Loss of TRPV2 Homeostatic Control of Cell Proliferation Drives Tumor Progression


Abstract
:1. Introduction
2. Structure, Expression and Functional Activation of TRPV2 Channels.
3. Role of TRPV2 Expression in Different Cell Types of Tumor

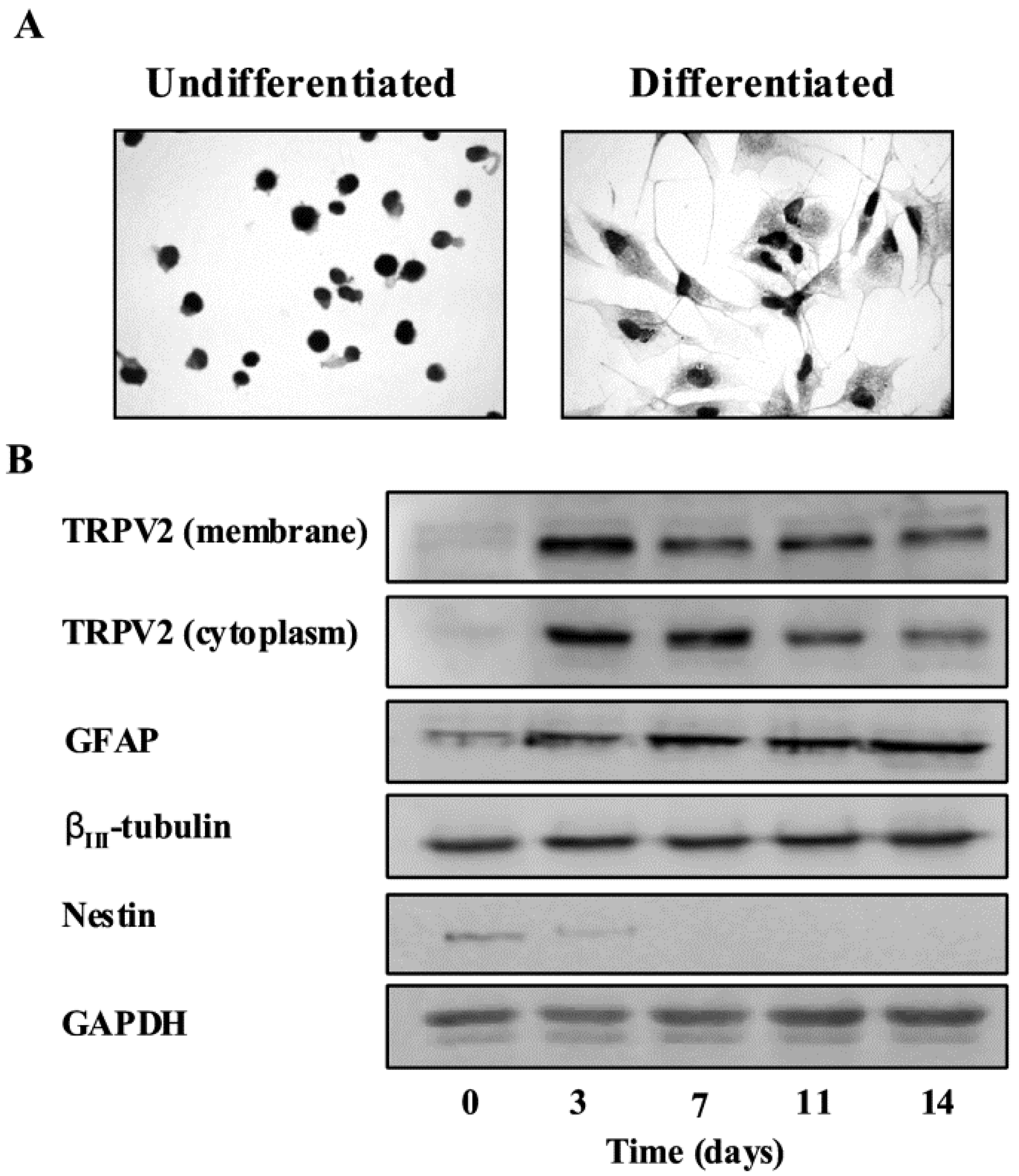{kind=link}
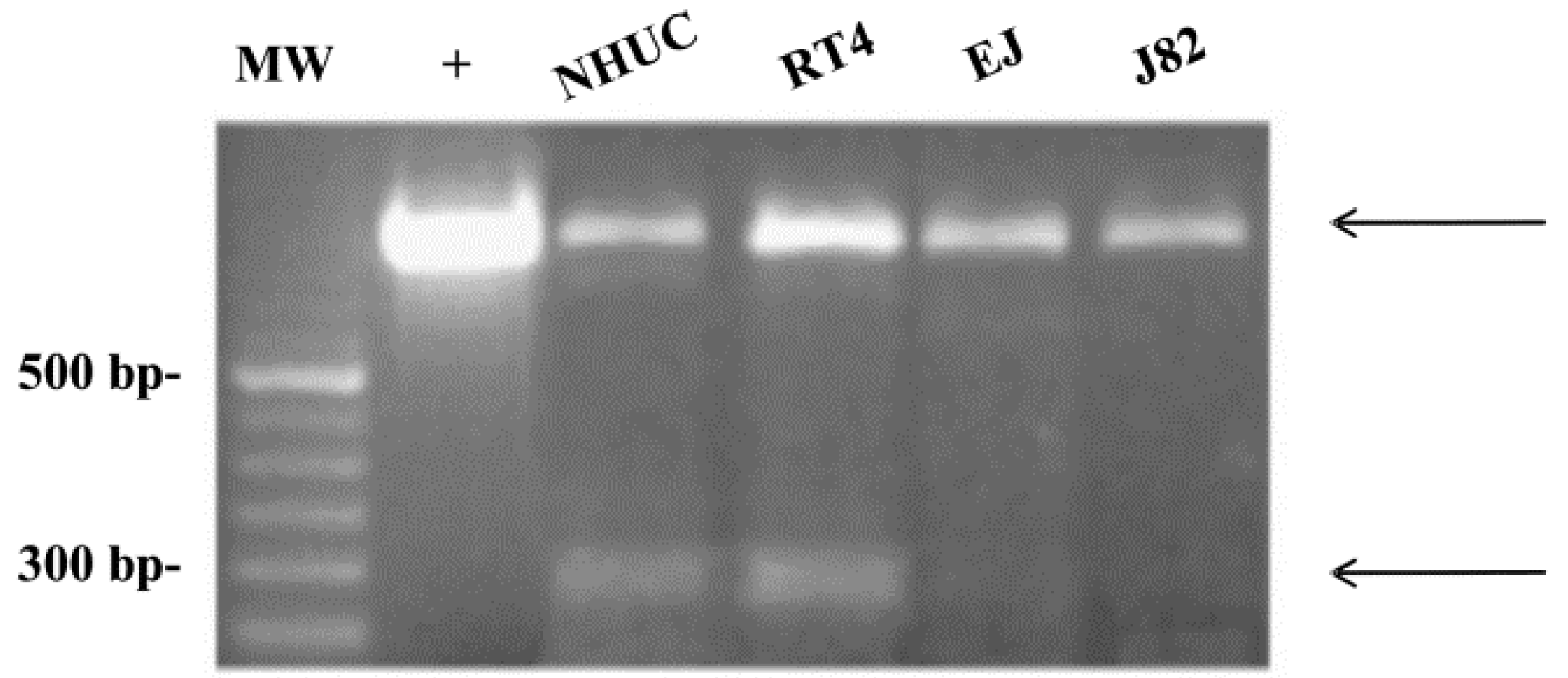{kind=link}
| Tumor | Oncogenic | Tumor suppressor | Reference |
|---|---|---|---|
| Mantle Cell Lymphoma | + | [23] | |
| Multiple Myeloma | + | [24] | |
| Myeloid Acute Leukemia | − | [25] | |
| Glioblastoma | − | [26] | |
| Bladder Carcinoma | + | [27] | |
| Prostate Adenocarcinoma | + | [28] | |
| Hepatocarcinoma | + | [29] |
3.1. TRPV2 in Lymphomas, Leukemias, and Multiple Myelomas
3.2. TRPV2 Expression in Gliomas

3.3. TRPV2 Expression in Bladder Tumor Cells

3.4. TRPV2 Expression in Prostate Cancers
3.5. TRPV2 Expression in Hepatocarcinoma
4. Conclusion
List of Abbreviations
| CBD | Cannabidiol |
| DOXO | Doxorubicin |
| GBM | glioblastoma multiforme |
| GSC | glioblastoma stem-like cells |
| HCC | hepatocellular carcinoma |
| MCL | mantle cell lymphoma |
| MMP | matrix metalloproteinase |
| PI-K | phosphatidylinositol kinase |
| TRP | Transient receptor potential |
| TRPV | TRP Vanilloid |
| UC | urothelial cancer cells |
Acknowledgments
Conflicts of Interest
References
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Nilius, B.; Voets, T. TRP channels: A TR(I)P through a world of multifunctional cation channels. Pflugers. Arch. 2005, 451, 1–10. [Google Scholar] [CrossRef]
- Owsianik, G.; D'hoedt, D.; Voets, T.; Nilius, B. Structure-function relationship of the TRP channel superfamily. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 61–90. [Google Scholar]
- Kiselyov, K.; Soyombo, A.; Muallem, S. TRPpathies. J. Physiol. 2007, 578, 641–653. [Google Scholar] [CrossRef]
- Gkika, D.; Prevarskaya, N. Molecular mechanisms of TRP regulation in tumor growth and metastasis. Biochim. Biophys. Acta 2009, 1793, 953–958. [Google Scholar] [CrossRef]
- Santoni, G.; Farfariello, V. TRP channels and cancer: new targets for diagnosis and chemotherapy. Endocr. Metab. Immune. Disord. Drug Targets 2011, 11, 54–67. [Google Scholar]
- Hoeft, B.; Linseisen, J.; Beckmann, L.; Müller-Decker, K.; Canzian, F.; Hüsing, A.; Kaaks, R.; Vogel, U.; Jakobsen, M.U.; Overvad, K.; et al. Polymorphisms in fatty-acid-metabolism-related genes are associated with colorectal cancer risk. Carcinogenesis 2010, 3, 466–472. [Google Scholar]
- Kalogris, C.; Caprodossi, S.; Amantini, C.; Lambertucci, F.; Nabissi, M.; Morelli, M.B.; Farfariello, V.; Filosa, A.; Emiliozzi, M.C.; Mammana, G.; et al. Expression of transient receptor potential vanilloid-1 (TRPV1) in urothelial cancers of human bladder: Relation to clinicopathological and molecular parameters. Histopathology 2010, 57, 744–752. [Google Scholar] [CrossRef]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef]
- Perálvarez-Marín, A.; Doñate-Macian, P.; Gaudet, R. What do we know about the transient receptor potential vanilloid 2 (TRPV2) ion channel? FEBS J. 2013, 280, 5471–5487. [Google Scholar] [CrossRef]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef]
- Huynh, K.W.; Cohen, M.R.; Chakrapani, S.; Holdaway, H.A.; Stewart, P.L.; Moissenkova-Bell, V.Y. Structural insigh into the assembly of TRPV channels. Structure 2014, 22, 1–9. [Google Scholar] [CrossRef]
- Qin, N.; Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Lubin, M.L.; Flores, C.M. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J. Neurosci. 2008, 28, 6231–6238. [Google Scholar] [CrossRef]
- Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Wang, Y.; Flores, C.M.; Qin, N. Activation properties of heterologously expressed mammalian TRPV2: Evidence for species dependence. J. Biol. Chem. 2007, 282, 15894–15902. [Google Scholar]
- Yamada, T.; Ueda, T.; Shibata, Y.; Ikegami, Y.; Saito, M.; Ishida, Y.; Ugawa, S.; Kohri, K.; Shimada, S. TRPV2 activation induces apoptotic cell death in human T24 bladder cancer cells: a potential therapeutic target for bladder cancer. Urology 2010, 76, 509.e1–509.e7. [Google Scholar]
- Saito, M.; Hanson, P.I.; Schlesinger, P. Luminal chloride-dependent activation of endosome calcium channels: Patch clamp study of enlarged endosomes. J. Biol. Chem. 2007, 282, 27327–27333. [Google Scholar] [CrossRef]
- Abe, K.; Puertollano, R. Role of TRP Channels in the Regulation of the Endosomal Pathway. Physiology 2011, 26, 14–22. [Google Scholar] [CrossRef]
- Kanzaki, M.; Zhang, Y.Q.; Mashima, H.; Li, L.; Shibata, H.; Kojima, I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat. Cell. Biol. 1999, 1, 165–170. [Google Scholar] [CrossRef]
- Penna, A.; Juvin, V.; Chemin, J.; Compan, V.; Monet, M.; Rassendren, F.A. PI3-kinase promotes TRPV2 activity independently of channel translocation to the plasma membrane. Cell Calcium 2006, 39, 495–507. [Google Scholar] [CrossRef]
- Aoyagi, K.; Ohara-Imaizumi, M.; Nishiwaki, C.; Nakamichi, Y.; Nagamatsu, S. Insulin/phosphoinositide 3-kinase pathway accelerates the glucose-induced first-phase insulin secretion through TrpV2 recruitment in pancreatic β-cells. Biochem. J. 2010, 432, 375–386. [Google Scholar] [CrossRef]
- Boels, K.; Glassmeier, G.; Herrmann, D.; Riedel, I.B.; Hampe, W.; Kojima, I.; Schwarz, J.R.; Schaller, H.C. The neuropeptide head activator induces activation and translocation of the growth-factor-regulated Ca(2+)-permeable channel GRC. J. Cell Sci. 2001, 114, 3599–3606. [Google Scholar]
- Cohen, M.R.; Huynh, K.W.; Cawley, D.; Moiseenkova-Bell, V.Y. Understanding the cellular function of TRPV2 chennel through generation of specific monoclonal antibodies. PLoS One 2013, 8, e85392. [Google Scholar]
- Boyd, R.S.; Jukes-Jones, R.; Walewska, R.; Brown, D.; Dyer, M.J.; Cain, K. Protein profiling of plasma membranes defines aberrant signaling pathways in mantle cell lymphoma. Mol. Cell. Proteomics. 2009, 8, 1501–1515. [Google Scholar] [CrossRef]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2013. [Google Scholar] [CrossRef]
- Zatkova, A.; Merk, S.; Wendehack, M.; Bilban, M.; Muzik, E.M.; Muradyan, A.; Haferlach, C.; Haferlach, T.; Wimmer, K.; Fonatsch, C.; et al. AML/MDS with 11q/MLL amplification show characteristic gene expression signature and interplay of DNA copy number changes. Genes Chromosomes Cancer 2009, 48, 510–520. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R.; et al. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef]
- Caprodossi, S.; Lucciarini, R.; Amantini, C.; Nabissi, M.; Canesin, G.; Ballarini, P.; di Spilimbergo, A.; Cardarelli, M.A.; Servi, L.; Mammana, G.; et al. Transient receptor potential vanilloid type 2 (TRPV2) expression in normal urothelium and in urothelial carcinoma of human bladder: correlation with the pathologic stage. Eur. Urol. 2007, 54, 612–620. [Google Scholar]
- Monet, M.; Gkika, D.; Lehen'kyi, V.; Pourtier, A.; Vanden Abeele, F.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarsakaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta 2009, 1793, 528–539. [Google Scholar] [CrossRef]
- Li, N.; Long, Y.; Fan, X.; Liu, H.; Li, C.; Chen, L.; Wang, Z. Proteomic analysis of differentially expressed proteins in hepatitis B virus-related hepatocellular carcinoma tissues. J. Exp. Clin. Cancer Res. 2009, 28, 122. [Google Scholar] [CrossRef]
- Park, K.S.; Pang, B.; Park, S.J.; Lee, Y.G.; Bae, J.Y.; Park, S.; Kim, I.; Kim, S.J. Identification and functional characterization of ion channels in CD34(+) hematopoietic stem cells from human peripheral blood. Mol. Cells 2011, 32, 181–188. [Google Scholar] [CrossRef]
- Chubb, D.; Weinhold, N.; Broderick, P.; Chen, B.; Johnson, D.C.; Försti, A.; Vijayakrishnan, J.; Migliorini, G.; Dobbins, S.E.; Holroyd, A.; et al. Common variation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myeloma risk. Nat. Genet. 2013, 45, 1221–1225. [Google Scholar] [CrossRef]
- Boyd, R.S.; Dyer, M.J.; Cain, K. Proteomic analysis of B-cell malignancies. J. Proteomics 2010, 73, 1804–1822. [Google Scholar] [CrossRef]
- Capasso, M.; Bhamrah, M.; Boyd, R.S.; Cain, K.; Pulford, K.; Musset, B.; Cherny, V.V.; Morgan, D.; DeCoursey, T.E.; Gascoyne, R.D.; et al. The voltage-gated proton channel HVCN1 co-localises with B-cell receptor and is involved in class switch recombination [abstract]. Blood 2000, 112, 707. [Google Scholar]
- Fabris, S.; Todoerti, K.; Mosca, L.; Agnelli, L.; Intini, D.; Lionetti, M.; Guerneri, S.; Lambertenghi-Deliliers, G.; Bertoni, F.; Neri, A. Molecular and transcriptional characterization of the novel 17p11.2-p12 amplicon in multiple myeloma. Genes Chromosomes Cancer 2007, 46, 1109–1118. [Google Scholar] [CrossRef]
- Nagasawa, M.; Nakagawa, Y.; Tanaka, S.; Kojima, I. Chemotactic peptide fMetLeuPhe induces translocation of the TRPV2 channel in macrophages. J. Cell Physiol. 2007, 210, 692–702. [Google Scholar] [CrossRef]
- Yuan, J.; Cahir-McFarland, E.; Zhao, B.; Kieff, E. Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 2006, 80, 2548–2565. [Google Scholar] [CrossRef]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–229. [Google Scholar]
- Giese, A.; Westphal, M. Glioma invasion in the central nervous system. Neurosurgery 1996, 39, 235–252. [Google Scholar] [CrossRef]
- Castro, M.G.; Cowen, R.; Williamson, I.K.; David, A.; Jimenez-Dalmaroni, M.J.; Yuan, X.; Bigliari, A.; Williams, J.C.; Hu, J.; Lowenstein, P.R. Current and future strategies for the treatment of malignant brain tumors. Pharmacol. Ther. 2003, 98, 71–108. [Google Scholar] [CrossRef]
- Yount, G.L.; Levine, K.S.; Kuriyama, H.; Haas-Kogan, D.A.; Israel, M.A. Fas (APO-1/CD95) signaling pathway is intact in radioresistant human glioma cells. Cancer Res. 1999, 59, 1362–1365. [Google Scholar]
- Giraud, S.; Bessette, B.; Boda, C.; Lalloué, F.; Petit, D.; Mathonnet, M.; Jauberteau, M.O. In vitro apoptotic induction of human glioblastoma cells by Fas ligand plus etoposide and in vivo antitumour activity of combined drugs in xenografted nude rats. Int. J. Oncol. 2007, 30, 273–281. [Google Scholar]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; di Martino, S.; Pallini, R.; Larocca, L.M.; Caprodossi, S.; Santoni, M.; et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int. J. Cancer. 2012, 131, E1067–E1077. [Google Scholar] [CrossRef]
- McLendon, R.E.; Friedman, A.H.; Gray, L. Glioblastoma, 7th ed.; Hodder Arnold: London, UK, 2006. [Google Scholar]
- Kawano, H.; Kubota, T.; Sato, K.; Goya, T.; Arikawa, S.; Wakisaka, S. Immunohistochemical study of giant cell in glioblastoma. Clin. Neuropathol. 1995, 14, 118–123. [Google Scholar]
- Ohgaki, H.; Biernat, W.; Reis, R.; Hegi, M.; Kleihues, P. Pathology and Genetics of Tumours of the Nervous System, World Health Organization Classification of Tumours; Kleihues, P., Cavenee, W.K., Eds.; IARC Press: Lyon, France, 2000. [Google Scholar]
- Wang, C.; Hu, H.Z.; Colton, C.K.; Wood, J.D.; Zhu, M.X. An alternative splicing product of the murine TRPV1 gene dominant negatively modulates the activity of TRPV1 channels. J. Biol. Chem. 2004, 279, 37423–37430. [Google Scholar] [CrossRef]
- Thalmann, G.N. Prognostic markers for bladder cancer — Are we there yet? Eur. Urol. 2007, 51, 591–592. [Google Scholar] [CrossRef]
- Yamada, T.; Ueda, T.; Shibata, Y.; Ikegami, Y.; Saito, M.; Ishida, Y.; Ugawa, S.; Kohri, K.; Shimada, S. TRPV2 activation induces apoptotic cell death in human T24 bladder cancer cells: a potential therapeutic target for bladder cancer. Urology 2010, 76, 509.e1–509.e7. [Google Scholar]
- Liu, Q.; Wang, X. Effect of TRPV2 cation channels on the proliferation, migration and invasion of 5637 bladder cancer cells. Exp. Ther. Med. 2013, 6, 1277–1282. [Google Scholar]
- Bödding, M. TRP proteins and cancer. Cell. Signal. 2007, 19, 617–624. [Google Scholar] [CrossRef]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of insulin-like growth factor 1 receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Dunn, S.E.; Hardman, R.A.; Kari, F.W.; Barret, J.C. Insulin-like growth factor 1 (IGF-1) alters drug sensitivity of HBL100 human breast cancer cells by inhibition of apoptosis induced by diverse anticancer drugs. Cancer Res. 1997, 57, 2687–2693. [Google Scholar]
- Galvin, D.J.; Watson, R.W.G.; Gillespie, J.I.; Brady, H.; Fitzpatrick, J.M. Mechanical stretch regulates cell survival in human bladder smooth muscle cells in vitro. Am. J. Physiol. Renal. Physiol. 2002, 283, 1192–1199. [Google Scholar]
- Zhao, H.; Grossman, H.B.; Spitz, M.R.; Lerner, S.P.; Zhang, K.; Wu, X. Plasma levels of insulin-like growth factor-1 and binding protein-3, and their association with bladder cancer risk. J. Urol. 2003, 169, 714–717. [Google Scholar] [CrossRef]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochem. Biophys. Acta 2007, 1772, 989–1003. [Google Scholar]
- Birder, L.A. More than just a barrier: Urothelium as a drugtarget for urinary bladder pain. Am. J. Physiol. Renal. Physiol. 2005, 285, F489–F495. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Pisanti, S.; Gazzerro, P. Cannabinoids and cancer: pros and cons of an antitumour strategy. Br. J. Pharmacol. 2006, 148, 123–135. [Google Scholar] [CrossRef]
- Gkika, D.; Prevarskaya, N. TRP channels in prostate cancer: the good, the bad and the ugly? Asian J. Androl. 2011, 13, 673–676. [Google Scholar] [CrossRef]
- Monet, M.; Lehen'kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef]
- Salvia, R.; Singal, A.G. Hepatocellular carcinoma and other liver lesions. Med. Clin. North Am. 2014, 98, 103–118. [Google Scholar] [CrossRef]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhu, Y.; Huang, D.R.; Wang, Z.Q. Constructing the HBV-human protein interaction network to understand the relationship between HBV andhepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2010, 29, 146. [Google Scholar]
- Velázquez, R.F.; Rodríguez, M.; Navascués, C.A.; Linares, A.; Pérez, R.; Sotorríos, N.G.; Martínez, I.; Rodrigo, L. Prospective analysis of risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Hepatology 2003, 37, 520–527. [Google Scholar] [CrossRef]
- Chisari, F.V. Viruses, immunity and cancer: Lessons from hepatitis B. Am. J. Pathol. 2000, 156, 1117–1132. [Google Scholar] [CrossRef]
- Shimosato, G.; Amaya, F.; Ueda, M.; Tanaka, Y.; Decosterd, I.; Tanaka, M. Peripheral inflammation induces up-regulation of TRPV2 epression in rat DRG. Pain 2005, 119, 225–232. [Google Scholar] [CrossRef]
- Frederick, J.; Buck, M.E.; Matson, D.J.; Cortright, D.N. Increased TRPA1, TRPM8, and TRPV2 expression in dorsal root ganglia by nerve injury. Biochem. Biophys. Res. Commun. 2007, 358, 1058–1064. [Google Scholar] [CrossRef]
- Li, Q.; Xu, B.; Fu, L.; Hao, X.S. Correlation of four vascular specific growth factors with carcinogenesis and portal vein tumor thrombus formation in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2006, 25, 403–409. [Google Scholar]
- Takizawa, D.; Kakizaki, S.; Sohara, N.; Sato, K.; Takagi, H.; Arai, H.; Katakai, K.; Kojima, A.; Matsuzaki, Y.; Mori, M. Hepatocellular carcinoma with portal vein tumor thrombosis: Clinical characteristics, prognosis, and patient survival analysis. Dig. Dis. Sci. 2007, 52, 3290–3295. [Google Scholar] [CrossRef]
- Martins, A.; Cortez-Pinto, H.; Marques-Vidal, P.; Mendes, N.; Silva, S.; Fatela, N.; Glória, H.; Marinho, R.; Távora, I.; Ramalho, F.; de Moura, M.C. Treatment and prognostic factors in patients with hepatocellular carcinoma. Liver Int. 2006, 26, 680–687. [Google Scholar] [CrossRef]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liberati, S.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Santoni, M.; Conti, A.; Nabissi, M.; Cascinu, S.; Santoni, G. Loss of TRPV2 Homeostatic Control of Cell Proliferation Drives Tumor Progression. Cells 2014, 3, 112-128. https://doi.org/10.3390/cells3010112
Liberati S, Morelli MB, Amantini C, Farfariello V, Santoni M, Conti A, Nabissi M, Cascinu S, Santoni G. Loss of TRPV2 Homeostatic Control of Cell Proliferation Drives Tumor Progression. Cells. 2014; 3(1):112-128. https://doi.org/10.3390/cells3010112
Chicago/Turabian StyleLiberati, Sonia, Maria Beatrice Morelli, Consuelo Amantini, Valerio Farfariello, Matteo Santoni, Alessandro Conti, Massimo Nabissi, Stefano Cascinu, and Giorgio Santoni. 2014. "Loss of TRPV2 Homeostatic Control of Cell Proliferation Drives Tumor Progression" Cells 3, no. 1: 112-128. https://doi.org/10.3390/cells3010112