
Genomic Engineering of Oral Keratinocytes to Establish In Vitro Oral Potentially Malignant Disease Models as a Platform for Treatment Investigation

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. Processing of Oral Leukoplakia and Tumor-Adjacent Biopsies
2.3. Cell Culturing
2.4. DNA Library Preparation
2.5. Somatic Copy Number Calling
2.6. Somatic Mutation Calling
2.7. CDKN2A Methylation Assay
2.8. Genomic Engineering
2.9. Western Blot
2.10. Dose–Response Analysis
2.11. Telomere Length and Telomerase Activity Assays
3. Results
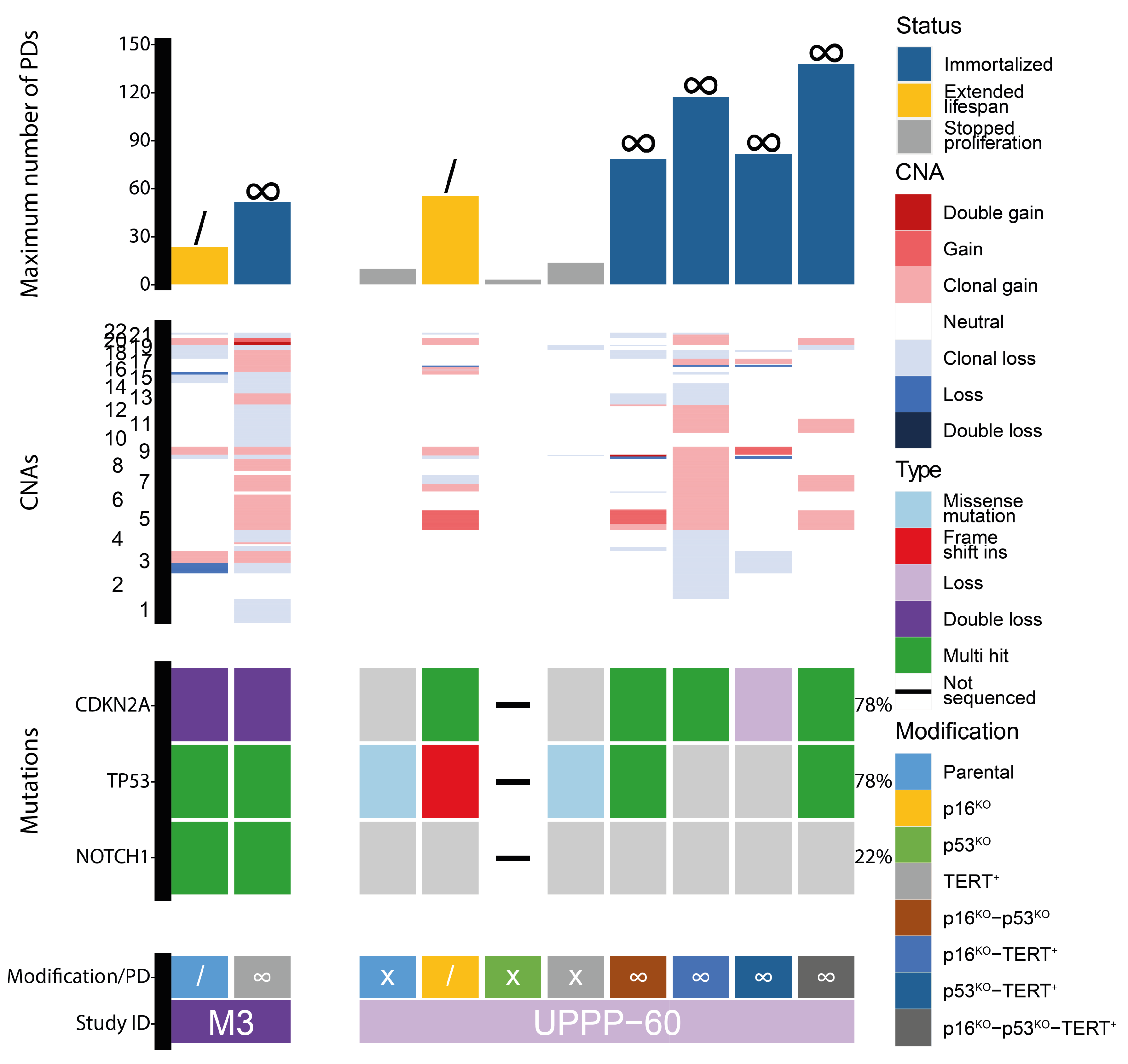
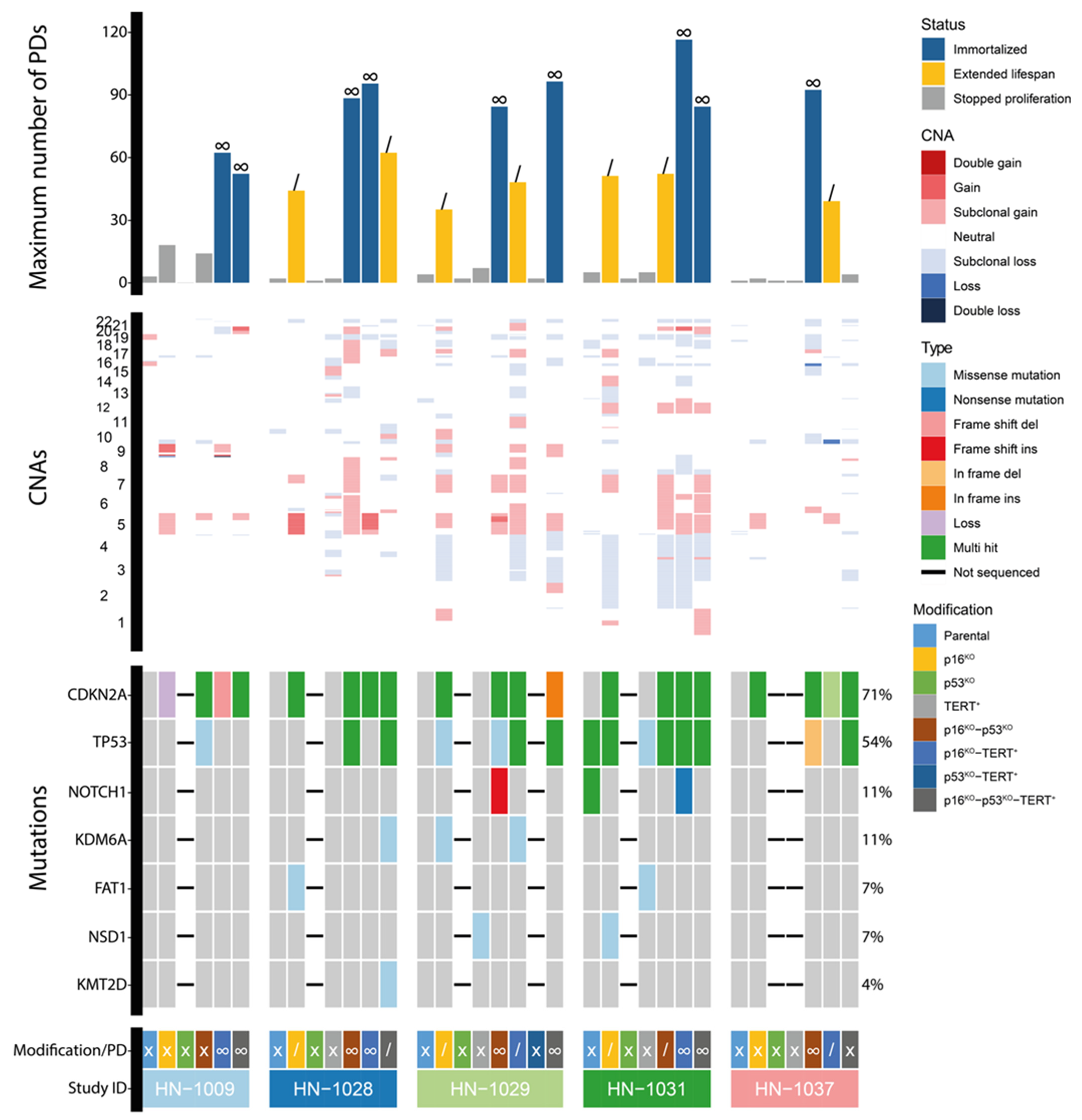
3.1. Immortalization of Oral Keratinocytes by Genomic Engineering of Selected Target Genes
3.2. Genetic Aberrations Introduced in oral Keratinocytes by Genomic Modification and/or Prolonged Culturing
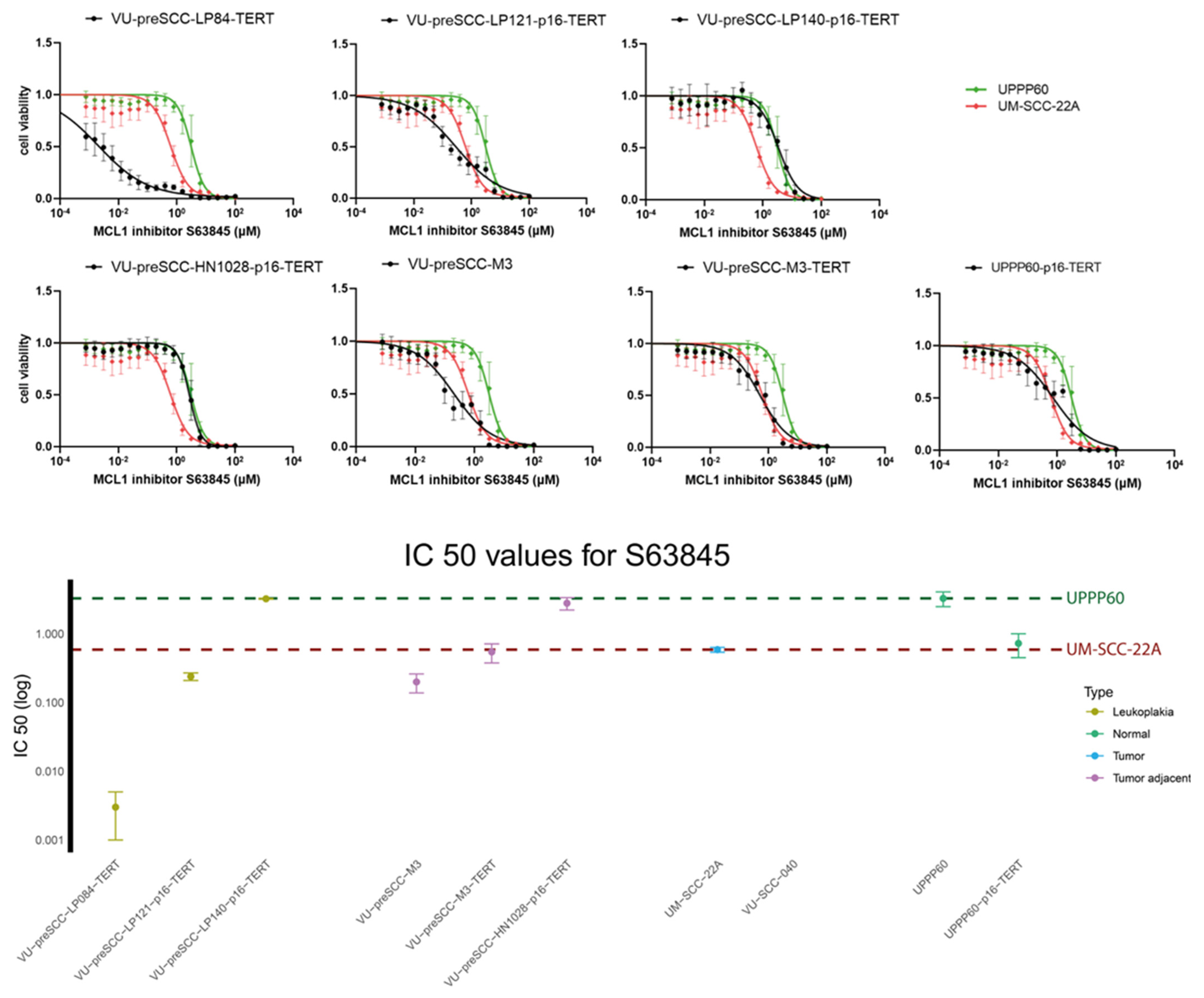
3.3. Assessment of Drug Effectivity in the Oral Cell Culture Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tabor, M.P.; Brakenhoff, R.H.; Van Houten, V.M.M.; Kummer, J.A.; Snel, M.H.J.; Snijders, P.J.F.; Snow, G.B.; Leemans, C.R.; Braakhuis, B.J.M. Persistence of Genetically Altered Fields in Head and Neck Cancer Patients: Biological and Clinical Implications. Clin. Cancer Res. 2001, 7, 1523–1532. [Google Scholar]
- Poell, J.B.; Wils, L.J.; Brink, A.; Dietrich, R.; Krieg, C.; Velleuer, E.; Evren, I.; Brouns, E.R.; De Visscher, J.G.; Bloemena, E.; et al. Oral Cancer Prediction by Noninvasive Genetic Screening. Int. J. Cancer 2022, 152, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Tabor, M.P.; Brakenhoff, R.H.; Ruijter-Schippers, H.J.; Kummer, J.A.; Leemans, C.R.; Braakhuis, B.J.M. Genetically Altered Fields as Origin of Locally Recurrent Head and Neck Cancer: A Retrospective Study. Clin. Cancer Res. 2004, 10, 3607–3613. [Google Scholar] [CrossRef]
- Warnakulasuriya, S.; Kujan, O.; Bagan, J.V.; González-moles, M.Á.; Kerr, A.R.; Lodi, G.; Weber, F.; Luis, M.; Ogden, G.R.; Sloan, P.; et al. Oral Potentially Malignant Disorders: Nomenclature and Classification. Oral Dis. 2021, 27, 1862–1880. [Google Scholar] [CrossRef]
- Reibel, J.; Gale, N.; Hille, J.; Hunt, J.L.; Lingen, M.; Muller, S.; Sloan, P.; Tilakaratne, W.M.; Westra, W.H.; Williams, M.D.; et al. Tumours of the Oral Cavity and Mobile Tongue. In WHO Classification of Head and Neck Tumours; El-Naggar, A.K., Chan, J.K.C., Grandis, J.R., Takata, T., Slootweg, P.J., Eds.; IARC Press: Lyon, France, 2017; pp. 112–114. ISBN 978-92-832-2483-9. [Google Scholar]
- Muller, S.; Tilakaratne, W.M. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Tumours of the Oral Cavity and Mobile Tongue. Head Neck Pathol. 2022, 16, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Petti, S. Pooled Estimate of World Leukoplakia Prevalence: A Systematic Review. Oral Oncol. 2003, 39, 770–780. [Google Scholar] [CrossRef]
- Lodi, G.; Franchini, R.; Warnakulasuriya, S.; Varoni, E.M.; Sardella, A.; Kerr, A.R.; Carrassi, A.; MacDonald, L.C.; Worthington, H. V Interventions for Treating Oral Leukoplakia to Prevent Oral Cancer. Cochrane Database Syst. Rev. 2016, 7, CD001829. [Google Scholar] [CrossRef]
- Evren, I.; Brouns, E.R.; Wils, L.J.; Poell, J.B.; Peeters, C.F.W.; Brakenhoff, R.H.; Bloemena, E.; de Visscher, J.G.A.M. Annual Malignant Transformation Rate of Oral Leukoplakia Remains Consistent: A Long-Term Follow-up Study. Oral Oncol. 2020, 110, 105014. [Google Scholar] [CrossRef]
- Evren, I.; Brouns, E.R.; Poell, J.B.; Wils, L.J.; Brakenhoff, R.H.; Bloemena, E.; de Visscher, J.G.A.M. Associations between Clinical and Histopathological Characteristics in Oral Leukoplakia. Oral Dis. 2021, 29, 696–706. [Google Scholar] [CrossRef]
- Lodi, G.; Sardella, A.; Bez, C.; Demarosi, F.; Carrassi, A. Interventions for Treating Oral Leukoplakia. Cochrane Database Syst. Rev. 2006, 4, CD001829. [Google Scholar] [CrossRef]
- Holmstrup, P.; Dabelsteen, E. Oral Leukoplakia-to Treat or Not to Treat. Oral Dis. 2016, 22, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Schepman, K.P.; Van Der Meij, E.H.; Smeele, L.E.; Van Der Waal, I. Malignant Transformation of Oral Leukoplakia: A Follow-up Study of a Hospital-Based Population of 166 Patients with Oral Leukoplakia from The Netherlands. Oral Oncol. 1998, 34, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Brouns, E.R.; Baart, J.; Karagozoglu, K.; Aartman, I.; Bloemena, E.; van der Waal, I. Malignant Transformation of Oral Leukoplakia in a Well-Defined Cohort of 144 Patients. Oral Dis. 2014, 20, e19–e24. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The Molecular Landscape of Head and Neck Cancer. Nat. Rev. 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.; Buse, J.; Davidson, M.; Ferrannini, E.; Holman, R.; Sherwin, R.; Zinman, B. Medical Management of Hyperglycemia in Type 2 Diabetes: A Consensus Algorithm for the Initiation and Adjustment of Therapy. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Madera, D.; Vitale-cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; Mchugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of Tumor Growth Driven by PIK3CA and HPV Oncogenes by Targeting MTOR Signaling with Metformin in Oral Squamous Carcinomas Expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C. Metformin May Reduce Oral Cancer Risk in Patients with Type 2 Diabetes. Oncotarget 2015, 7, 2000–2008. [Google Scholar] [CrossRef]
- Gutkind, J.S.; Molinolo, A.A.; Wu, X.; Wang, Z.; Nachmanson, D.; Harismendy, O.; Alexandrov, L.B.; Wuertz, B.R.; Ondrey, F.G.; Laronde, D.; et al. Inhibition of MTOR Signaling and Clinical Activity of Metformin in Oral Premalignant Lesions. JCI Insight 2021, 6, e147096. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; William, W.N.; Dannenberg, A.J.; Lippman, S.M.; Lee, J.J.; Ondrey, F.G.; Peterson, D.E.; Feng, L.; Atwell, A.; El-naggar, A.K.; et al. Cancer Therapy: Clinical Pilot Randomized Phase II Study of Celecoxib in Oral Premalignant Lesions. Clin. Cancer Res. 2008, 14, 2095–2101. [Google Scholar] [CrossRef]
- Wirth, L.J.; Krane, J.F.; Li, Y.; Othus, M.; Moran, A.E.; Dorfman, D.M.; Norris, C.M.; Goguen, L.; Posner, M.R.; Haddad, R.I.; et al. A Pilot Surrogate Endpoint Biomarker Study of Celecoxib in Oral Premalignant Lesions. Cancer Prev. Res. 2008, 1, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Nankivell, P.; Dunn, J.; Langman, M.; Mehanna, H. Feasibility of Recruitment to an Oral Dysplasia Trial in the United Kingdom. Head Neck Oncol. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Mulshine, J.L.; Atkinson, J.C.; Greer, R.O.; Papadimitrakopoulou, V.A.; Van Waes, C.; Rudy, S.; Martin, J.W.; Steinberg, S.M.; Liewehr, D.J.; Avis, I.; et al. Randomized, Double-Blind, Placebo-Controlled Phase IIB Trial of the Cyclooxygenase Inhibitor Ketorolac as an Oral Rinse in Oropharyngeal Leukoplakia. Clin. Cancer Res. 2004, 10, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Kujan, O.; Van Schaijik, B.; Farah, C.S. Immune Checkpoint Inhibitors in Oral Cavity Squamous Cell Carcinoma and Oral Potentially Malignant Disorders: A Systematic Review. Cancers 2020, 12, 1937. [Google Scholar] [CrossRef] [PubMed]
- Yagyuu, T.; Hatakeyama, K.; Imada, M.; Kurihara, M.; Matsusue, Y.; Yamamoto, K.; Obayashi, C.; Kirita, T. Programmed Death Ligand 1 (PD-L1) Expression and Tumor Microenvironment: Implications for Patients with Oral Precancerous Lesions. Oral Oncol. 2017, 68, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Wong, F.; Millner, A.; Le, N. Topical Bleomycin Treatment of Oral Leukoplakia: A Randomized Double-Blind Clinical Trial. Head Neck 1994, 16, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, Z.; Mao, F.; Cai, L.; Dan, H.; Jiang, L.; Zeng, X.; Li, T.; Zhou, Y.; Chen, Q. PD-1 Blockade Prevents the Progression of Oral Carcinogenesis. Carcinogenesis 2021, 42, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.; Jabara, J.T.; Lai, W.; Svider, P.F.; Warner, B.M.; Lin, H.; Raza, S.N.; Fribley, A.M. Topical Agents for Oral Cancer Chemoprevention: A Systematic Review of the Literature. Oral Oncol. 2017, 67, 153–159. [Google Scholar] [CrossRef]
- Mak, M.; William, W., Jr. Targeting the Epidermal Growth Factor Receptor for Head and Neck Cancer Chemoprevention. Oral Oncol. 2015, 50, 918–923. [Google Scholar] [CrossRef]
- Califano, J.; Ferris, R.; Epstein, J.; Gillespie, M.; Feldman, L.; Gibson, M.; Pytynia, K.; Khan, Z. A Phase II Trial of Cetuximab in High-Risk Premalignant Lesions of the Upper Aerodigestive Tract. J. Clin. Oncol. 2012, 30, 5528. [Google Scholar] [CrossRef]
- William, W., Jr.; Papadimitrakopoulou, V.; Lee, J.; Mao, L.; Cohen, E.; Lin, H.; Gillenwater, A.; Martin, J.; Lingen, M.; Boyle, J.; et al. Erlotinib and the Risk of Oral Cancer: The Erlotinib Prevention of Oral Cancer (EPOC) Randomized Clinical Trial. JAMA Oncol. 2016, 2, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Saba, N.F.; Hurwitz, S.J.; Kono, S.A.; Yang, C.S.; Zhao, Y.; Chen, A.Y.; Moore, C.E.; Owonikoko, T.K.; Ramalingam, S.; Beitler, J.; et al. Chemoprevention of Head and Neck Cancer with Celecoxib and Erlotinib: Results of a Phase Ib and Pharmacokinetic Study. Cancer Prev. Res. 2015, 7, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.P.; Strong, E.; DeCosse, J.; Itri, L.; Sellers, P. Effect of Retinoids on Oral Leukoplakia. Am. J. Surg. 1983, 146, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Braakhuis, B.J.M. Anticancer Activity and Mechanism of Action of Retinoids in Oral and Pharyngeal Cancer. Oral Oncol. 2002, 38, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Wils, L.J.; Poell, J.B.; Brink, A.; Evren, I.; Brouns, E.R.; de Visscher, J.G.A.M.; Bloemena, E.; Brakenhoff, R.H. Elucidating the Genetic Landscape of Oral Leukoplakia to Predict Malignant Transformation. Clin. Cancer Res. 2023, 29, 602–613. [Google Scholar] [CrossRef] [PubMed]
- van Harten, A.; Brakenhoff, R.H. Targeted Treatment of Head and Neck (Pre)Cancer: Preclinical Target Identification and Development of Novel Therapeutic Applications. Cancers 2021, 13, 2774. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, A.M.; De Boer, D.V.; Kemp, S.R.M.; Bui, M.; Ganzevles, S.H.; Hunter, K.D.; Leemans, C.R.; Van Beusechem, V.W.; Wolthuis, R.M.F.; De Menezes, R.X.; et al. Chemopreventive Targeted Treatment of Head and Neck Precancer by Wee1 Inhibition. Sci. Rep. 2020, 10, 2330. [Google Scholar] [CrossRef]
- De Boer, D.V.; Kemp, S.R.M.; Buijze, M.; Stigter-, M.; Walsum, V.; Bloemena, E.; Dietrich, R.; Leemans, C.R.; Victor, W.; Beusechem, V.; et al. Targeting PLK1 as a Novel Chemopreventive Approach to Eradicate Preneoplastic Mucosal Changes in the Head and Neck. Oncotarget 2017, 8, 97928–97940. [Google Scholar] [CrossRef]
- van Harten, A.M.; Buijze, M.; van der Mast, R.; Rooimans, M.A.; Martens-de Kemp, S.R.; Bachas, C.; Brink, A.; Stigter-van Walsum, M.; Wolthuis, R.M.F.; Brakenhoff, R.H. Targeting the Cell Cycle in Head and Neck Cancer by Chk1 Inhibition: A Novel Concept of Bimodal Cell Death. Oncogenesis 2019, 8, 38. [Google Scholar] [CrossRef]
- Olmos, D.; Barker, D.; Sharma, R.; Brunetto, A.T.; Yap, T.A.; Taegtmeyer, A.B.; Barriuso, J.; Medani, H.; Degenhardt, Y.Y.; Allred, A.J.; et al. Phase I Study of GSK461364, a Specific and Competitive Polo-like Kinase 1 Inhibitor, in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2011, 17, 3420–3430. [Google Scholar] [CrossRef]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients with Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Hahn, W.C.; Ramsey, M.R.; Wu, J.Y.; Guo, Z.; Tsao, H.; De Luca, M.; Catricala, C.; Toole, K.M.O. A Two-Stage, P16 INK4A- and P53-Dependent Keratinocyte Senescence Mechanism That Limits Replicative Potential Independent of Telomere Status †. Mol. Cell. Biol. 2002, 22, 5157–5172. [Google Scholar] [CrossRef] [PubMed]
- Mcgregor, F.; Wagner, E.; Felix, D.; Soutar, D.; Harrison, P.R. Inappropriate Retinoic Expression in Oral Dysplasias: Correlation with Acquisition of the Immortal Phenotype1. Cancer Res. 1997, 57, 3886–3889. [Google Scholar] [PubMed]
- Mcgregor, F.; Muntoni, A.; Fleming, J.; Brown, J.; Felix, D.H.; Macdonald, D.G.; Parkinson, E.K.; Harrison, P.R. Molecular Changes Associated with Oral Dysplasia Progression and Acquisition of Immortality: Potential for Its Reversal by 5-Azacytidine 1. Cancer Res. 2002, 62, 4757–4766. [Google Scholar] [PubMed]
- Hunter, K.D.; Thurlow, J.K.; Fleming, J.; Drake, P.J.H.; Vass, J.K.; Kalna, G.; Higham, D.J.; Herzyk, P.; Macdonald, D.G.; Parkinson, E.K.; et al. Divergent Routes to Oral Cancer. Cancer Res. 2006, 66, 7405–7413. [Google Scholar] [CrossRef] [PubMed]
- De Boer, D.V.; Brink, A.; Buijze, M.; Walsum, M.S.; Hunter, K.D.; Ylstra, B.; Bloemena, E.; Leemans, C.R.; Brakenhoff, R.H. Establishment and Genetic Landscape of Precancer Cell Model Systems from the Head and Neck Mucosal Lining. Mol. Cancer Res. 2019, 17, 12–15. [Google Scholar] [CrossRef] [PubMed]
- van Zeeburg, H.J.T.; Graveland, A.P.; Brink, A.; Nguyen, M.; Leemans, C.R.; Bloemena, E.; Braakhuis, B.J.M.; Brakenhoff, R.H. Generation of Precursor Cell Lines from Preneoplastic Fields Surrounding Head and Neck Cancers. Head Neck 2013, 35, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Hermsen, M.A.J.A.; Joenje, H.; Arwert, F.; Welters, M.J.P.; Braakhuis, B.J.M.; Bagnay, M.; Westerveld, A.; Slater, R. Centromeric Breakage as a Major Cause of Cytogenetic Abnormalities in Oral Squamous Cell Carcinoma. Genes Chromosom. Cancer 1996, 15, 1–9. [Google Scholar] [CrossRef]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Adult Height and Head and Neck Cancer: A Pooled Analysis within the INHANCE Consortium. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef]
- Soule, H.D.; Vazquez, J.; Long, A.; Albert, S.; Brennan, M. A Human Cell Line from a Pleural Effusion Derived from a Breast Carcinoma1,2. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, I.; Sie, D.; Bengtsson, H.; Van De Wiel, M.A.; Olshen, A.B.; Van Thuijl, H.F.; Van Essen, H.F.; Eijk, P.P.; Rustenburg, F.; Meijer, G.A.; et al. DNA Copy Number Analysis of Fresh and Formalin-Fixed Specimens by Shallow Whole-Genome Sequencing with Identification and Exclusion of Problematic Regions in the Genome Assembly. Genome Res. 2014, 24, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- van de Wiel, M.A.; Brosens, R.; Eilers, P.H.C.; Kumps, C.; Meijer, G.A.; Menten, B.; Sistermans, E.; Speleman, F.; Timmerman, M.E.; Ylstra, B. Smoothing Waves in Array CGH Tumor Profiles. Bioinformatics 2009, 25, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.; Poppelaars, J.; van der Horst, M.; Beekman, A.T.F.; Brug, J.; van Tilburg, T.G.; Deeg, D.J.H. Cohort Profile: The Longitudinal Aging Study Amsterdam. Int. J. Epidemiol. 2011, 40, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Poell, J.B.; Mendeville, M.; Sie, D.; Brink, A.; Brakenhoff, R.H.; Ylstra, B.; Birol, I. ACE: Absolute Copy Number Estimation from Low-Coverage Whole-Genome Sequencing Data. Bioinformatics 2019, 35, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Foox, J.; Tighe, S.W.; Nicolet, C.M.; Zook, J.M.; Byrska-Bishop, M.; Clarke, W.E.; Khayat, M.M.; Mahmoud, M.; Laaguiby, P.K.; Herbert, Z.T.; et al. Performance Assessment of DNA Sequencing Platforms in the ABRF Next-Generation Sequencing Study. Nat. Biotechnol. 2021, 39, 1129–1140. [Google Scholar] [CrossRef]
- Maruya, S.I.; Issa, J.P.J.; Weber, R.S.; Rosenthal, D.I.; Haviland, J.C.; Lotan, R.; El-Naggar, A.K. Differential Methylation Status of Tumor-Associated Genes in Head and Neck Squamous Carcinoma: Incidence and Potential Implications. Clin. Cancer Res. 2004, 10, 3825–3830. [Google Scholar] [CrossRef]
- Martens-de Kemp, S.R.; Nagel, R.; Stigter-van Walsum, M.; Van Der Meulen, I.H.; Van Beusechem, V.W.; Braakhuis, B.J.M.; Brakenhoff, R.H. Functional Genetic Screens Identify Genes Essential for Tumor Cell Survival in Head and Neck and Lung Cancer. Clin. Cancer Res. 2013, 19, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh-Van Der Plas, M.; Martens-De Kemp, S.R.; De Maaker, M.; Van Wieringen, W.N.; Ylstra, B.; Agami, R.; Cerisoli, F.; Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. Identification of Lethal MicroRNAs Specific for Head and Neck Cancer. Clin. Cancer Res. 2013, 19, 5647–5657. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in Cancer: Current Status and Perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, E.; Omobono, J.D.; Guo, Z.; Hopkinson, S.; Lazar, A.J.F.; Brenn, T.; Jones, J.C.; Rheinwald, J.G. A Keratinocyte Hypermotility/Growth-Arrest Response Involving Laminin 5 and P16INK4A Activated in Wound Healing and Senescence. Am. J. Pathol. 2006, 168, 1821–1837. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G. Immortalization Eliminates a Roadblock during Cellular Reprogramming into IPS Cells. Nature 2014, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, I.; Erkul, B.E.; Ozturk Sari, S.; Issin, G.; Tural, E.; Terzi Kaya Terzi, N.; Karatay, H.; Celik, M.; Ulusan, M.; Bilgic, B. Promoter Region Mutations of the Telomerase Reverse Transcriptase (TERT) Gene in Head and Neck Squamous Cell Carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2020, 130, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-P.; Wang, C.-I.; Pickering, C.R.; Huang, Y.; Tsai, C.-N.; Tsang, C.-M.; Kao, H.-K.; Cheng, M.-H.; Myers, J.N. Prevalence of Promoter Mutations in the TERT Gene in Oral Cavity Squamous Cell Carcinoma. Head Neck 2017, 39, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic Analysis of Telomere Length and Somatic Alterations in 31 Cancer Types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous Occurrence of Telomeric DNA Damage Response in the Absence of Chromosome Fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Chen, Y.J.; Hakin-Smith, V.; Teo, M.; Xinarianos, G.E.; Jellinek, D.A.; Carroll, T.; McDowell, D.; MacFarlane, M.R.; Boet, R.; Baguley, B.C.; et al. Association of Mutant TP53 with Alternative Lengthening of Telomeres and Favorable Prognosis in Glioma. Cancer Res. 2006, 66, 6473–6476. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Lee, J.; Lee, H. Alternative Paths to Telomere Elongation. Semin. Cell Dev. Biol. 2021, 113, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef] [PubMed]
- Darbro, B.W.; Schneider, G.B.; Klingelhutz, A.J. Co-Regulation of P16INK4a and Migratory Genes in Culture Conditions That Lead to Premature Senescence in Human Keratinocytes. J. Investig. Dermatol. 2005, 125, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Rossello, F.J.; Tothill, R.W.; Britt, K.; Marini, K.D.; Falzon, J.; Thomas, D.M.; Peacock, C.D.; Marchionni, L.; Li, J.; Bennett, S.; et al. Next-Generation Sequence Analysis of Cancer Xenograft Models. PLoS ONE 2013, 8, e74432. [Google Scholar] [CrossRef] [PubMed]
- Niklander, S.E.; Hunter, K.D. A Protocol to Produce Genetically Edited Primary Oral Keratinocytes Using the CRISPR-Cas9 System. In Oral Biology: Molecular Techniques and Applications; Humana: New York, NY, USA, 2023; pp. 217–229. [Google Scholar]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional Reprogramming and Long-Term Expansion of Normal and Tumor Cells from Human Biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Suryaprakash, R.T.C.; Kujan, O.; Shearston, K.; Farah, C.S. Three-Dimensional Cell Culture Models to Investigate Oral Carcinogenesis: A Scoping Review. Int. J. Mol. Sci. 2020, 21, 9520. [Google Scholar] [CrossRef]
- Zhao, H.; Jiang, E.; Shang, Z. 3D Co-Culture of Cancer-Associated Fibroblast with Oral Cancer Organoids. J. Dent. Res. 2021, 100, 201–208. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability an Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Ghadikar, M.A.; Zhang, J.; Shen, L.; Rao, X.; Wang, J.; Zhao, M.; Kalu, N.N.; Johnson, F.M.; Byers, L.A.; Heymach, J.; et al. CDKN2A/P16 Deletion in Head and Neck Cancer Cells Is Associated with Cdk2 Activation, Replication Stress, and Vulnerability to Chk1 Inhibition. Cancer Res. 2018, 78, 781–797. [Google Scholar] [CrossRef]
- Sethi, N.S.; Kikuchi, O.; Duronio, G.; Stachler, M.D.; McFarland, J.M.; Ferrer-Luna, R.; Zhang, Y.; Bao, C.; Bronson, R.T.; Patil, D.; et al. Early TP53 Alterations Engage Environmental Exposures to Promote Gastric Premalignancy in an Integrative Mouse Model. Nat. Genet. 2020, 52, 219–230. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a P53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 Genome Editing Induces Megabase-Scale Chromosomal Truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.; Ambrosetti, D.; Boyer, J.; Bozec, A.; Doyen, J.; Chamorey, E.; He, X.; Bourget, I.; Rousset, J.; Saada, E.; et al. The Polo-like Kinase 1 Inhibitor Onvansertib Represents a Relevant Treatment for Head and Neck Squamous Cell Carcinoma Resistant to Cisplatin and Radiotherapy. Theranostics 2021, 11, 9571–9586. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.S.; Deutsch, E.; Mehmet, A.; Fayette, J.; TAO, Y.; Nabell, L.; Spencer, S.A.; Wang, X.A.; Spoljoric, E.A.; Zhang, W.; et al. A Phase 1b Trial of Prexasertib in Combination with Chemoradiation in Patients with Locally Advanced Head and Neck Squamous Cell Carcinoma. Radiother. Oncol. 2021, 157, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Chera, B.S.; Sheth, S.H.; Patel, S.A.; Goldin, D.; Douglas, K.E.; Green, R.L.; Shen, C.J.; Gupta, G.P.; Moore, D.T.; Grilley Olson, J.E.; et al. Phase 1 Trial of Adavosertib (AZD1775) in Combination with Concurrent Radiation and Cisplatin for Intermediate-Risk and High-Risk Head and Neck Squamous Cell Carcinoma. Cancer 2021, 127, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Serpico, A.F.; D’alterio, G.; Vetrei, C.; Della Monica, R.; Nardella, L.; Visconti, R.; Grieco, D. Wee1 Rather than Plk1 Is Inhibited by AZD1775 at Therapeutically Relevant Concentrations. Cancers 2019, 11, 819. [Google Scholar] [CrossRef]
- Gelderblom, H.; Jalving, M.; Desar, I.; Saavedra, O.; Gietema, J.A.; van Ravensteijn, S.; Ajmone Marsan, N.; Bellon, A.; Micallef, S.; Luong, N.; et al. Debio 0123-101: A Phase 1 Trial of Debio 0123 in Combination with Carboplatin in Advanced Solid Tumors—Safety, Pharmacokinetic, and Preliminary Antitumor Activity Data. J. Clin. Oncol. 2023, 41, 3012. [Google Scholar] [CrossRef]
- Tolcher, A.; Mamdani, H.; Chalasani, P.; Meric-Bernstam, F.; Gazdoiu, M.; Makris, L.; Pultar, P.; Voliotis, D. Abstract CT016: Clinical Activity of Single-Agent ZN-C3, an Oral WEE1 Inhibitor, in a Phase 1 Dose-Escalation Trial in Patients with Advanced Solid Tumors. Cancer Res. 2021, 81, CT016. [Google Scholar] [CrossRef]
- Sobol, B.; Azzam Nieto, O.; Eberlein, E.L.; Scherr, A.L.; Ismail, L.; Kessler, A.; Nader, L.; Schwab, M.; Hoffmeister, P.; Schmitt, N.; et al. Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors. Int. J. Mol. Sci. 2022, 23, 7850. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.; Przybilla, M.J.; Kotler, E.; Khan, A.; Xu, H.; Karagyozova, K.; Sockell, A.; Wong, W.H.; Liu, K.; Mah, A.; et al. Deterministic Evolution and Stringent Selection during Preneoplasia. Nature 2023, 618, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Usman, O.H.; Zhang, L.; Xie, G.; Kocher, H.M.; Hwang, C.i.; Wang, Y.J.; Mallory, X.; Irianto, J. Genomic Heterogeneity in Pancreatic Cancer Organoids and Its Stability with Culture. npj Genom. Med. 2022, 7, 71. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Age at Biopsy | Location | TNM | Mutations Present in Parental Culture | Modifications |
|---|---|---|---|---|---|---|
| LP084 | Female | 74 | Floor of mouth | - | None | TERT+ |
| LP121 | Male | 51 | Tongue | - | FAT1 | p53KO, p16KO, p16KO + TERT+ |
| LP140 | Male | 62 | Tongue | - | KMT2D, TGFBR2, KDM6A | p53KO, p16KO, p16KO + TERT+ |
| M3 | Male | 67 | Tumor-adjacent tissue of a larynx carcinoma | pT4aN0 (TNM7) | TP53, NOTCH1, focal 9p double loss | TERT+ |
| HN1009 | Male | 65 | Tumor-adjacent tissue of a hypopharynx carcinoma | pT4aN3b | None | p53KO, p16KO, p16KO + p53KO, p16KO + TERT+, p16KO + p53KO + TERT+ |
| HN1028 | Male | 85 | Tumor-adjacent tissue of a larynx carcinoma | pT4aN0 | None | p53KO, p16KO, TERT+, p16KO + p53KO, p16KO + TERT+, p16KO + p53KO + TERT+ |
| HN1029 | Male | 67 | Tumor-adjacent tissue of a larynx carcinoma | pT4aN1 | None | p53KO, p16KO, TERT+, p16KO + p53KO, p53KO + TERT+, p16KO + TERT+, p16KO + p53KO + TERT+ |
| HN1031 | Male | 58 | Tumor-adjacent tissue of a hypopharynx carcinoma | pT4aN3b | TP53, NOTCH1 | p53KO, p16, TERT+, p16KO + p53KO, p16KO + TERT+, p16KO + p53KO + TERT+ |
| HN1037 | Female | 57 | Tumor-adjacent tissue of a lip and oral cavity carcinoma | pT3N2b | None | p53KO, p16KO, TERT+, p16KO + p53KO, p16KO + TERT+, p16KO + p53KO + TERT+ |
| UPPP-60 | Male | Unknown | Uvula | - | TP53 | p53KO, p16KO, TERT+, p16KO + p53KO, p53KO + TERT+, p16KO + TERT+, p16KO + p53KO + TERT+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wils, L.J.; Buijze, M.; Stigter-van Walsum, M.; Brink, A.; van Kempen, B.E.; Peferoen, L.; Brouns, E.R.; de Visscher, J.G.A.M.; van der Meij, E.H.; Bloemena, E.; et al. Genomic Engineering of Oral Keratinocytes to Establish In Vitro Oral Potentially Malignant Disease Models as a Platform for Treatment Investigation. Cells 2024, 13, 710. https://doi.org/10.3390/cells13080710
Wils LJ, Buijze M, Stigter-van Walsum M, Brink A, van Kempen BE, Peferoen L, Brouns ER, de Visscher JGAM, van der Meij EH, Bloemena E, et al. Genomic Engineering of Oral Keratinocytes to Establish In Vitro Oral Potentially Malignant Disease Models as a Platform for Treatment Investigation. Cells. 2024; 13(8):710. https://doi.org/10.3390/cells13080710
Chicago/Turabian StyleWils, Leon J., Marijke Buijze, Marijke Stigter-van Walsum, Arjen Brink, Britt E. van Kempen, Laura Peferoen, Elisabeth R. Brouns, Jan G. A. M. de Visscher, Erik H. van der Meij, Elisabeth Bloemena, and et al. 2024. "Genomic Engineering of Oral Keratinocytes to Establish In Vitro Oral Potentially Malignant Disease Models as a Platform for Treatment Investigation" Cells 13, no. 8: 710. https://doi.org/10.3390/cells13080710
APA StyleWils, L. J., Buijze, M., Stigter-van Walsum, M., Brink, A., van Kempen, B. E., Peferoen, L., Brouns, E. R., de Visscher, J. G. A. M., van der Meij, E. H., Bloemena, E., Poell, J. B., & Brakenhoff, R. H. (2024). Genomic Engineering of Oral Keratinocytes to Establish In Vitro Oral Potentially Malignant Disease Models as a Platform for Treatment Investigation. Cells, 13(8), 710. https://doi.org/10.3390/cells13080710