
Targeting Integrin α3 Blocks β1 Maturation, Triggers Endoplasmic Reticulum Stress, and Sensitizes Glioblastoma Cells to TRAIL-Mediated Apoptosis
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Transfection of Short-Interfering RNAs and Overexpression Plasmids
2.4. Cell Viability and Death Assays
2.5. Protein Extraction and Western Blotting
2.6. Detection of Apoptotic Nuclei
2.7. Reverse Transcriptase PCR (RT-PCR)
2.8. mRNA Stability Assay
2.9. RNA-Seq
2.10. Immunoprecipitation
2.11. Statistical Considerations
2.12. Bioinformatics Analysis
2.13. Illustration
3. Results
3.1. Integrin α3β1 Is Overexpressed in Cancer and Specific Silencing of Integrin α3 Induces Apoptosis
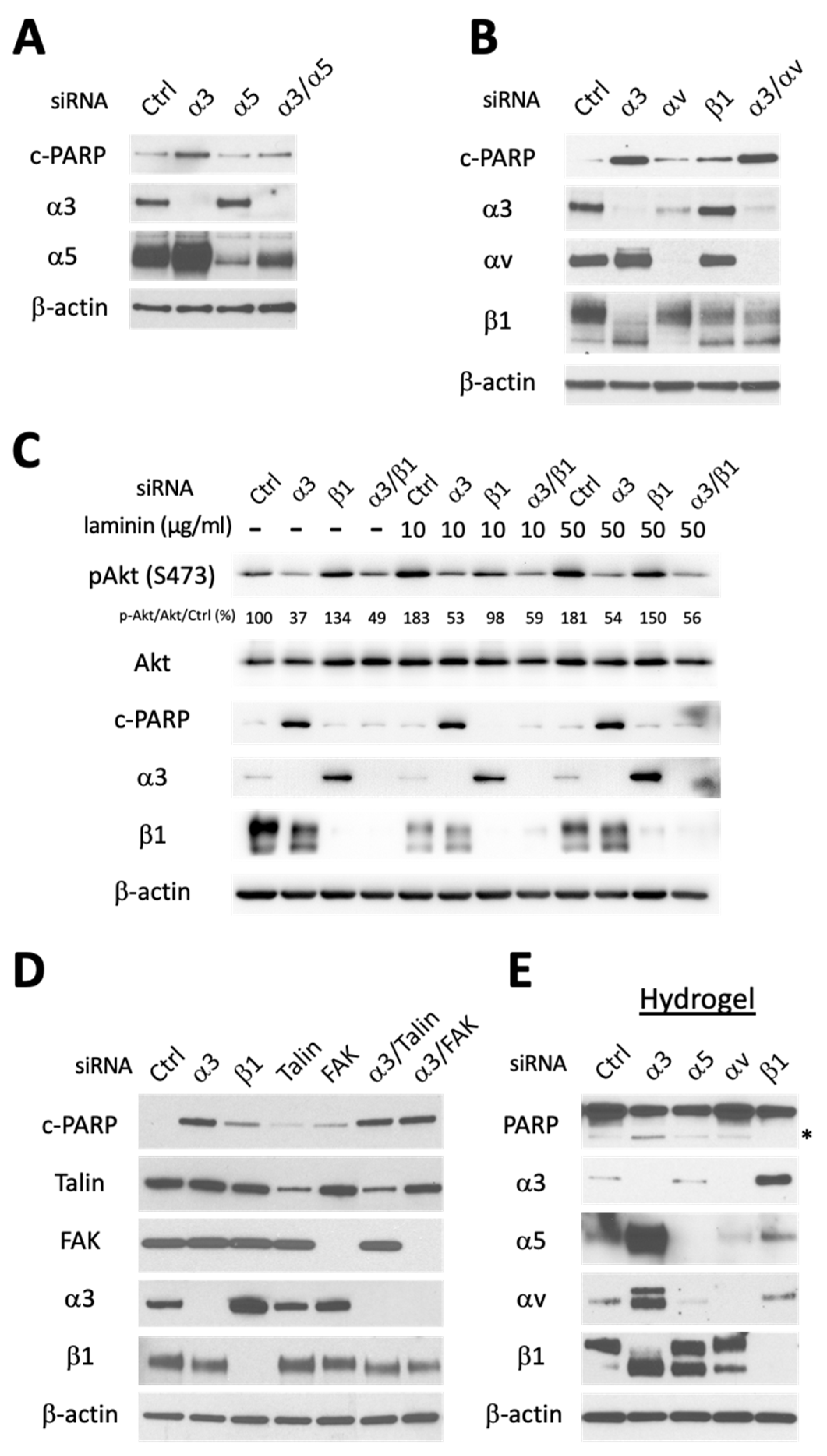
3.2. Apoptosis Induced by Integrin α3 Silencing Is Not Dependent on Activation of Other Integrins and Is Independent of Integrin-Mediated Survival Signaling
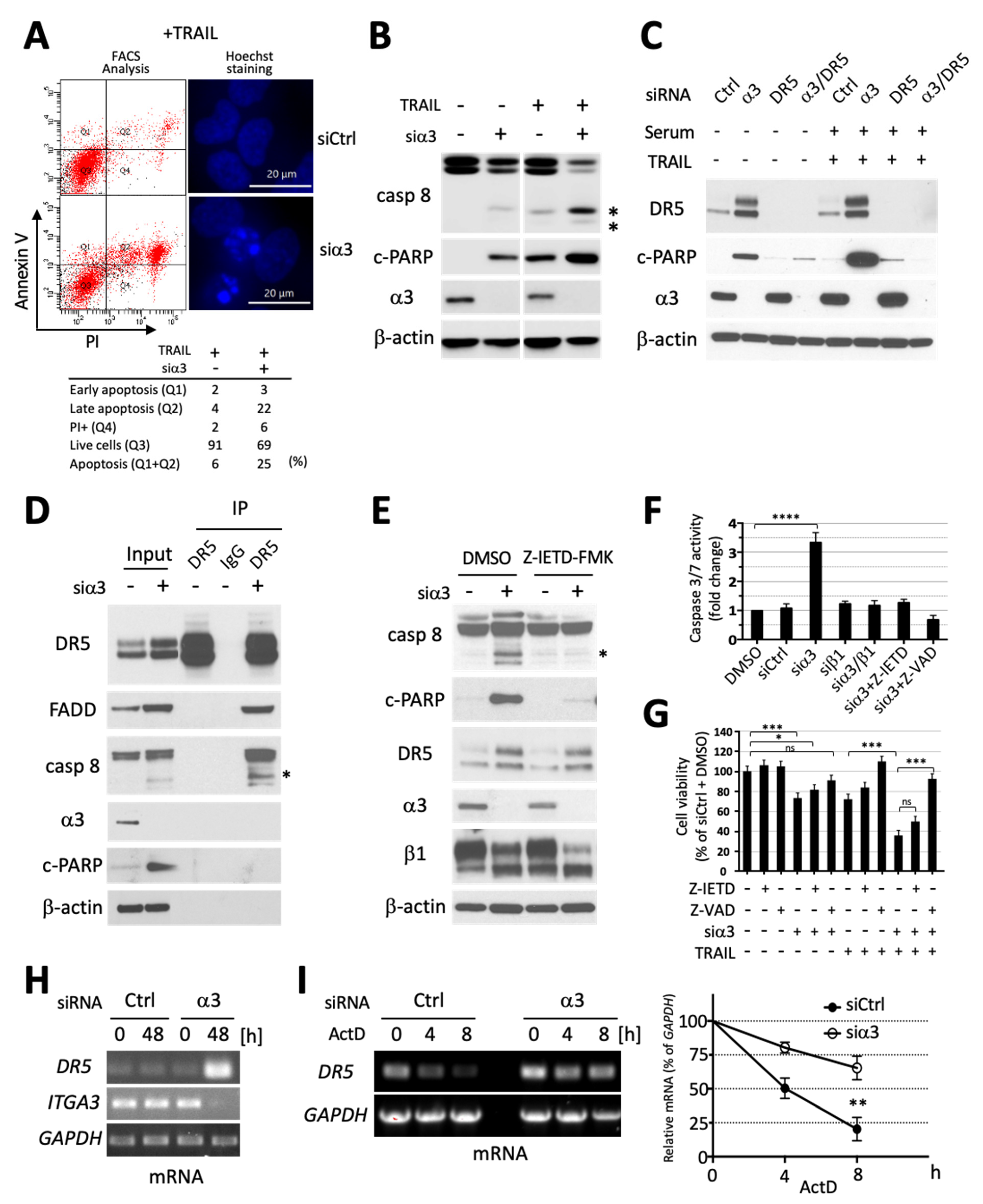
3.3. Silencing of Integrin α3 Increases Death Receptor 5 Expression and Extrinsic Caspase 8-Initiated Apoptosis Cascade
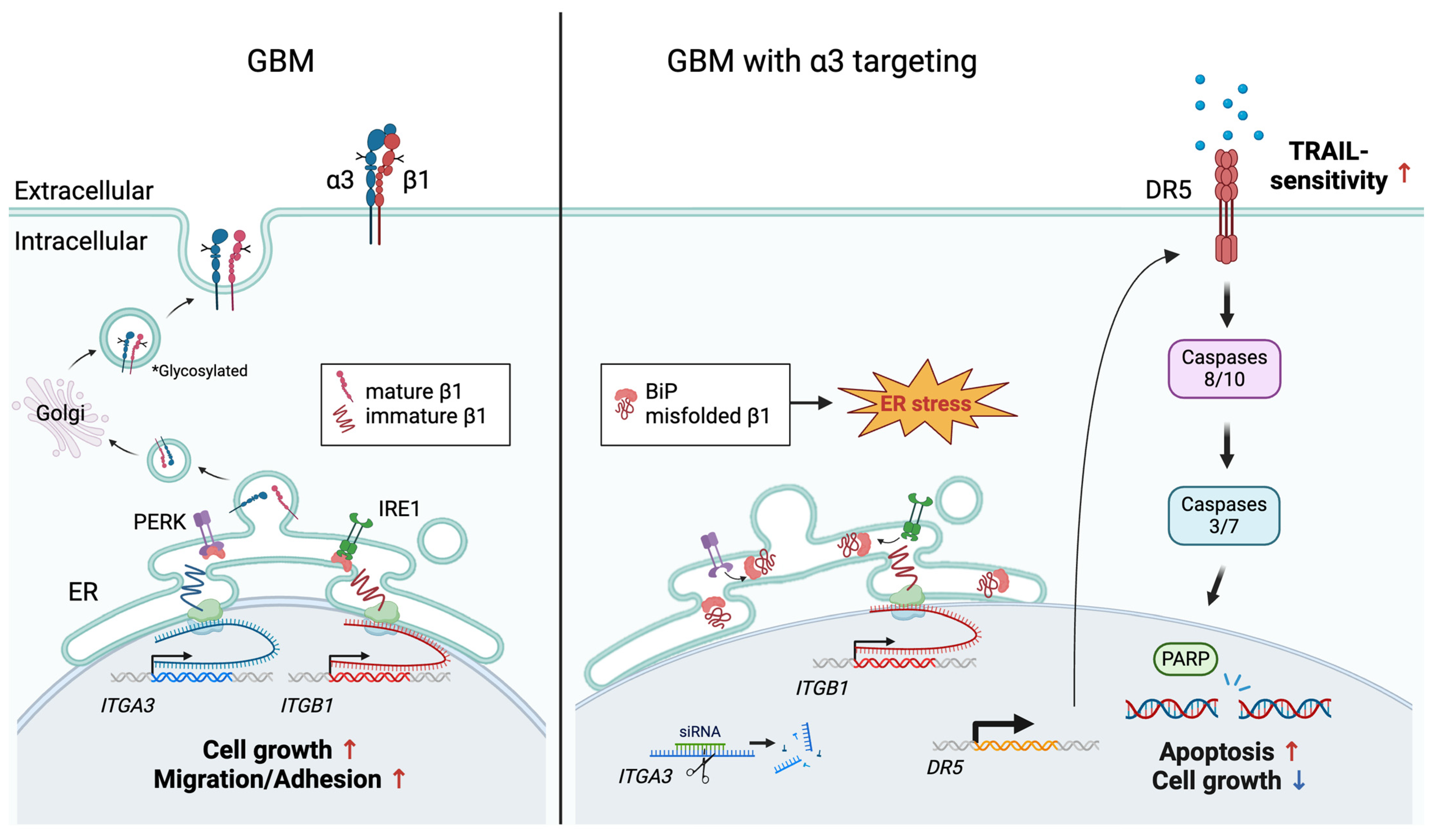
3.4. SiRNA-Mediated Targeting of Integrin α3 Triggers an ER Stress Response Due to Interference with Integrin β1 Maturation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsu, E.; Keene, D.; Ventureyra, E.; Matzinger, M.A.; Jimenez, C.; Wang, H.S.; Grimard, L. Bone marrow metastasis in astrocytic gliomata. J. Neurooncol. 1998, 37, 285–293. [Google Scholar] [CrossRef]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef]
- Piper, R.J.; Senthil, K.K.; Yan, J.L.; Price, S.J. Neuroimaging classification of progression patterns in glioblastoma: A systematic review. J. Neurooncol. 2018, 139, 77–88. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef]
- Simon, L.G.; Martin, P. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef]
- Goel, H.L.; Breen, M.; Zhang, J.; Das, I.; Aznavoorian-Cheshire, S.; Greenberg, N.M.; Elgavish, A.; Languino, L.R. beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005, 65, 6692–6700. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Fang, C.; Xu, H.; Yuan, L.; Du, Y.; Ni, Y.; Xu, Y.; Shao, A.; Zhang, A.; Lou, M. Anoikis resistance in diffuse glioma: The potential therapeutic targets in the future. Front. Oncol. 2022, 12, 976557. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef]
- Lee, J.W.; Juliano, R. Mitogenic signal transduction by integrin- and growth factor receptor-mediated pathways. Mol. Cells 2004, 17, 188–202. [Google Scholar] [CrossRef]
- Bianconi, D.; Unseld, M.; Prager, G.W. Integrins in the Spotlight of Cancer. Int. J. Mol. Sci. 2016, 17, 2307. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Subbaram, S.; Dipersio, C.M. Integrin α3β1 as a breast cancer target. Expert Opin. Ther. Targets 2011, 15, 1197–1210. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Yoshino, H.; Yonemori, M.; Miyamoto, K.; Sugita, S.; Matsushita, R.; Itesako, T.; Tatarano, S.; Nakagawa, M.; Enokida, H. Regulation of ITGA3 by the dual-stranded microRNA-199 family as a potential prognostic marker in bladder cancer. Br. J. Cancer 2017, 116, 1077–1087. [Google Scholar] [CrossRef]
- Kurozumi, A.; Goto, Y.; Matsushita, R.; Fukumoto, I.; Kato, M.; Nishikawa, R.; Sakamoto, S.; Enokida, H.; Nakagawa, M.; Ichikawa, T.; et al. Tumor-suppressive microRNA-223 inhibits cancer cell migration and invasion by targeting ITGA3/ITGB1 signaling in prostate cancer. Cancer Sci. 2016, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.I.; et al. Integrin alpha3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Ljubimova, J.Y.; Fugita, M.; Khazenzon, N.M.; Das, A.; Pikul, B.B.; Newman, D.; Sekiguchi, K.; Sorokin, L.M.; Sasaki, T.; Black, K.L. Association between laminin-8 and glial tumor grade, recurrence, and patient survival. Cancer 2004, 101, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Christopher, S.S. Laminin-binding integrins and their tetraspanin partners as potential antimetastatic targets. Expert Rev. Mol. Med. 2010, 12, e3. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Albertoni, M.; Daub, D.M.; Arden, K.C.; Viars, C.S.; Powell, C.; Van Meir, E.G. Genetic instability leads to loss of both p53 alleles in a human glioblastoma. Oncogene 1998, 16, 321–326. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, R.T. β1 Integrin Cell-surface Immunoprecipitation (Selective Immunoprecipitation). Bio-Protocol 2013, 3, e962. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, F.; Møller, M.; Fu, C.; Clokie, S.J.; Zykovich, A.; Coon, S.L.; Klein, D.C.; Rath, M.F. The Lhx9 homeobox gene controls pineal gland development and prevents postnatal hydrocephalus. Brain Struct. Funct. 2015, 220, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhao, Y.; Wei, Y.; Ding, X.; Tan, C.; Wang, C. Identification and validation of an anoikis-associated gene signature to predict clinical character, stemness, IDH mutation, and immune filtration in glioblastoma. Front. Immunol. 2022, 13, 939523. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef]
- Morizot, A.; Merino, D.; Lalaoui, N.; Jacquemin, G.; Granci, V.; Iessi, E.; Lanneau, D.; Bouyer, F.; Solary, E.; Chauffert, B.; et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell Death Differ. 2011, 18, 700–711. [Google Scholar] [CrossRef]
- Song, J.H.; Song, D.K.; Pyrzynska, B.; Petruk, K.C.; Van Meir, E.G.; Hao, C. TRAIL triggers apoptosis in human malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 2003, 13, 539–553. [Google Scholar] [CrossRef]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef]
- Lee, S.H.; Khwaja Rehman, F.; Tyler, K.C.; Yu, B.; Zhang, Z.; Osuka, S.; Zerrouqi, A.; Kaluzova, M.; Hadjipanayis, C.G.; Cummings, R.D.; et al. A Chimeric Signal Peptide-Galectin-3 Conjugate Induces Glycosylation-Dependent Cancer Cell-Specific Apoptosis. Clin. Cancer Res. 2020, 26, 2711–2724. [Google Scholar] [CrossRef]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Hemmer, B.; Wiendl, H.; Roth, K.; Wessels, H.; Höfler, J.; Hornuss, C.; Liedert, B.; Selmaj, K. Efficacy and Safety of Proposed Biosimilar Natalizumab (PB006) in Patients With Relapsing-Remitting Multiple Sclerosis: The Antelope Phase 3 Randomized Clinical Trial. JAMA Neurol. 2023, 80, 298–307. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Nwagwu, C.D.; Immidisetti, A.V.; Bukanowska, G.; Vogelbaum, M.A.; Carbonell, A.M. Convection-Enhanced Delivery of a First-in-Class Anti-β1 Integrin Antibody for the Treatment of High-Grade Glioma Utilizing Real-Time Imaging. Pharmaceutics 2020, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Rauschenbach, L.; Wieland, A.; Reinartz, R.; Kebir, S.; Till, A.; Darkwah Oppong, M.; Dobersalske, C.; Ullrich, V.; Ahmad, A.; Jabbarli, R.; et al. Drug repositioning of antiretroviral ritonavir for combinatorial therapy in glioblastoma. Eur. J. Cancer 2020, 140, 130–139. [Google Scholar] [CrossRef]
- Yu, D.; Khan, O.F.; Suva, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Askari, J.A.; Humphries, M.J.; Bulleid, N.J. Divalent cations regulate the folding and activation status of integrins during their intracellular trafficking. J. Cell Sci. 2011, 124, 1672–1680. [Google Scholar] [CrossRef]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuranaga, Y.; Yu, B.; Osuka, S.; Zhang, H.; Devi, N.S.; Bae, S.; Van Meir, E.G. Targeting Integrin α3 Blocks β1 Maturation, Triggers Endoplasmic Reticulum Stress, and Sensitizes Glioblastoma Cells to TRAIL-Mediated Apoptosis. Cells 2024, 13, 753. https://doi.org/10.3390/cells13090753
Kuranaga Y, Yu B, Osuka S, Zhang H, Devi NS, Bae S, Van Meir EG. Targeting Integrin α3 Blocks β1 Maturation, Triggers Endoplasmic Reticulum Stress, and Sensitizes Glioblastoma Cells to TRAIL-Mediated Apoptosis. Cells. 2024; 13(9):753. https://doi.org/10.3390/cells13090753
Chicago/Turabian StyleKuranaga, Yuki, Bing Yu, Satoru Osuka, Hanwen Zhang, Narra S. Devi, Sejong Bae, and Erwin G. Van Meir. 2024. "Targeting Integrin α3 Blocks β1 Maturation, Triggers Endoplasmic Reticulum Stress, and Sensitizes Glioblastoma Cells to TRAIL-Mediated Apoptosis" Cells 13, no. 9: 753. https://doi.org/10.3390/cells13090753