
Fluvastatin Converts Human Macrophages into Foam Cells with Increased Inflammatory Response to Inactivated Mycobacterium tuberculosis H37Ra
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mtb Strain Selection
2.2. Isolation of Peripheral Blood Mononuclear Cells (PBMC) and Culture Conditions
2.3. Transmission Electron Microscopy
2.4. Immunolocalization of NLRP3 and ASC
2.5. FAM-FLICA Assay
2.6. Dynamics of Caspase-1 Activation
2.7. Kinetic Analysis of Cytokines Emission
2.8. Lactate Dehydrogenase (LDH) Determination
2.9. Contribution of IL-12, IL-1β, and IL-18 to IFNγ Production
2.10. Intracellular IFNγ Determination by Flow Cytometry
2.11. Rabbit Treatment and Immunization
2.12. Statistical Analysis
3. Results
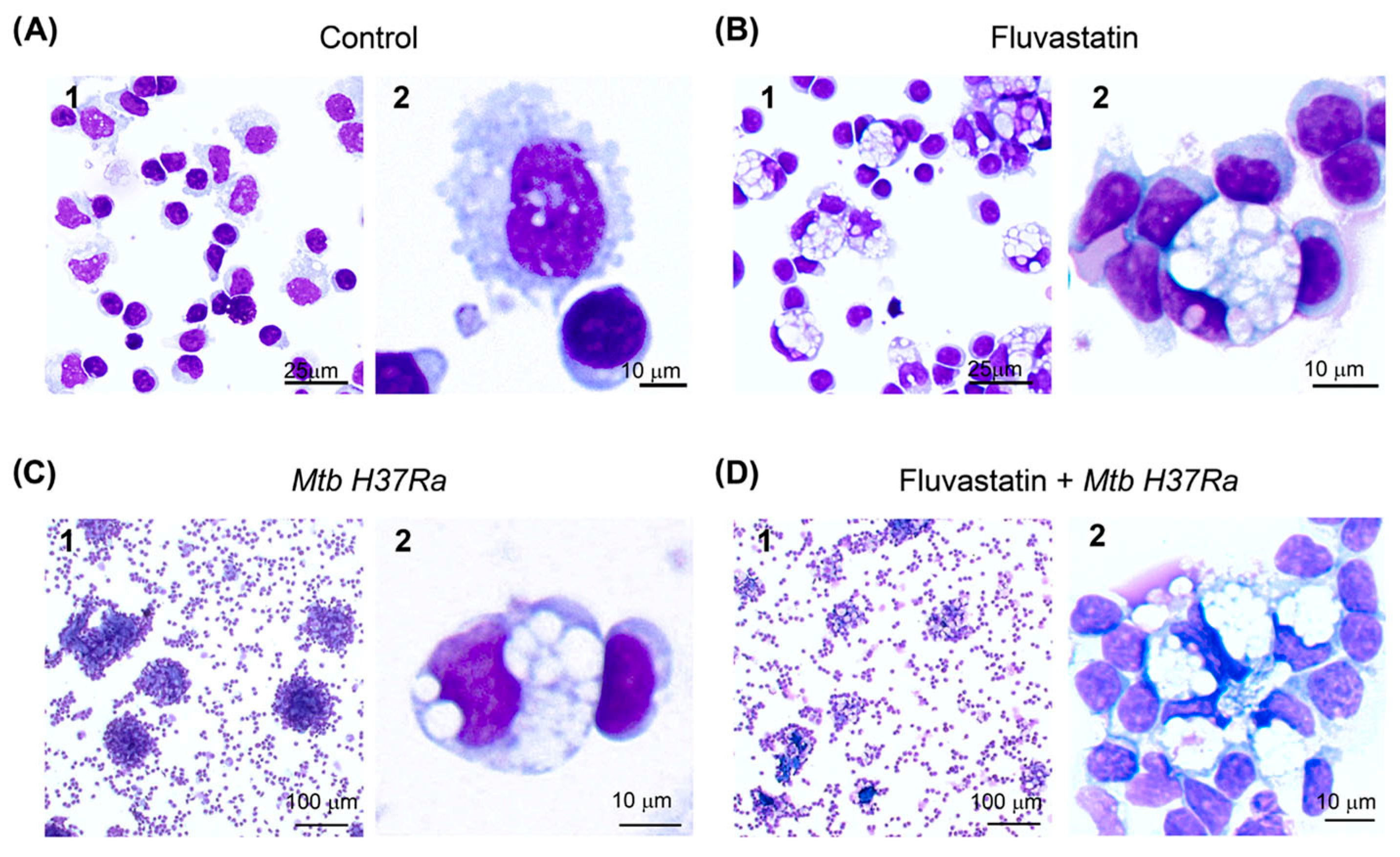
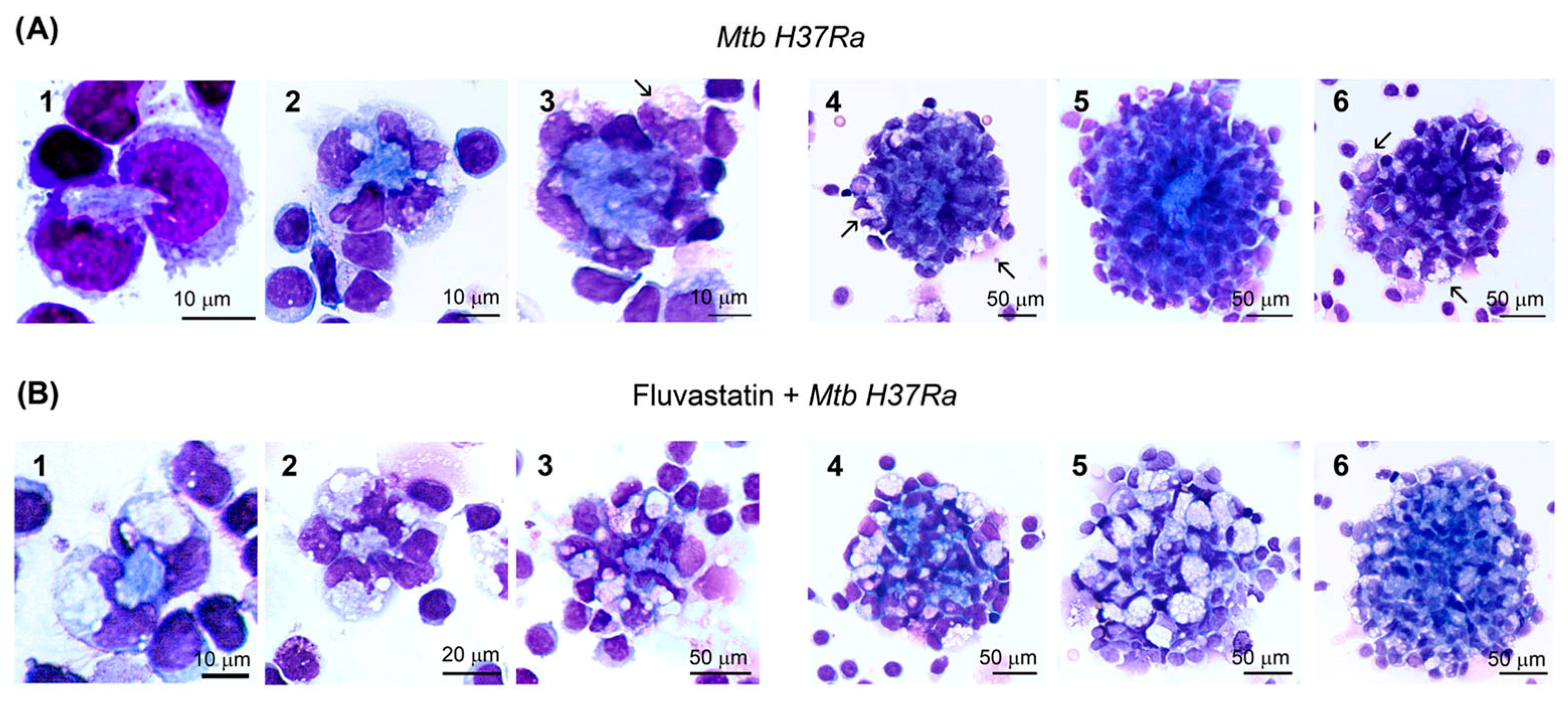
3.1. Fluvastatin Converts Macrophages to Foamy Cells Unable to Form Granulomatous Structures in Response to Inactivated Mtb H37Ra
3.2. Ultrastructural Study of Macrophages
3.3. FFMs Overproduce NLRP3 and ASC and the Bacteria Promotes the Co-Localization of Both Proteins
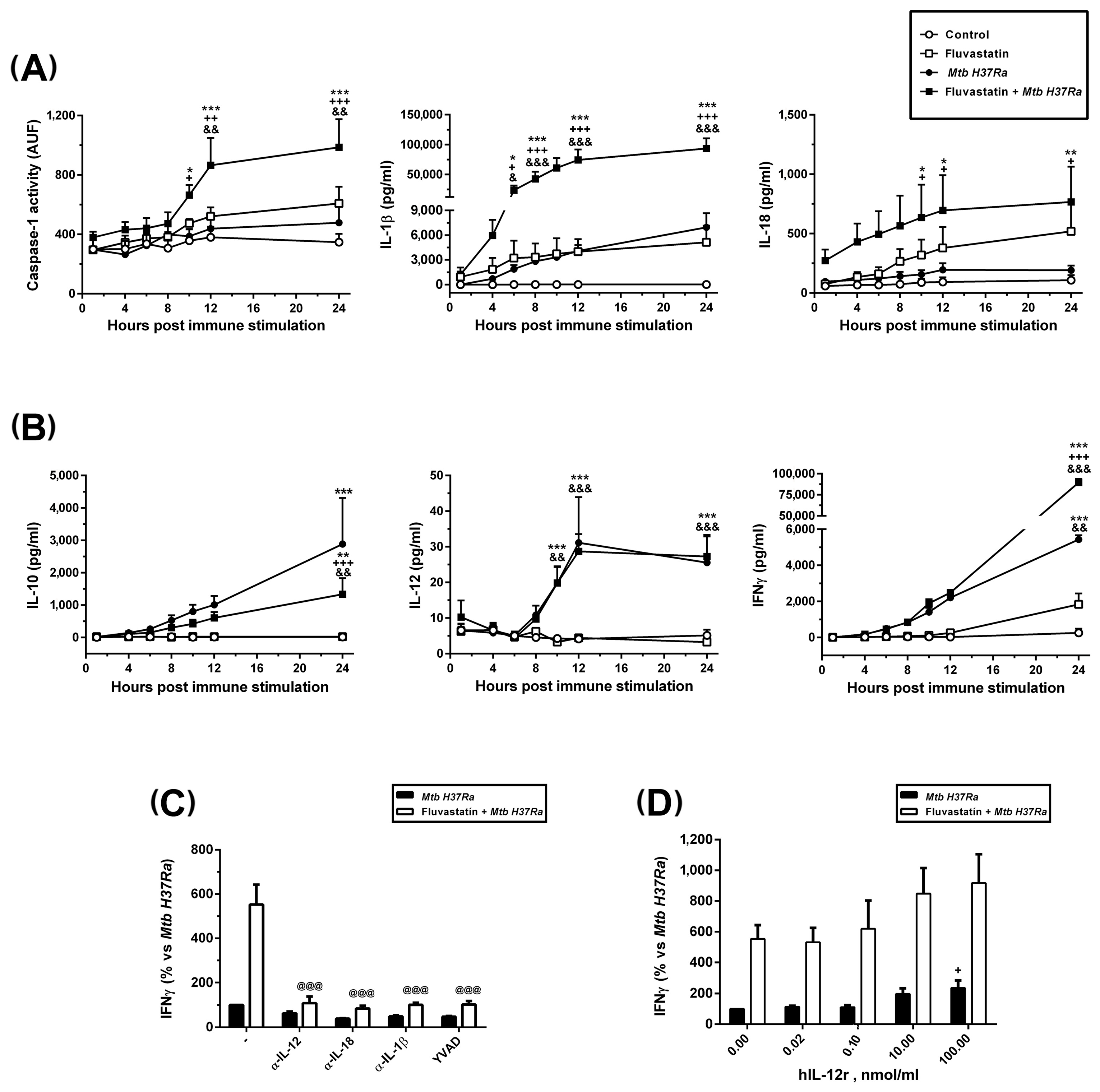
3.4. Fluvastatin Exacerbates Caspase-1 Activation in Response to Mtb PAMPs
3.5. Fluvastatin Exacerbates the Cellular Response against Inactivated Mtb H37Ra
4. Discussion
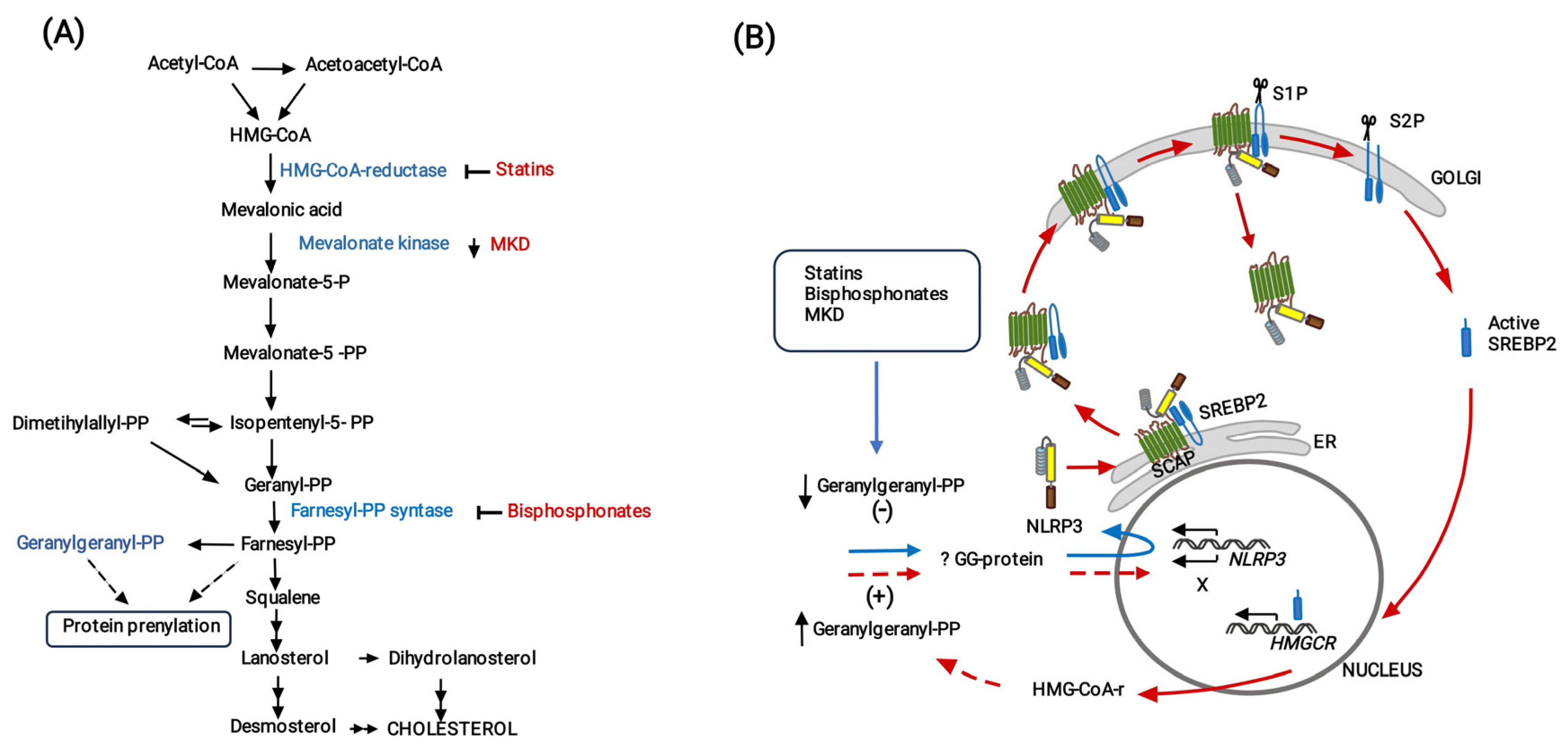
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- World Health Organization. Global Tuberculosis Reports. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 4 September 2023).
- Cohen, S.B.; Gern, B.H.; Urdahl, K.B. The Tuberculous Granuloma and Preexisting Immunity. Annu. Rev. Immunol. 2022, 40, 589–614. [Google Scholar] [CrossRef]
- Pagan, A.J.; Ramakrishnan, L. Immunity and Immunopathology in the Tuberculous Granuloma. Cold Spring Harb. Perspect. Med. 2014, 5, a018499. [Google Scholar] [CrossRef]
- Soldevilla, P.; Vilaplana, C.; Cardona, P.J. Mouse Models for Mycobacterium tuberculosis Pathogenesis: Show and Do Not Tell. Pathogens 2022, 12, 49. [Google Scholar] [CrossRef]
- Wilburn, K.M.; Fieweger, R.A.; VanderVen, B.C. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathog. Dis. 2018, 76, fty021. [Google Scholar] [CrossRef]
- Pawelczyk, J.; Brzostek, A.; Minias, A.; Plocinski, P.; Rumijowska-Galewicz, A.; Strapagiel, D.; Zakrzewska-Czerwinska, J.; Dziadek, J. Cholesterol-dependent transcriptome remodeling reveals new insight into the contribution of cholesterol to Mycobacterium tuberculosis pathogenesis. Sci. Rep. 2021, 11, 12396. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Dartois, V. Caseum: A Niche for Mycobacterium tuberculosis Drug-Tolerant Persisters. Clin. Microbiol. Rev. 2020, 33, e00159-19. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 2020, 11, 910. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.; Marwitz, S.; Tazoll, S.C.; Waldow, F.; Kalsdorf, B.; Vierbuchen, T.; Scholzen, T.; Gross, A.; Goldenbaum, S.; Holscher, A.; et al. WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J. Clin. Investig. 2021, 131, e141833. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Bhaskar, A.; Dwivedi, V.P. Repurposing Immunomodulatory Drugs to Combat Tuberculosis. Front. Immunol. 2021, 12, 645485. [Google Scholar] [CrossRef] [PubMed]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shin, S.J. Revolutionizing control strategies against Mycobacterium tuberculosis infection through selected targeting of lipid metabolism. Cell. Mol. Life Sci. 2023, 80, 291. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sheng, L.; Lou, L. Statin Use May Be Associated With Reduced Active Tuberculosis Infection: A Meta-Analysis of Observational Studies. Front. Med. 2020, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Su, V.Y.; Pan, S.W.; Yen, Y.F.; Feng, J.Y.; Su, W.J.; Chen, Y.M. Statin use and impact on tuberculosis risk. Expert. Rev. Anti-Infect. Ther. 2021, 19, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, T.; Zhang, X.; Yu, A.; Cao, Y. Statin use and risk of tuberculosis: A systemic review of observational studies. Int. J. Infect. Dis. 2020, 93, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Parihar, S.P.; Guler, R.; Brombacher, F. Statins: A viable candidate for host-directed therapy against infectious diseases. Nat. Rev. Immunol. 2019, 19, 104–117. [Google Scholar] [CrossRef]
- Tahir, F.; Bin Arif, T.; Ahmed, J.; Shah, S.R.; Khalid, M. Anti-tuberculous Effects of Statin Therapy: A Review of Literature. Cureus 2020, 12, e7404. [Google Scholar] [CrossRef]
- Guerra-De-Blas, P.D.C.; Torres-Gonzalez, P.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Ponce-De-Leon-Garduno, A.; Sifuentes-Osornio, J. Potential Effect of Statins on Mycobacterium tuberculosis Infection. J. Immunol. Res. 2018, 2018, 7617023. [Google Scholar] [CrossRef]
- Sun, H.Y.; Singh, N. Potential role of statins for the management of immune reconstitution syndrome. Med. Hypotheses 2011, 76, 307–310. [Google Scholar] [CrossRef]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Reviews. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Bifulco, M. Role of the isoprenoid pathway in ras transforming activity, cytoskeleton organization, cell proliferation and apoptosis. Life Sci. 2005, 77, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Montero, M.T.; Hernandez, O.; Suarez, Y.; Matilla, J.; Ferruelo, A.J.; Martinez-Botas, J.; Gomez-Coronado, D.; Lasuncion, M.A. Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine release in peripheral blood mononuclear cells. Atherosclerosis 2000, 153, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhao, S.; Gao, X.; Wang, R.; Liu, J.; Zhou, X.; Zhou, Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens 2021, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Cyster, J.G. Loss of sterol metabolic homeostasis triggers inflammasomes—How and why. Curr. Opin. Immunol. 2019, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Ross, C.; Chan, A.H.; von Pein, J.B.; Maddugoda, M.P.; Boucher, D.; Schroder, K. Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annu. Rev. Immunol. 2022, 40, 249–269. [Google Scholar] [CrossRef]
- Master, S.S.; Rampini, S.K.; Davis, A.S.; Keller, C.; Ehlers, S.; Springer, B.; Timmins, G.S.; Sander, P.; Deretic, V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 2008, 3, 224–232. [Google Scholar] [CrossRef]
- Wawrocki, S.; Druszczynska, M. Inflammasomes in Mycobacterium tuberculosis-Driven Immunity. Can. J. Infect. Dis. Med. Microbiol. 2017, 2017, 2309478. [Google Scholar] [CrossRef]
- Rastogi, S.; Briken, V. Interaction of Mycobacteria With Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136. [Google Scholar] [CrossRef] [PubMed]
- Eklund, D.; Welin, A.; Andersson, H.; Verma, D.; Soderkvist, P.; Stendahl, O.; Sarndahl, E.; Lerm, M. Human Gene Variants Linked to Limit Intramacrophage Growth of Mycobacterium tuberculosis. J. Infect. Dis. 2014, 209, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.; Perucha, E. Cholesterol metabolism: A new molecular switch to control inflammation. Clin. Sci. 2021, 135, 1389–1408. [Google Scholar] [CrossRef] [PubMed]
- Malen, H.; De Souza, G.A.; Pathak, S.; Softeland, T.; Wiker, H.G. Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains. BMC Microbiol. 2011, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gong, J.; Lin, Y.; Barnes, P.F. Growth of virulent and avirulent Mycobacterium tuberculosis strains in human macrophages. Infect. Immun. 1998, 66, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669–681. [Google Scholar] [CrossRef]
- Youmans, G.P.; Youmans, A.S. An Acute Pulmonary Granulomatous Response in Mice Produced by Mycobacterial Cells and Its Relation to Increased Resistance and Increased Susceptibility to Experimental Tuberculous Infection. J. Infect. Dis. 1964, 114, 135–151. [Google Scholar] [CrossRef]
- Boyum, A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand. J. Clin. Lab. Investig. Suppl. 1968, 97, 77–89. [Google Scholar]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhao, M.M.; Sun, S.; Guo, X.L.; Wang, Q.; Li, S.A.; Lee, W.H.; Zhang, Y. A high concentration of DMSO activates caspase-1 by increasing the cell membrane permeability of potassium. Cytotechnology 2018, 70, 313–320. [Google Scholar] [CrossRef]
- Patino, S.; Alamo, L.; Cimino, M.; Casart, Y.; Bartoli, F.; Garcia, M.J.; Salazar, L. Autofluorescence of mycobacteria as a tool for detection of Mycobacterium tuberculosis. J. Clin. Microbiol. 2008, 46, 3296–3302. [Google Scholar] [CrossRef]
- Montero, M.T.; Matilla, J.; Gomez-Mampaso, E.; Lasuncion, M.A. Geranylgeraniol regulates negatively caspase-1 autoprocessing: Implication in the Th1 response against Mycobacterium tuberculosis. J. Immunol. 2004, 173, 4936–4944. [Google Scholar] [CrossRef]
- Ivessa, N.E.; Gravotta, D.; De Lemos-Chiarandini, C.; Kreibich, G. Functional protein prenylation is required for the brefeldin A-dependent retrograde transport from the Golgi apparatus to the endoplasmic reticulum. J. Biol. Chem. 1997, 272, 20828–20834. [Google Scholar] [CrossRef]
- Huang, Z.; Luo, Q.; Guo, Y.; Chen, J.; Xiong, G.; Peng, Y.; Ye, J.; Li, J. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PLoS ONE 2015, 10, e0129744. [Google Scholar] [CrossRef] [PubMed]
- Naeim, F.; Rao, P.N.; Song, S.X.; Phan, R.T. Atlas of Hematopathology: Morphology, Immunophenotype, Cytogenetics, and Molecular Approaches. In Histiocytic Disorders; Academic Press: Cambridge, MA, USA, 2018; pp. 829–849. [Google Scholar] [CrossRef]
- MyPathologyReport/Foamy Histiocytes. Available online: https://www.mypathologyreport.ca/es/pathology-dictionary/foamy-histiocytes/ (accessed on 2 May 2023).
- Adams, D.O. The structure of mononuclear phagocytes differentiating in vivo. I. Sequential fine and histologic studies of the effect of Bacillus Calmette-Guerin (BCG). Am. J. Pathol. 1974, 76, 17–48. [Google Scholar] [PubMed]
- Armstrong, J.A.; Hart, P.D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 1971, 134, 713–740. [Google Scholar] [CrossRef] [PubMed]
- Westman, J.; Grinstein, S.; Marques, P.E. Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front. Immunol. 2019, 10, 3030. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.C.; Dvorak, A.M. Lipid body-phagosome interaction in macrophages during infectious diseases: Host defense or pathogen survival strategy? PLoS Pathog. 2012, 8, e1002729. [Google Scholar] [CrossRef]
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 2009, 182, 3173–3182. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Corleis, B.; Dorhoi, A. Early dynamics of innate immunity during pulmonary tuberculosis. Immunol. Lett. 2020, 221, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Cadena, A.M.; Flynn, J.L.; Fortune, S.M. The Importance of First Impressions: Early Events in Mycobacterium tuberculosis Infection Influence Outcome. mBio 2016, 7, e00342-16. [Google Scholar] [CrossRef] [PubMed]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis replicates within necrotic human macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Divangahi, M.; Remold, H.G. Evasion of innate immunity by Mycobacterium tuberculosis: Is death an exit strategy? Nat. Rev. Microbiol. 2010, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.R. The Formation of Macrophages, Epithelioid Cells and Giant Cells from Leucocytes in Incubated Blood. Am. J. Pathol. 1925, 1, 91–100.1. [Google Scholar] [PubMed]
- Cosma, C.L.; Humbert, O.; Sherman, D.R.; Ramakrishnan, L. Trafficking of superinfecting Mycobacterium organisms into established granulomas occurs in mammals and is independent of the Erp and ESX-1 mycobacterial virulence loci. J. Infect. Dis. 2008, 198, 1851–1855. [Google Scholar] [CrossRef]
- Moule, M.G.; Cirillo, J.D. Mycobacterium tuberculosis Dissemination Plays a Critical Role in Pathogenesis. Front. Cell. Infect. Microbiol. 2020, 10, 65. [Google Scholar] [CrossRef]
- Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/foam-cell (accessed on 25 July 2023).
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends Immunol. 2019, 40, 1163–1179. [Google Scholar] [CrossRef]
- Guerrini, V.; Prideaux, B.; Khan, R.; Subbian, S.; Wang, Y.; Sadimin, E.; Pawar, S.; Ukey, R.; Singer, E.A.; Xue, C.; et al. Heterogeneity of foam cell biogenesis across diseases. bioRxiv 2023. [Google Scholar] [CrossRef]
- Rossi, G.; Cavazza, A.; Spagnolo, P.; Bellafiore, S.; Kuhn, E.; Carassai, P.; Caramanico, L.; Montanari, G.; Cappiello, G.; Andreani, A.; et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series “Pathology for the clinician” Edited by Peter Dorfmuller and Alberto Cavazza. Eur. Respir. Rev. 2017, 26, 170009. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, V.; Prideaux, B.; Blanc, L.; Bruiners, N.; Arrigucci, R.; Singh, S.; Ho-Liang, H.P.; Salamon, H.; Chen, P.Y.; Lakehal, K.; et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 2018, 14, e1007223. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Gordon, S.; Martinez, F.O. Foam Cell Macrophages in Tuberculosis. Front. Immunol. 2021, 12, 775326. [Google Scholar] [CrossRef] [PubMed]
- Cardona, P.J.; Llatjos, R.; Gordillo, S.; Diaz, J.; Ojanguren, I.; Ariza, A.; Ausina, V. Evolution of granulomas in lungs of mice infected aerogenically with Mycobacterium tuberculosis. Scand. J. Immunol. 2000, 52, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Pagan, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P. The immune response in tuberculosis. Annu. Rev. Immunol. 2013, 31, 475–527. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes. Dev. 2001, 15, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef]
- Mahmoudi, A.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Identification of key upregulated genes involved in foam cell formation and the modulatory role of statin therapy. Int. Immunopharmacol. 2023, 119, 110209. [Google Scholar] [CrossRef]
- Pontillo, A.; Paoluzzi, E.; Crovella, S. The inhibition of mevalonate pathway induces upregulation of NALP3 expression: New insight in the pathogenesis of mevalonate kinase deficiency. Eur. J. Hum. Genet. 2010, 18, 844–847. [Google Scholar] [CrossRef]
- Kaneko, J.; Okinaga, T.; Hikiji, H.; Ariyoshi, W.; Yoshiga, D.; Habu, M.; Tominaga, K.; Nishihara, T. Zoledronic acid exacerbates inflammation through M1 macrophage polarization. Inflamm. Regen. 2018, 38, 16. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Piscianz, E.; Zweyer, M.; Bortul, R.; Loganes, C.; Girardelli, M.; Baj, G.; Monasta, L.; Celeghini, C. Geranylgeraniol and Neurological Impairment: Involvement of Apoptosis and Mitochondrial Morphology. Int. J. Mol. Sci. 2016, 17, 365. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, W.; Lee, S.; Xu, Q.; Naji, A.; Le, A.D. Bisphosphonate Induces Osteonecrosis of the Jaw in Diabetic Mice via NLRP3/Caspase-1-Dependent IL-1beta Mechanism. J. Bone Miner. Res. 2015, 30, 2300–2312. [Google Scholar] [CrossRef]
- Mandey, S.H.; Kuijk, L.M.; Frenkel, J.; Waterham, H.R. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum. 2006, 54, 3690–3695. [Google Scholar] [CrossRef]
- Politiek, F.A.; Waterham, H.R. Compromised Protein Prenylation as Pathogenic Mechanism in Mevalonate Kinase Deficiency. Front. Immunol. 2021, 12, 724991. [Google Scholar] [CrossRef]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 2018, 49, 842–856. [Google Scholar] [CrossRef]
- Jeyaratnam, J.; Frenkel, J. Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. Front. Immunol. 2020, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Pizzuto, M.; Pelegrin, P.; Ruysschaert, J.M. Lipid-protein interactions regulating the canonical and the non-canonical NLRP3 inflammasome. Prog. Lipid Res. 2022, 87, 101182. [Google Scholar] [CrossRef]
- Pandey, A.; Shen, C.; Feng, S.; Man, S.M. Cell biology of inflammasome activation. Trends Cell Biol. 2021, 31, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Ta, A.; Vanaja, S.K. Inflammasome activation and evasion by bacterial pathogens. Curr. Opin. Immunol. 2021, 68, 125–133. [Google Scholar] [CrossRef]
- Gideon, H.P.; Hughes, T.K.; Tzouanas, C.N.; Wadsworth, M.H., 2nd; Tu, A.A.; Gierahn, T.M.; Peters, J.M.; Hopkins, F.F.; Wei, J.R.; Kummerlowe, C.; et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity 2022, 55, 827–846. [Google Scholar] [CrossRef]
- Tominaga, K.; Yoshimoto, T.; Torigoe, K.; Kurimoto, M.; Matsui, K.; Hada, T.; Okamura, H.; Nakanishi, K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int. Immunol. 2000, 12, 151–160. [Google Scholar] [CrossRef]
- Cardona, P.J. Revisiting the natural history of tuberculosis. The inclusion of constant reinfection, host tolerance, and damage-response frameworks leads to a better understanding of latent infection and its evolution towards active disease. Arch. Immunol. Et. Ther. Exp. 2010, 58, 7–14. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Bruiners, N.; Zimmerman, M.D.; Tan, S.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Adjunctive Host-Directed Therapy With Statins Improves Tuberculosis-Related Outcomes in Mice. J. Infect. Dis. 2020, 221, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Adewole, O.O.; Omotoso, B.A.; Ogunsina, M.; Aminu, A.; Odeyemi, A.O.; Awopeju, O.F.; Ayoola, O.; Adedeji, T.; Sogaolu, O.M.; Adewole, T.O.; et al. Atorvastatin accelerates Mycobacterium tuberculosis clearance in pulmonary TB: A randomised phase IIA trial. Int. J. Tuberc. Lung Dis. 2023, 27, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. J. Inflamm. Res. 2020, 13, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Mansi, I.A.; Chansard, M.; Lingvay, I.; Zhang, S.; Halm, E.A.; Alvarez, C.A. Association of Statin Therapy Initiation With Diabetes Progression: A Retrospective Matched-Cohort Study. JAMA Intern. Med. 2021, 181, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Lanzafame, M.; Vento, S. Tuberculosis-immune reconstitution inflammatory syndrome. J. Clin. Tuberc. Other Mycobact. Dis. 2016, 3, 6–9. [Google Scholar] [CrossRef]
- Hunter, R.L. The Pathogenesis of Tuberculosis-The Koch Phenomenon Reinstated. Pathogens 2020, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. The End of the Binary Era: Revisiting the Spectrum of Tuberculosis. J. Immunol. 2018, 201, 2541–2548. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montero-Vega, M.T.; Matilla, J.; Bazán, E.; Reimers, D.; De Andrés-Martín, A.; Gonzalo-Gobernado, R.; Correa, C.; Urbano, F.; Gómez-Coronado, D. Fluvastatin Converts Human Macrophages into Foam Cells with Increased Inflammatory Response to Inactivated Mycobacterium tuberculosis H37Ra. Cells 2024, 13, 536. https://doi.org/10.3390/cells13060536
Montero-Vega MT, Matilla J, Bazán E, Reimers D, De Andrés-Martín A, Gonzalo-Gobernado R, Correa C, Urbano F, Gómez-Coronado D. Fluvastatin Converts Human Macrophages into Foam Cells with Increased Inflammatory Response to Inactivated Mycobacterium tuberculosis H37Ra. Cells. 2024; 13(6):536. https://doi.org/10.3390/cells13060536
Chicago/Turabian StyleMontero-Vega, María Teresa, Joaquín Matilla, Eulalia Bazán, Diana Reimers, Ana De Andrés-Martín, Rafael Gonzalo-Gobernado, Carlos Correa, Francisco Urbano, and Diego Gómez-Coronado. 2024. "Fluvastatin Converts Human Macrophages into Foam Cells with Increased Inflammatory Response to Inactivated Mycobacterium tuberculosis H37Ra" Cells 13, no. 6: 536. https://doi.org/10.3390/cells13060536