3.3. Design of AQUA Peptides
The selection of AQUA peptides for quantification is based on several rules [
22]. From the chemical point of view, AQUA peptides should be stable under all treatment conditions used for proteolytic digestion and mass spectrometry. Amino acid residues which can easily oxidize such as methionine, cysteine and tryptophan should be excluded. N-terminal glutamine residues can undergo pyroglutamate formation to a certain extent and should therefore be avoided. Likewise, sequences with labile amide bonds such as the Asp-Pro bond can undergo hydrolysis and should not be selected.
From the mass spectrometry point of view peptides should be synthesized in such a way, that the isotopic clusters in the mass spectra do not overlap. We have incorporated either [13C6,15N2] lysine or [13C6,15N4] arginine at the C-termini of the selected heavy AQUA peptides thus resulting in a mass increase of 8 and 10 Da, respectively, for the singly charged molecules in relation to their light counterparts.
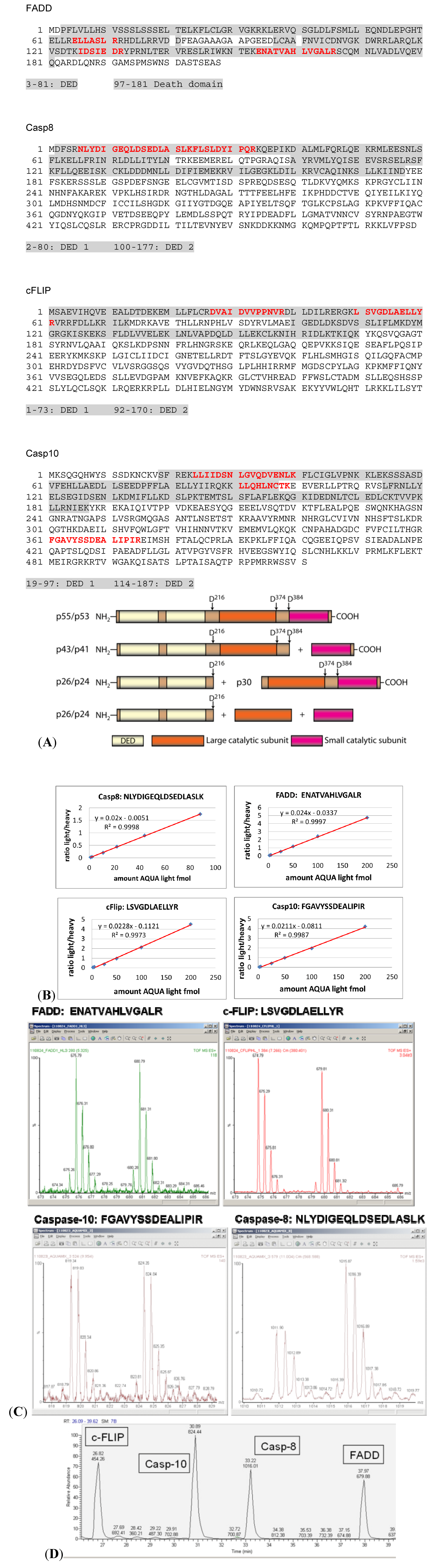
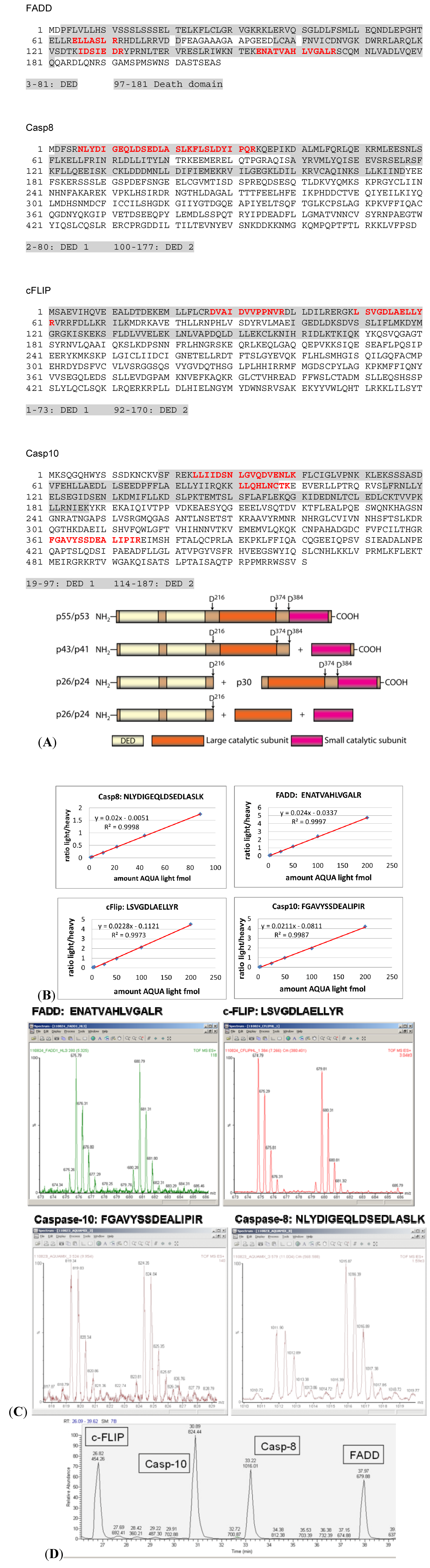
For a reliable quantification, proteins should ideally be represented by at least two different peptides. In the case of FADD, three peptides were selected, spanning amino acid (aa) 65–71, aa 126–132 and aa 154–166, respectively (
Figure 2A). The first peptide is located within the death effector domain (DED); the other two peptides are within the death domain (DD). In our previous mass spectrometric analysis FADD was detected in only one single band at a molecular weight range between 25–28 kDa. Furthermore, phosphorylation of FADD at Ser 194 has been observed [
20] as well as cleavage at the C-terminal Asp201 (unpublished observations). However, since neither phosphorylation nor cleavage is affecting the region of the selected AQUA peptides they should give comparable results.
Whereas FADD was found at its expected molecular weight in the 1D gel caspase-8 and caspase-10 as well as c-FLIP were detected in gel slices corresponding not only to their respective molecular weights in the mass range of 38–60 kDa, but also in the lower mass ranges of 20–35 kDa. The additional bands at lower molecular weights represent the different cleavage products of these proteins. This had to be considered in the selection of suitable AQUA peptides. Procaspase-8a/b undergoes processing at the DISC yielding N-terminal procaspase-8a/b prodomains (p26/p24), which remain bound to the DISC [
23] (
Figure 2A, lower part). Therefore, two peptides within the prodomain covering aa 6–23 and aa 24–33 were selected for quantification and were synthesized as heavy AQUA standards (
Figure 2A).
For procaspase-10 we selected three peptides, two of which were located within DED and one within the subunit p23/p17 (
Figure 2A).
c-FLIP
L is cleaved to p43-FLIP and only this part remains at the DISC. In addition, there are two short c-FLIP isoforms reported: Short (c-FLIP
S, 26 kDa) and Raji (c-FLIP
R, 25 kDa) that are bound to the DISC also
via DED interactions [
9]. In SKW6.4 cells c-FLIP
R is the only short isoform detected. Therefore, we chose two AQUA peptides from the first DED, namely aa 27–38 and aa 50–62 (
Figure 2A).
Finally, it is essential that the chosen peptide is unique for the protein of interest. Therefore, all selected peptides were analyzed using the BLAST algorithm to avoid any interference or overlap with other protein sequences.
In total, 10 heavy AQUA peptides were chemically synthesized. Details of all heavy and light peptide pairs are summarized in
Table 1.
Figure 2.
Selection and validation of AQUA peptides. (A) Amino acid sequences of the major components FADD, procaspase-8/10 and c-FLIP of the CD95 DISC. Selected AQUA peptides are highlighted in red and DED and DD are marked in grey. The scheme of procaspase-8a/b processing is shown below. (B) Basic calibration of corresponding light and heavy AQUA peptide pairs. Variable amounts of the light peptide ranging from 1 to 200 fmol were added to a constant amount of 50 fmol of the corresponding heavy peptide. All AQUA peptide pairs showed linearity in this concentration range when analyzed by mass spectrometry on an LTQ Orbitrap instrument. (C) Quality control of AQUA peptides. Light and heavy AQUA peptides pairs were mixed in a 1:1 ratio and analyzed by mass spectrometry on a QToF instrument. Heavy to light ratios were close to 1:1 or 1:2 confirming the specified peptide concentrations. (D) Chromatographic behavior of AQUA peptides. Extracted ion-chromatogram of heavy and light AQUA peptides are shown for c-FLIP, FADD, procaspase-8, and procaspase-10. Heavy and light peptides were mixed and analyzed by nanoLC-ESI-MS1 on an Orbitrap mass spectrometer.
Figure 2.
Selection and validation of AQUA peptides. (A) Amino acid sequences of the major components FADD, procaspase-8/10 and c-FLIP of the CD95 DISC. Selected AQUA peptides are highlighted in red and DED and DD are marked in grey. The scheme of procaspase-8a/b processing is shown below. (B) Basic calibration of corresponding light and heavy AQUA peptide pairs. Variable amounts of the light peptide ranging from 1 to 200 fmol were added to a constant amount of 50 fmol of the corresponding heavy peptide. All AQUA peptide pairs showed linearity in this concentration range when analyzed by mass spectrometry on an LTQ Orbitrap instrument. (C) Quality control of AQUA peptides. Light and heavy AQUA peptides pairs were mixed in a 1:1 ratio and analyzed by mass spectrometry on a QToF instrument. Heavy to light ratios were close to 1:1 or 1:2 confirming the specified peptide concentrations. (D) Chromatographic behavior of AQUA peptides. Extracted ion-chromatogram of heavy and light AQUA peptides are shown for c-FLIP, FADD, procaspase-8, and procaspase-10. Heavy and light peptides were mixed and analyzed by nanoLC-ESI-MS1 on an Orbitrap mass spectrometer.
![Cells 02 00476 g002]()
Table 1.
Sequences of AQUA peptides. Heavy peptides are labeled either with [13C6,15N2] lysine or with [13C6,15N4] arginine at their respective C-terminus.
Table 1.
Sequences of AQUA peptides. Heavy peptides are labeled either with [13C6,15N2] lysine or with [13C6,15N4] arginine at their respective C-terminus.
| Protein | Sequence | Light | Heavy |
|---|
| m/z (charge state) | m/z (charge state) |
|---|
| FADD | ENATVAHLVGALR | 675.8781 (2+) | 680.8822 (2+) |
| | | 450.9211 (3+) | 454.2572 (3+) |
| | IDSIEDR | 424.2114 (2+) | 429.2156 (2+) |
| | ELLASLR | 401.2451 (2+) | 406.2492 (2+) |
| Caspase-8 | NLYDIGEQLDSEDLASLK | 1011.9969 (2+) | 1016.0040 (2+) |
| | FLSLDYIPQR | 626.3402 (2+) | 631.3444 (2+) |
| c-FLIP | LSVGDLAELLYR | 674.8772 (2+) | 679.8813 (2+) |
| | DVAIDVVPPNVR | 647.3617 (2+) | 652.3658 (2+) |
| Caspase-10 | LLIIDSNLGVQDVENLK | 942.0279 (2+) | 946.0350 (2+) |
| | LLQHLNCTK | 563.8055 (2+) | 567.8126 (2+) |
| | FGAVYSSDEALIPIR | 819.4303 (2+) | 824.4345 (2+) |
3.4. Validation of AQUA Peptides
Although the non-labeled “light” AQUA peptides are not necessarily required for the quantification itself they are valuable tools for basic calibration purposes as well as for the determination of the detection limit and sensitivity. To test the linearity of the quantification in the expected concentration range and the recovery rate, we used 50 fmol of the heavy peptide and added variable amounts of the light counterpart. This is exemplified for one peptide each of FADD, procaspase-8, procaspase-10 and c-FLIP (
Figure 2B). Sequences of the four selected AQUA peptides are indicated in
Figure 2B. The calibration curves showed good linearity in the range from 1 to 200 fmol of spiked-in light AQUA peptides (
Figure 2B). The detection limit of AQUA peptides was about 1 fmol for Orbitrap quantifications.
To test the quality of both, light and heavy AQUA peptides as supplied by the vendor, stock solutions were mixed in a 1:1 (v:v) ratio. The sequences of the peptide pairs were the same as above for the calibration curves. Mass spectrometric analysis of those mixtures was performed on a QToF instrument operated off-line in the MS1 modus (
Figure 2C). As illustrated in
Figure 2C the MS1 traces of peptide mixtures showed a very good 1:1 ratio of the m/z signals for the doubly protonated parent ions of FADD, c-FLIP and procaspase-10 and a 1:2 ratio for the m/z signals of procaspase-8. These analyses were repeatedly performed over a period of time to test whether the stock solutions changed in concentration due to adsorption or degradation.
Another important issue is the knowledge of chromatographic behavior of the selected peptides. Heavy and light AQUA peptides derived from FADD, c-FLIP, procaspase-8 and -10 were mixed and separated on a UPLC column using a linear one hour gradient of acetonitrile in 0.1% formic acid. The heavy and light peptide pairs were the same as used before.
Figure 2D shows the extracted ion chromatogram for c-FLIP, procaspase-10, procaspase-8 and FADD. All four peptide pairs were well separated and eluted in sharp and symmetric peaks. Importantly, the retention times for corresponding light and heavy peptide pairs were essentially the same. No retention time shifts were observed which can occur when deuterated peptides are used as heavy AQUA standards [
22].
3.5. Quantification Workflow
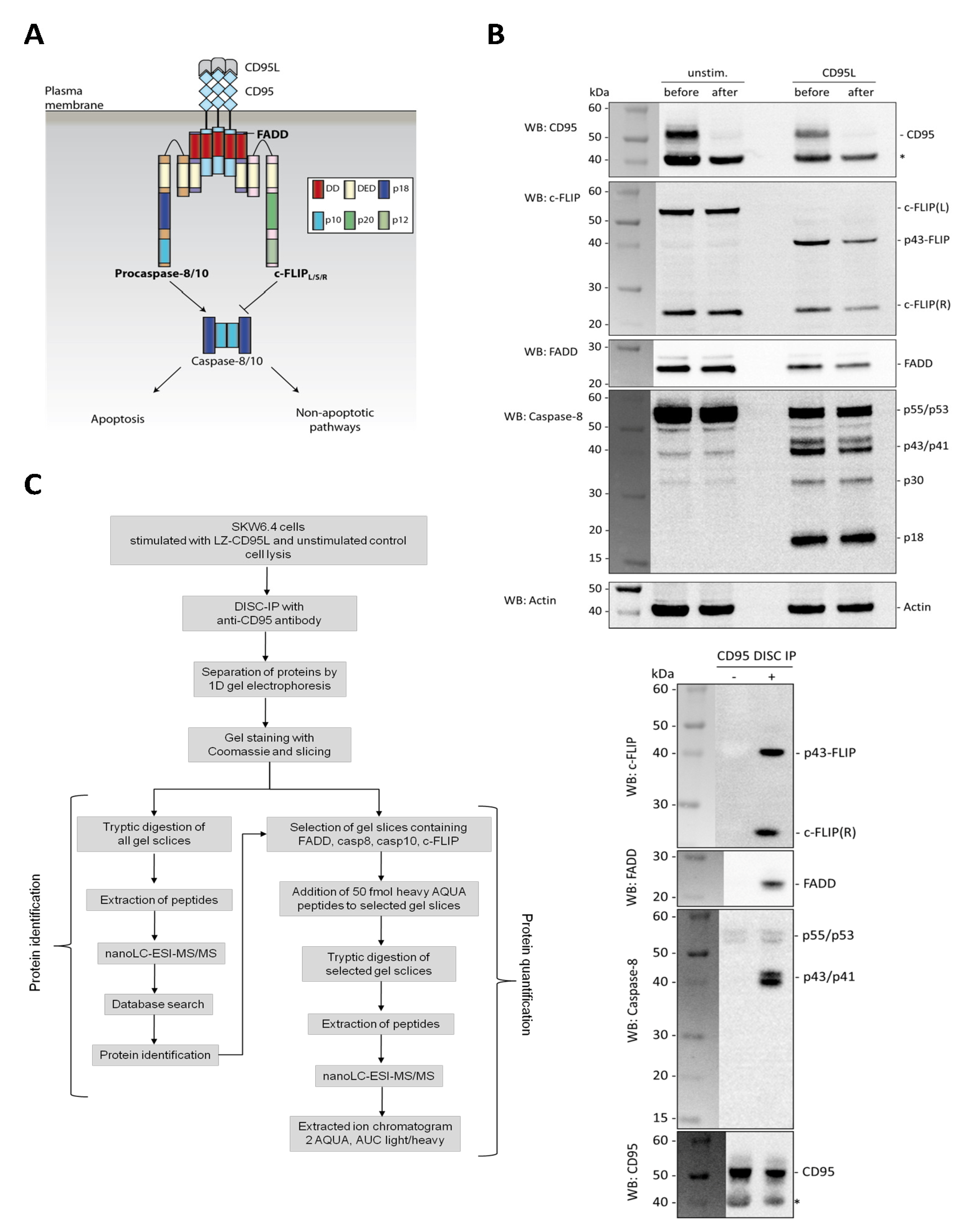
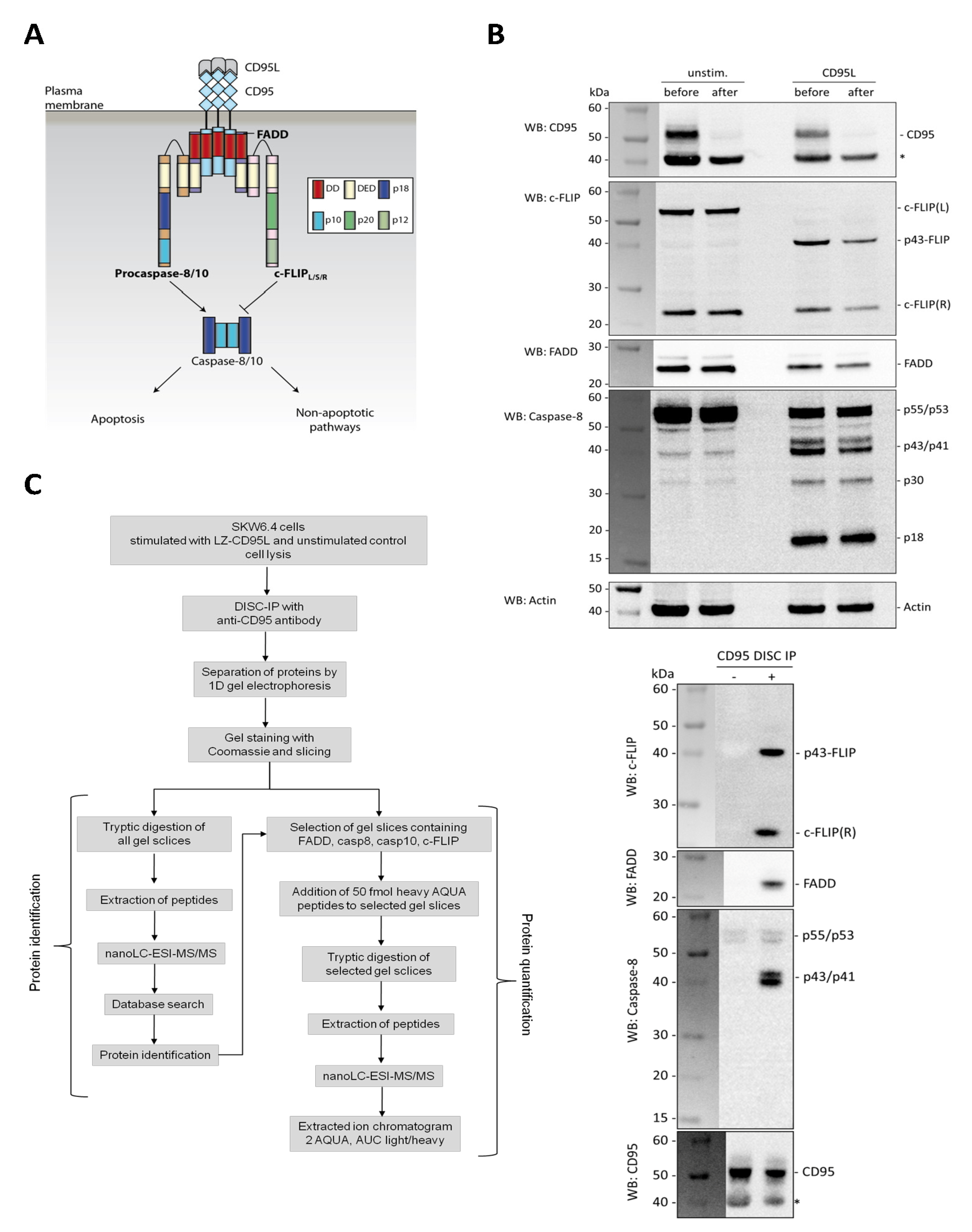
Our study mainly focused on the investigation of the stoichiometry of the DISC proteins FADD, c-FLIP, procaspase-8 and -10. Below we describe the quantification workflow in detail, though it has to be noted that the biological implications of these findings were reported in [
1]. Based on our previous results obtained from the identification workflow, we already knew the gel slices which contained our target molecules. Only these slices were included in the quantification workflow (
Figure 1C, right panel). FADD was detected only once in gel slices corresponding to a molecular weight of 25–28 kDa. In contrast, the three DED proteins procaspase-8, procaspase-10 and c-FLIP are further processed at the DISC and in addition have isoforms different in their molecular masses. They were detected in gel slices according to a mass range of 38–60 kDa, but also in gel slices corresponding to a lower molecular weight around 20–35 kDa.
After reduction and alkylation of proteins in the gel slices and several washing steps, the selected gel slices were spiked with 50 fmol of each heavy AQUA peptide prior to tryptic digestion. Thus, all 10 heavy peptides were present in each sample. Although all AQUA peptides used in our study represent tryptic peptides per se and cannot be further digested it is advisable to add them as early as possible in the workflow to minimize sample to sample variation. After incubation with trypsin, peptides were extracted from the gel pieces and analyzed by nanoLC-ESI-MS/MS.
Quantification of the proteins was achieved in several steps. Firstly, m/z values for the doubly and triply-charged ions of all 10 light and heavy AQUA peptides were calculated in silico. These m/z values were listed in an inclusion table. Secondly, during mass spectrometric analysis on the Orbitrap, the MS1 trace was recorded from all peptides above the detection limit derived from all proteins present in the corresponding gel slice. In contrast, the MS2 trace was only acquired from peptides specified in the inclusion table and reaching the data dependent acquisition threshold. The MS2 trace was later used for a correct assignment of the peptides. Thirdly, extracted ion chromatograms for the selected light and heavy peptide pairs were generated from the MS1 trace. Finally, the area under the curve (marked area, MA) was obtained by manual integration of the peak area of the light and heavy peptide, respectively. Since the spiked-in amount of the heavy peptide was known to be 50 fmol, the total amount of the corresponding light peptide could be calculated based on the light to heavy ratio.
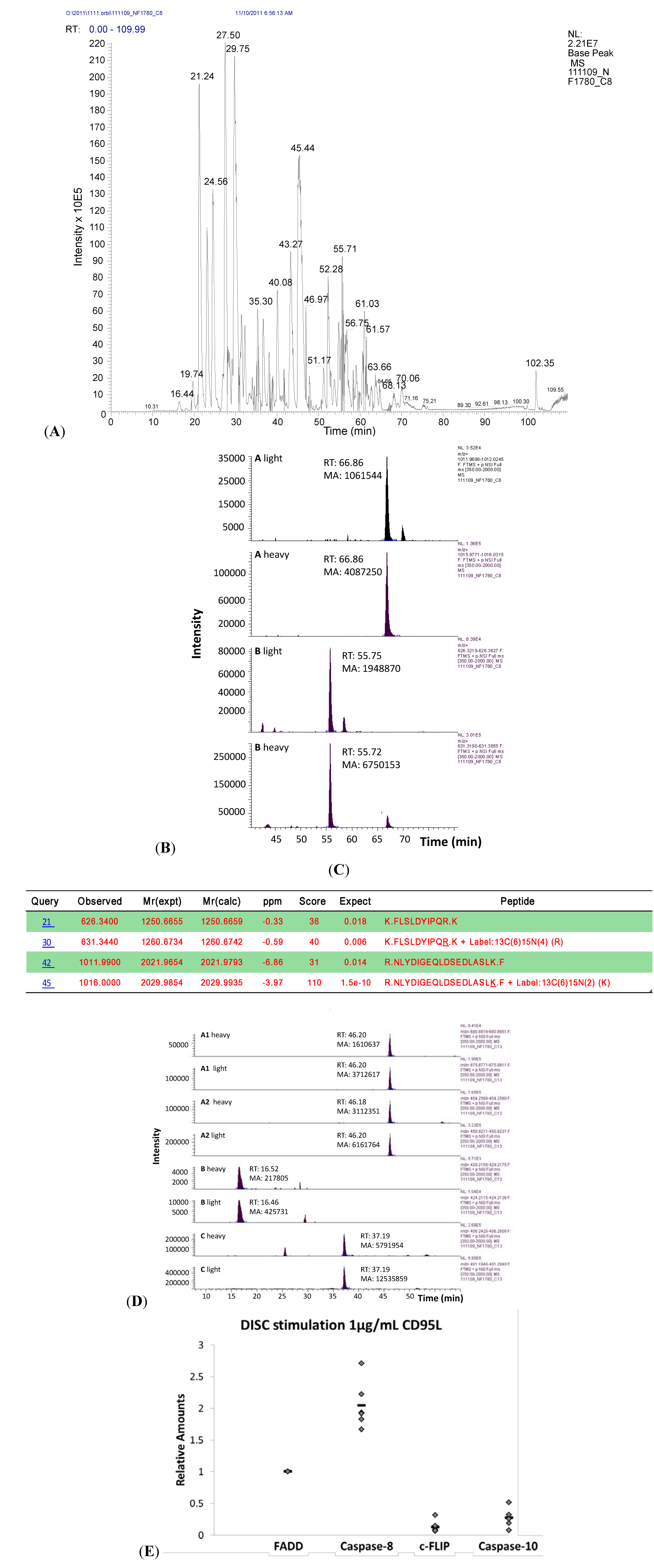
An example of this workflow is given in
Figure 3A–C for procaspase-8. The first panel (
Figure 3A) shows the base peak chromatogram for gel slice 8 and illustrates the high complexity of the sample. Extracted ion chromatograms for two different AQUA peptide pairs derived from procaspase-8 are shown in
Figure 3B. The first two chromatograms in
Figure 3B (A light and A heavy) represent the light and heavy peptide pairs with the sequence NLYDIGEQLDSEDLASLK and a retention time (RT) of about 66.9 min. The marked area (MA) of the heavy peptide corresponds to 50 fmol and is 3.85 times larger than the MA of the light peptide. Thus, the total amount of light peptide in this gel slice is about 14 fmol. The last two chromatograms in
Figure 3B (B light and B heavy) show the extracted ion chromatograms of the light and heavy peptide pairs FLSLDYIPQR with a RT of 55.7 min. The heavy to light ratio calculated from the MA is 3.46 which corresponds to 14.5 fmol of light peptide. This result is in good accordance with the first measurement using the first heavy peptide.
Figure 3.
Quantification of the DISC. (A–C) Workflow for the quantification as exemplified for caspase-8. (A) Base peak chromatogram of the mass spectrometric analysis of gel slice 8. A two hour gradient was applied. (B) Extracted ion chromatograms of the endogenous light and spiked-in heavy peptide NLYDIGEQLDSEDLASLK (A light and A heavy) and endogenous light and spiked-in heavy peptide FLSLDYIPQR. (B light and B heavy). RT: retention time; MA: marked area (AUC). The ratios of light and heavy areas are calculated and used for quantification of the light peptides. (C) Database search of MS2 data confirmed the sequence of the two light and heavy peptide pairs of procaspase-8. (D) Extracted ion chromatograms of corresponding peptide pairs for FADD in gel slice 13. A1 heavy/light and A2 heavy/light represent the same peptide sequence in two different charge states, 2+ and 3+, respectively, with the same retention time of 46 min. The second (B heavy/light) and third peptide (C heavy/light) are more hydrophilic and elute earlier from the reverse phase column. (E) Stoichiometry of the CD95 DISC. Total amounts of proteins were calculated from the extracted ion chromatograms and normalized to FADD. The stoichiometry was determined to be 1:2:0.1:0.3 for FADD, procaspase-8, c-FLIP and procaspase-10.
Figure 3.
Quantification of the DISC. (A–C) Workflow for the quantification as exemplified for caspase-8. (A) Base peak chromatogram of the mass spectrometric analysis of gel slice 8. A two hour gradient was applied. (B) Extracted ion chromatograms of the endogenous light and spiked-in heavy peptide NLYDIGEQLDSEDLASLK (A light and A heavy) and endogenous light and spiked-in heavy peptide FLSLDYIPQR. (B light and B heavy). RT: retention time; MA: marked area (AUC). The ratios of light and heavy areas are calculated and used for quantification of the light peptides. (C) Database search of MS2 data confirmed the sequence of the two light and heavy peptide pairs of procaspase-8. (D) Extracted ion chromatograms of corresponding peptide pairs for FADD in gel slice 13. A1 heavy/light and A2 heavy/light represent the same peptide sequence in two different charge states, 2+ and 3+, respectively, with the same retention time of 46 min. The second (B heavy/light) and third peptide (C heavy/light) are more hydrophilic and elute earlier from the reverse phase column. (E) Stoichiometry of the CD95 DISC. Total amounts of proteins were calculated from the extracted ion chromatograms and normalized to FADD. The stoichiometry was determined to be 1:2:0.1:0.3 for FADD, procaspase-8, c-FLIP and procaspase-10.
![Cells 02 00476 g003]()
To check whether the extraction ion chromatograms represent indeed the two peptide pairs, we performed a database search using the MS2 data. The search result is summarized in
Figure 3C. Both peptides in their light and heavy form were unambiguously identified in the mass spectrometric analysis of gel slice 8, thus confirming that our assignment was correct.
In total, six DISC IPs were performed. Procaspase-8a/b was detected in gel slices 5–15. However, the amount of procaspase-8a/b varied strongly in the different gel slices due to its cleavage to p43/p41, p26/p24, p30 and p18/p10. As can be seen from
Table 2 we observed two peaks of caspase-8, one in gel slices 6–9 and a second one in gel slices 13 and 14. In contrast, gel slices 10–12 contained almost no caspase-8. Since gel slices 6–9 correspond to a molecular weight of about 38–60 kDa we assume that these slices contain unprocessed procaspase-8 and the cleavage product p43/p41. Gel slices 13 and 14 correspond to a molecular weight region around 30 kDa and contain the processed procaspase-8a/b prodomains which are known to remain at the DISC.
Table 2.
Quantification of caspase-8 in different gel slices and total amounts in the six DISC IPs.
Table 2.
Quantification of caspase-8 in different gel slices and total amounts in the six DISC IPs.
| | IP 1 | IP 2 | IP 3 | IP 4 | IP 5 | IP 6 |
|---|
| Gel slice | fmol |
|---|
| 5 | n.d. | 2 | 3 | 4 | 12 | n.d. |
| 6 | 12 | 22 | 37 | 37 | 51 | n.d. |
| 7 | 34 | 83 | 8 | 23 | 58 | 8 |
| 8 | 7 | 14 | 7 | 28 | 26 | 8 |
| 9 | 10 | 22 | 7 | 48 | 47 | 15 |
| 10 | n.d. | 1 | n.d. | 2 | 2 | 1 |
| 11 | n.d. | 1 | n.d. | 1 | 2 | 1 |
| 12 | 2 | 4 | 6 | 8 | 6 | 4 |
| 13 | 21 | 54 | 8 | 55 | 45 | 33 |
| 14 | 7 | 14 | 1 | 51 | 44 | 15 |
| 15 | n.d. | n.d. | n.d. | 1 | 1 | n.d. |
| | | | | | |
| Total Caspase-8 | 91 | 217 | 76 | 259 | 293 | 84 |
FADD was quantified using three different peptides as depicted in
Table 1. The extracted ion chromatograms for all three peptides taken from the mass spectrometry analysis of gel slice 13 are depicted in
Figure 3D. Peptide ENATVAHLVGALR was detected in two different charge states. The A1 heavy/light chromatograms represent the charge state 2+ whereas the A2 heavy/light chromatograms represent the charge state 3+. The retention time is about 46 min and the heavy to light (H/L) ratio is calculated as 0.433 and 0.505, respectively, corresponding to 115 and 99 fmol of FADD in gel slice 13. The second FADD peptide IDSIEDR shown in
Figure 3D (B heavy and light) is the most hydrophilic one and eluted already at 16.5 min. The H/L ratio for this peptide is 0.512 corresponding to a total amount of 98 fmol of FADD in gel slice 13. The third peptide used for quantification of FADD was ELLASLR (
Figure 3D; C heavy and light). The H/L ratio was 0.46 corresponding to 108 fmol of FADD.
For procaspase-10 and c-FLIP the same procedure was applied. Two AQUA peptides were selected for c-FLIP and three AQUA peptides for procaspase-10 (
Table 1). For c-FLIP we could not detect the second AQUA peptide DVAIDVVPPNVR in all DISC IPs. In those cases quantification was based on one peptide only. For procaspase-10 the first two peptides in
Table 1 were derived from the DED whereas peptide FGAVYSSDEALIPIR was selected from the subunit p23/p17. Since procaspase-10 is also further processed at the DISC we could detect the latter peptide only in the gel slices corresponding to the intact protein.
In summary, six replicate DISC IPs were carried out from 5x10
7 SKW6.4 cells stimulated with 1 μg/mL LZ-CD95L.
Table 3 summarizes the results of the quantification of the four DISC proteins. Total amounts of the different DISC proteins were obtained by adding up the amounts in the individual gel slices. Although the total amounts of DISC proteins varied between the different DISC IPs, the ratios of the respective DED proteins to FADD were very reproducible. This, in turn, means that although the total amount of DISC which was immunoprecipitated differed in the six experiments, the stoichiometry of the complex did not change.
Table 3.
Absolute amounts of FADD, procaspase-8, c-FLIP and procaspase-10 detected in DISC IPs after stimulation of SKW6.4 cells with 1 µg/mL LZ-CD95L. The ratios of DISC proteins normalized to FADD are given below.
Table 3.
Absolute amounts of FADD, procaspase-8, c-FLIP and procaspase-10 detected in DISC IPs after stimulation of SKW6.4 cells with 1 µg/mL LZ-CD95L. The ratios of DISC proteins normalized to FADD are given below.
| | | IP 1 | IP 2 | IP 3 | IP 4 | IP 5 | IP 6 | Mean | SD |
|---|
| FADD | fmol | 134 | 132 | 46 | 34 | 113 | 46 | | |
| Procaspase-8 | fmol | 259 | 293 | 84 | 91 | 217 | 76 | | |
| c-FLIP | fmol | 9 | 11 | 3 | 5 | 8 | 14 | | |
| Procaspase-10 | fmol | 25 | 36 | 12 | 11 | 58 | 3 | | |
| Procaspase-8/FADD | | 1.9 | 2.2 | 1.8 | 2.7 | 1.9 | 1.7 | 2.0 | 0.4 |
| c-FLIP/FADD | | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 0.1 | 0.1 |
| Procaspase-10/FADD | | 0.2 | 0.3 | 0.3 | 0.3 | 0.5 | 0.1 | 0.3 | 0.1 |
To further illustrate the method described above we have obtained the following ratios of the proteins: Overall, a mean ratio of 2.0 was measured for procaspase-8 to FADD. For c-FLIP to FADD we calculated a ratio of 0.1 and for procaspase-10 to FADD a ratio of 0.3. As illustrated in
Figure 3E, our results determined a stoichiometry of 1:2:0.1:0.3 for FADD, procaspase-8, c-FLIP and procaspase-10 in the CD95 DISC. These data have been reported in [
1] and served as a basis for the development of the new model of procaspase-8 activation and stoichiometry of the DISC. This stoichiometry provided new insights into DISC organization and apoptosis induction [
1].


{kind=link}
{kind=link}
{kind=link}