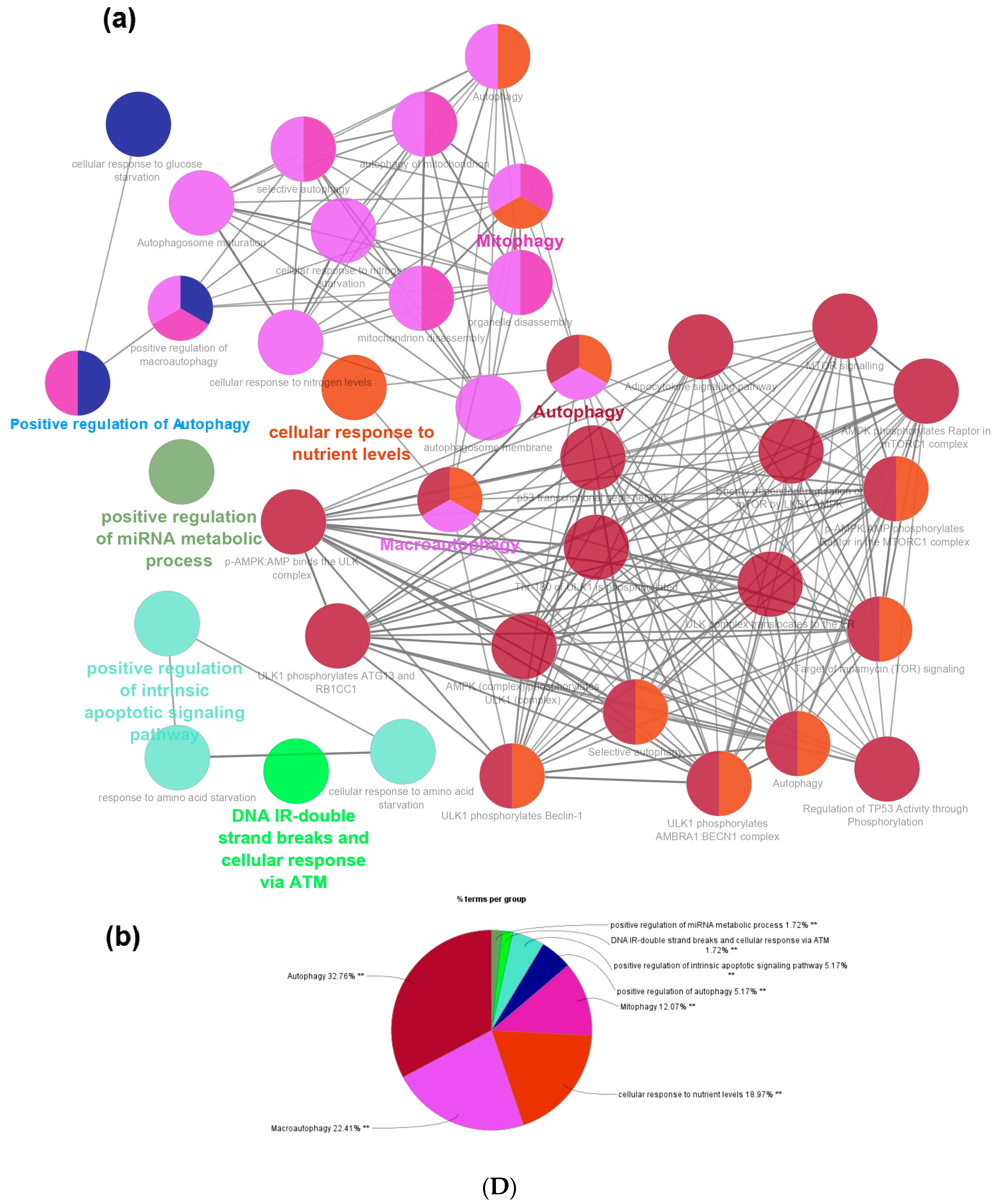
3.2. Identification of Major Biological Pathways Regulated by All Differentially Expressed Genes (DEGs)
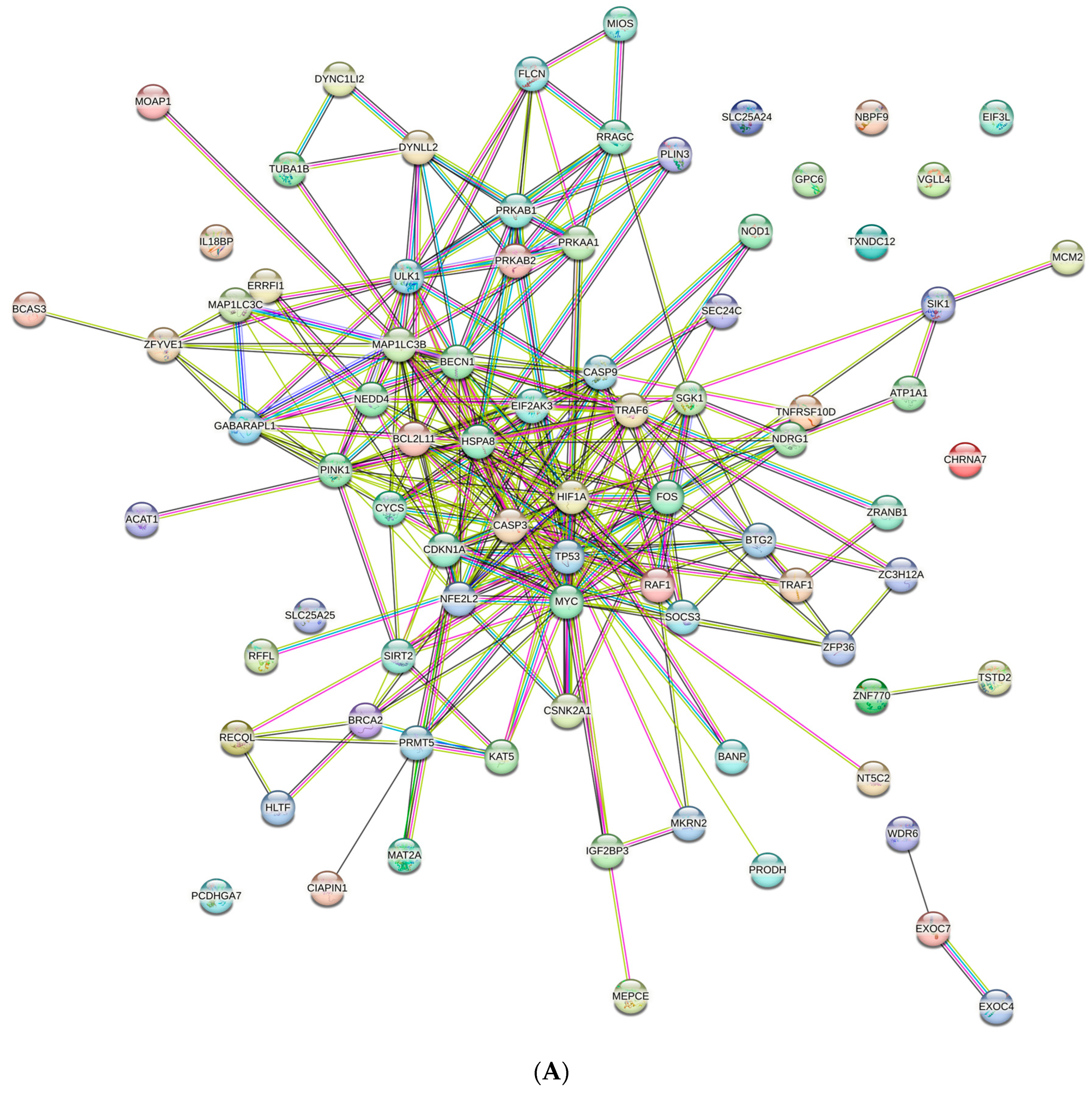
3.3. Protein–Protein Interaction (PPI) Network and Pathway Network Analysis
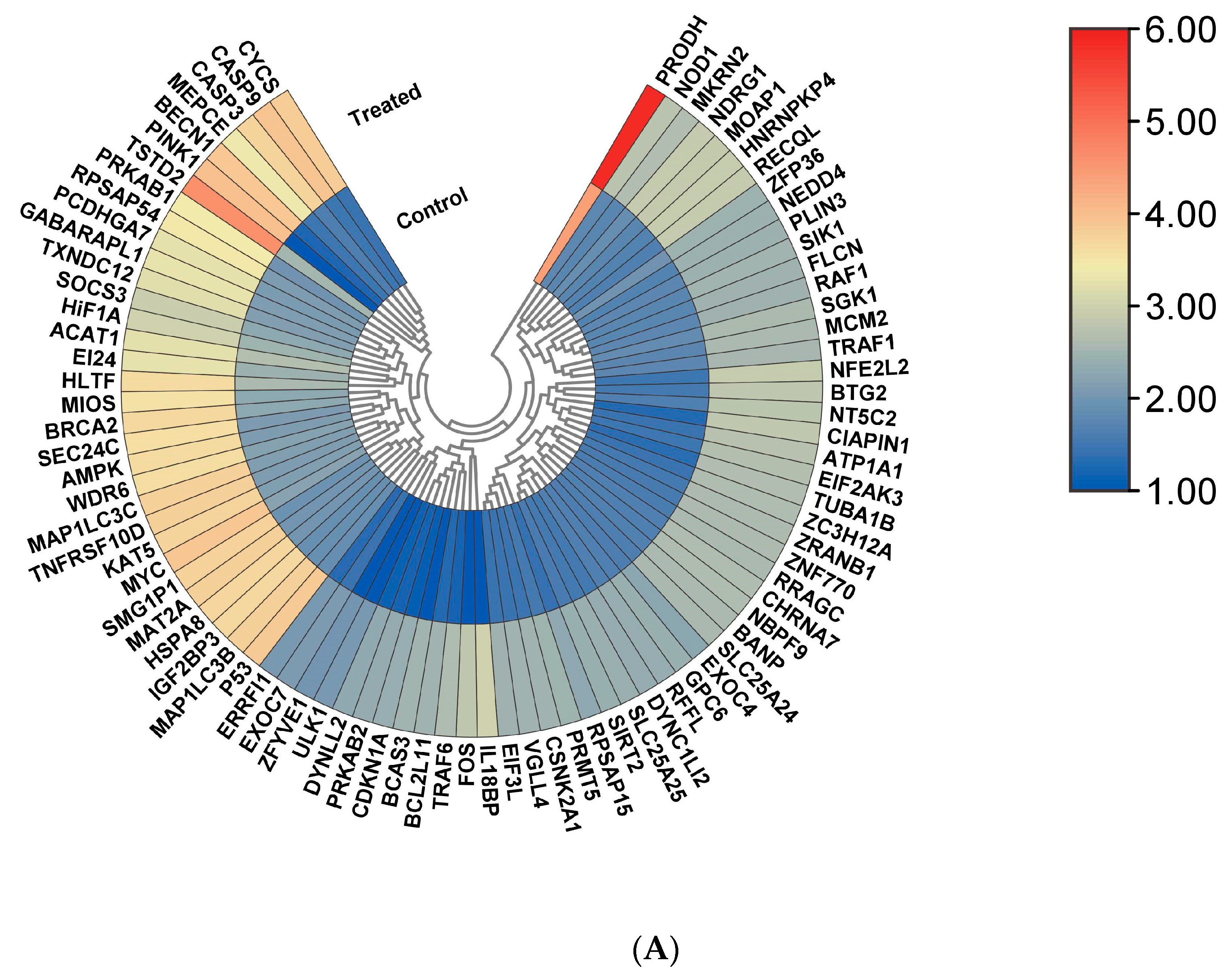
3.4. Major Regulatory Hub Genes Identification by Cytohubba Algorithm and Their Functional Analysis
3.5. Validation of Differentially Expressed Genes by Quantitative Real-Time PCR and Apoptosis Study for Determining Cell Death Induced by Cisplatin under an Autophagic Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bivona, T.G.; Robert, C.D. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Buono, R.; Longo, V.D. Starvation, stress resistance, and cancer. Trends Endocrinol. Metab. 2018, 29, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Pereira, G.J.; Jasiulionis, M.G.; Bincoletto, C.; Smaili, S.S. Nutritional shortage augments cisplatin-effects on murine melanoma cells. Chem. Biol. Interact. 2018, 281, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Omar, E.M.; Omran, G.A.; Mustafa, M.F.; El-Khodary, N.M. Intermittent fasting during adjuvant chemotherapy may promote differential stress resistance in breast cancer patients. J. Egypt. Nat. Cancer Inst. 2022, 34, 38. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, Y. JAK2/STAT3 inhibitor reduced 5-FU resistance and autophagy through ATF6-mediated ER stress. J. Recept. Signal Transduct. 2022, 42, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F. Autophagy modulation for cancer therapy. Cancer Biol. Ther. 2011, 11, 169–176. [Google Scholar] [CrossRef]
- Kim, E.A.; Jang, J.H.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Lee, T.J. MiR-1208 increases the sensitivity to cisplatin by targeting TBCK in renal cancer cells. Int. J. Mol. Sci. 2019, 20, 3540. [Google Scholar] [CrossRef]
- Kim, E.A.; Jang, J.H.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Sohn, H.Y.; Lee, T.J. Neferine increases sensitivities to multiple anticancer drugs via downregulation of Bcl-2 expression in renal cancer cells. Genes Genomics 2022, 44, 165–173. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Cleveland, J.L. Modulating autophagy for therapeutic benefit. Autophagy 2007, 3, 464–467. [Google Scholar] [CrossRef]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef]
- de Gruil, N.; Pijl, H.; van der Burg, S.H.; Kroep, J.R. Short-Term Fasting Synergizes with Solid Cancer Therapy by Boosting Antitumor Immunity. Cancers 2022, 14, 1390. [Google Scholar] [CrossRef]
- Basmaciyan, L.; Berry, L.; Gros, J.; Azas, N.; Casanova, M. Temporal analysis of the autophagic and apoptotic phenotypes in Leishmania parasites. Micro. Cell 2018, 5, 404. [Google Scholar] [CrossRef]
- Guo, S.; Liang, Y.; Murphy, S.F.; Huang, A.; Shen, H.; Kelly, D.F.; Pablo, S.; Sheng, Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015, 11, 560–572. [Google Scholar] [CrossRef]
- Oh, S.H.; Choi, Y.B.; Kim, J.H.; Weihl, C.C.; Ju, J.S. Quantification of autophagy flux using LC3 ELISA. Analyt. Biochem. 2017, 530, 57–67. [Google Scholar] [CrossRef]
- Kruger, N.J. The Bradford method for protein quantitation. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 17–24. [Google Scholar]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 1997, 21, A-3B. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold. Spr. Harb. Protoc. 2010, 20106, pdb-prot5439. [Google Scholar] [CrossRef] [PubMed]
- Takele Assefa, A.; Vandesompele, J.; Thas, O. On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genom. 2020, 21, 312. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acid Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, H.; Yang, Y.; Cheng, C.; Liu, Q. Identification of Potential Hub Genes Related to Aflatoxin B1, Liver Fibrosis and Hepatocellular Carcinoma via Integrated Bioinformatics Analysis. Biology 2023, 12, 205. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acid Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models. Genome Res. 1971, 13, 426. [Google Scholar]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef]
- Xiao, W.H.; Qu, X.L.; Li, X.M.; Sun, Y.L.; Zhao, H.X.; Wang, S.; Zhou, X. Identification of commonly dysregulated genes in colorectal cancer by integrating analysis of RNA-Seq data and qRT-PCR validation. Cancer Gene Ther. 2015, 22, 278–284. [Google Scholar] [CrossRef]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 50, e2597. [Google Scholar]
- Liu, H.; He, Z.; Simon, H.U. Targeting autophagy as a potential therapeutic approach for melanoma therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2013; Volume 23, pp. 352–360. [Google Scholar]
- Nazim, U.M.; Moon, J.H.; Lee, J.H.; Lee, Y.J.; Seol, J.W.; Eo, S.K.; Lee, J.H.; Park, S.Y. Activation of autophagy flux by metformin downregulates cellular FLICE–like inhibitory protein and enhances TRAIL-induced apoptosis. Oncotarget 2016, 7, 23468. [Google Scholar] [CrossRef]
- Sen, S.; Ganguli, S.; Chakraborty, R. What transcriptomics and proteomics can tell us about a high borate perturbed boron tolerant Bacilli strain. Mol. Omics 2023. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Safdie, F.M.; Raffaghello, L.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010, 70, 1564–1572. [Google Scholar] [CrossRef]
- de Groot, S.; Pijl, H.; van der Hoeven, J.J.; Kroep, J.R. Effects of short-term fasting on cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 209. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A.; et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 2015, 6, 11806. [Google Scholar] [CrossRef]
- Shim, H.S.; Wei, M.; Brandhorst, S.; Longo, V.D. Starvation promotes REV1 SUMOylation and p53-dependent sensitization of melanoma and breast cancer cells. Cancer Res. 2015, 75, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Ciro, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3β-MCL-1 axis. Cancer Cell 2019, 35, 798–815. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schindlmaier, K.; Galhuber, M.; Riahi, Z.; Berger, N.; Kupper, N.; Moyschewitz, E.; Auer, M.; Michenthaler, H.; et al. Fasting improves therapeutic response in hepatocellular carcinoma through p53-dependent metabolic synergism. Sci. Adv. 2022, 8, 2635. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Safirstein, R.; Dong, Z. Cisplatin nephrotoxicity: New insights and therapeutic implications. Nat. Rev. Nephrol. 2023, 19, 53–72. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Grasmann, G.; Mondal, A.; Leithner, K. Flexibility and adaptation of cancer cells in a heterogenous metabolic microenvironment. Int. J. Mol. Sci. 2021, 22, 1476. [Google Scholar] [CrossRef]
- Reyes-González, J.M.; Armaiz-Peña, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-MYC in platinum-resistant ovarian cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Grenier, A.; Poulain, L.; Mondesir, J.; Jacquel, A.; Bosc, C.; Stuani, L.; Mouche, S.; Larrue, C.; Sahal, A.; Birsen, R.; et al. AMPK-PERK axis represses oxidative metabolism and enhances apoptotic priming of mitochondria in acute myeloid leukemia. Cell Rep. 2022, 38, 110197. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Das, D.N.; Panda, P.K.; Sinha, N.; Naik, P.P.; Bissoyi, A.; Pramanik, K.; Bhutia, S.K. Autophagy protein Ulk1 promotes mitochondrial apoptosis through reactive oxygen species. Free Radic. Biol. Med. 2015, 89, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xiang, X.; Li, S.; Xie, P.; Gong, Q.; Goh, B.C.; Wang, L. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022; Volume 80, pp. 379–390. [Google Scholar]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Casola, S.; Foiani, M.; Pietrantonio, F.; de Braud, F.; Longo, V. Targeting cancer metabolism: Dietary and pharmacologic interventions. Cancer Discov. 2016, 6, 1315–1333. [Google Scholar] [CrossRef] [PubMed]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 2332. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, A.; Thakur, S.; Dey, D.K.; Kumar, A. Cisplatin and Starvation Differently Sensitize Autophagy in Renal Carcinoma: A Potential Therapeutic Pathway to Target Variegated Drugs Resistant Cancerous Cells. Cells 2024, 13, 471. https://doi.org/10.3390/cells13060471
Dutta A, Thakur S, Dey DK, Kumar A. Cisplatin and Starvation Differently Sensitize Autophagy in Renal Carcinoma: A Potential Therapeutic Pathway to Target Variegated Drugs Resistant Cancerous Cells. Cells. 2024; 13(6):471. https://doi.org/10.3390/cells13060471
Chicago/Turabian StyleDutta, Ankita, Subarna Thakur, Debasish Kumar Dey, and Anoop Kumar. 2024. "Cisplatin and Starvation Differently Sensitize Autophagy in Renal Carcinoma: A Potential Therapeutic Pathway to Target Variegated Drugs Resistant Cancerous Cells" Cells 13, no. 6: 471. https://doi.org/10.3390/cells13060471