Role of Receptor Tyrosine Kinases and Their Ligands in Glioblastoma

Abstract
:1. Introduction
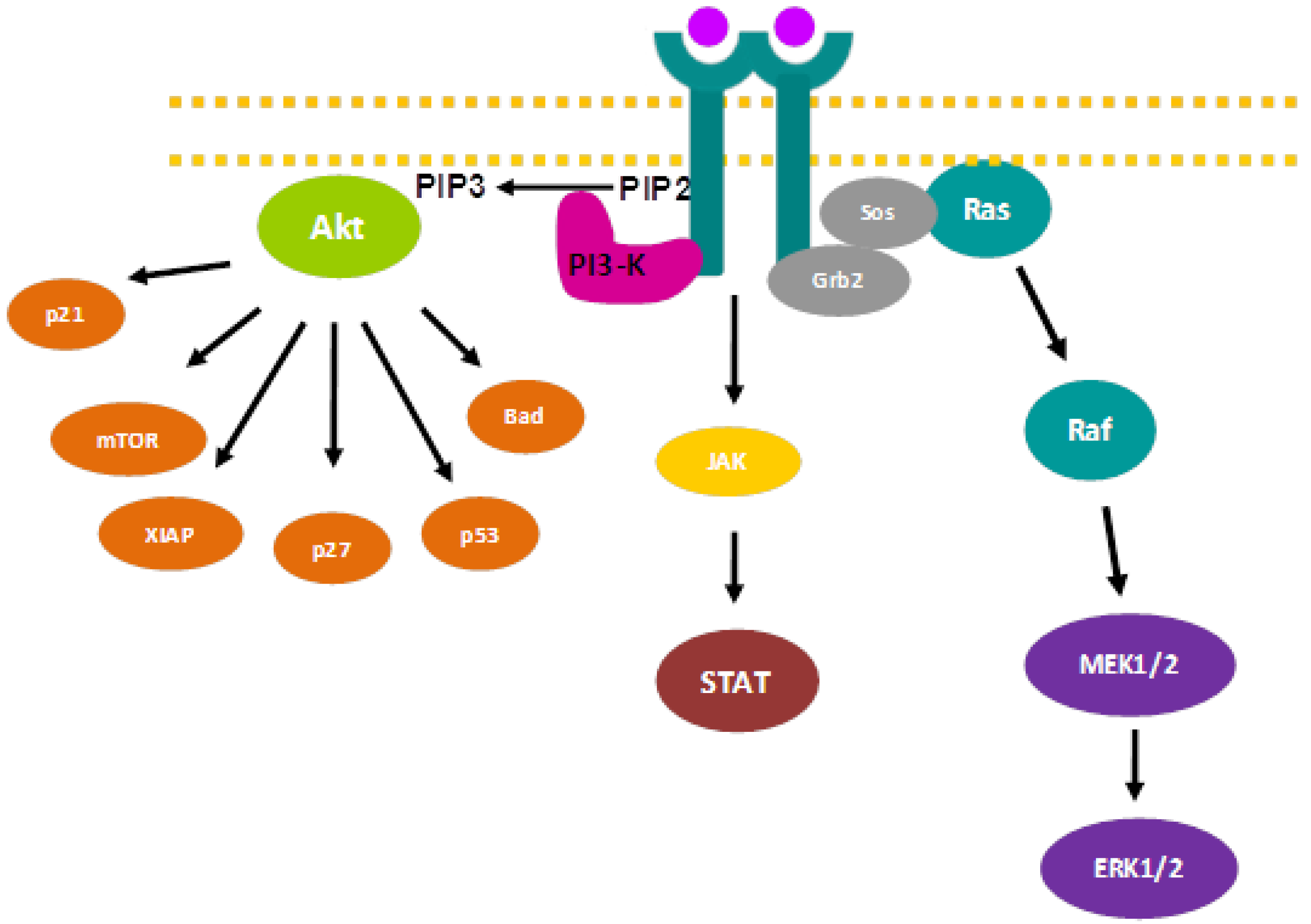
2. Signaling Pathways and Cancer Treatment

{kind=link}
{kind=link}
{kind=link}
| Drug | Company | Target | Indication |
|---|---|---|---|
| Gleevec® (Imatinib) | Novartis Pharma | PDGFR | Chronic myeloid leukemia (CML) and gastrointenstinal stromal tumor (GIST) |
| Erbitux® (Cetuximab/C225) | Merck | EGFR | Colorectal cancer and head and neck squamous cell tumors |
| Tykerb® (Lapatinib) | GlaxoSmithKline | ErbB-2/EGFR | ErbB-2 positive, advanced breast cancer, previously treated with anthracyclines, taxanes or Herceptin® |
| Iressa® (Gefitinib) | AstraZeneca | EGFR | Non-small cell lung cancer (NSCLC) |
| Tarceva® (Erlotinib) | Roche | EGFR | Non-small cell lung cancer (NSCLC) |
| Vectibix® (Panitumumab) | AMGEN | EGFR | Metastasic colorectal cancer |
| Herceptin® (Trastuzumab) | Merck | ErbB-2 | ErbB-2 positive metastasic breast cancer |
| Votrient® (Pazopanib) | GlaxoSmithKline | PDGFR VEGFR, c-KIT | Advanced renal carcinoma |
3. Transmembrane Receptor Tyrosine Kinases

3.1. Epidermal Growth Factor Receptor (EGFR)
3.1.1. Structure and Activation Mechanism
3.1.2. ErbB Ligands
| RECEPTOR | Locus | Protein size | Kinase activity | Ligands |
|---|---|---|---|---|
| EGFR | 7p13-q22 | 170 KDa | yes | EGF, TGFα, AR, HB-EGF, ERG and BTC |
| ErbB-2 | 17q21 | 185 KDa | yes | -- |
| ErbB-3 | 12q13 | 190 KDa | no | NRG 1-4 |
| ErbB-4 | 2q33 | 180 KDa | yes | NRG 1-4, HB-EGF, ERG and BTC |
3.1.3. EGFR and Cancer
3.2. Platelet-Derived Growth Factor Receptor (PDGFR)
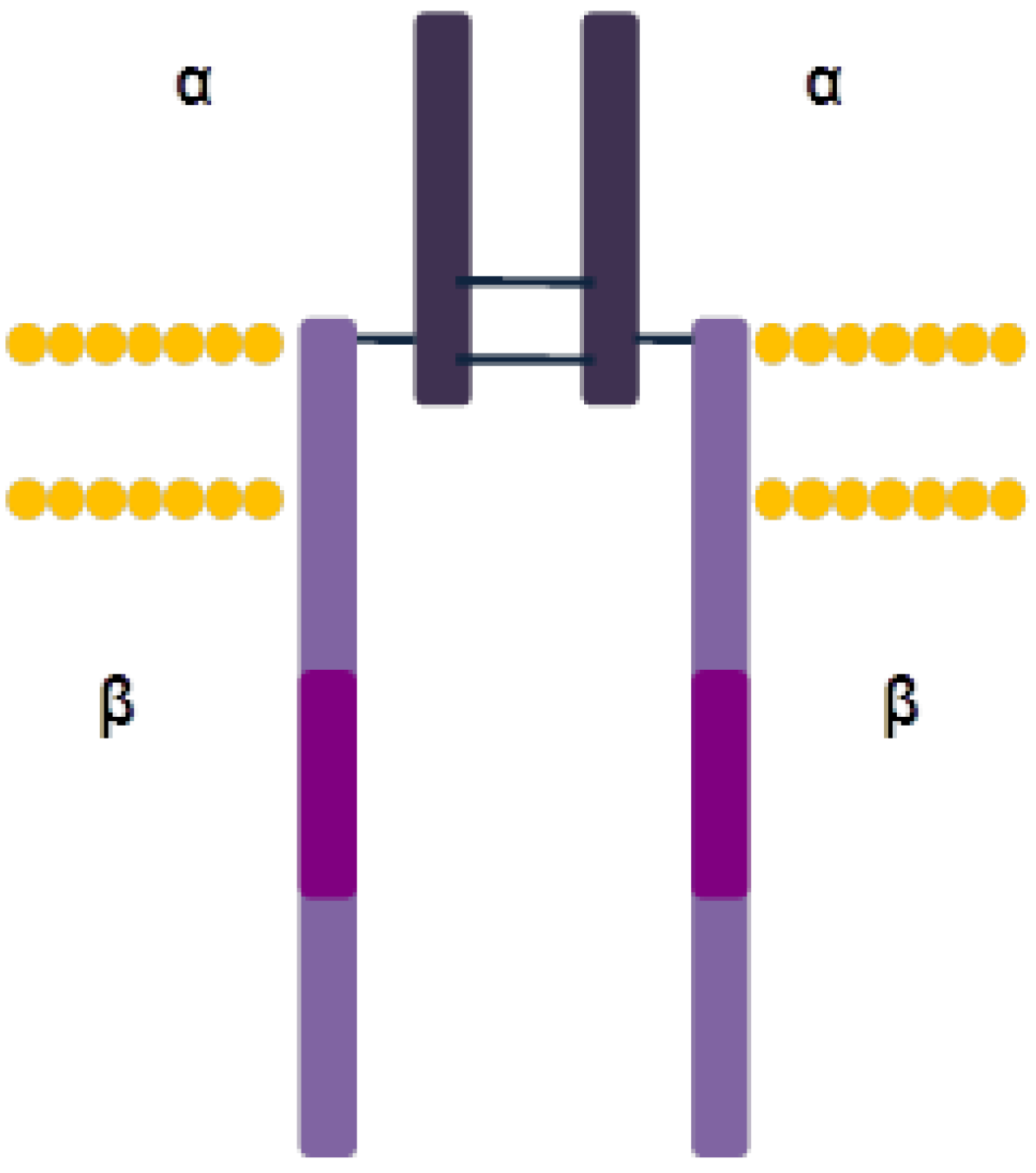
3.2.1. Structure and Activation Mechanism

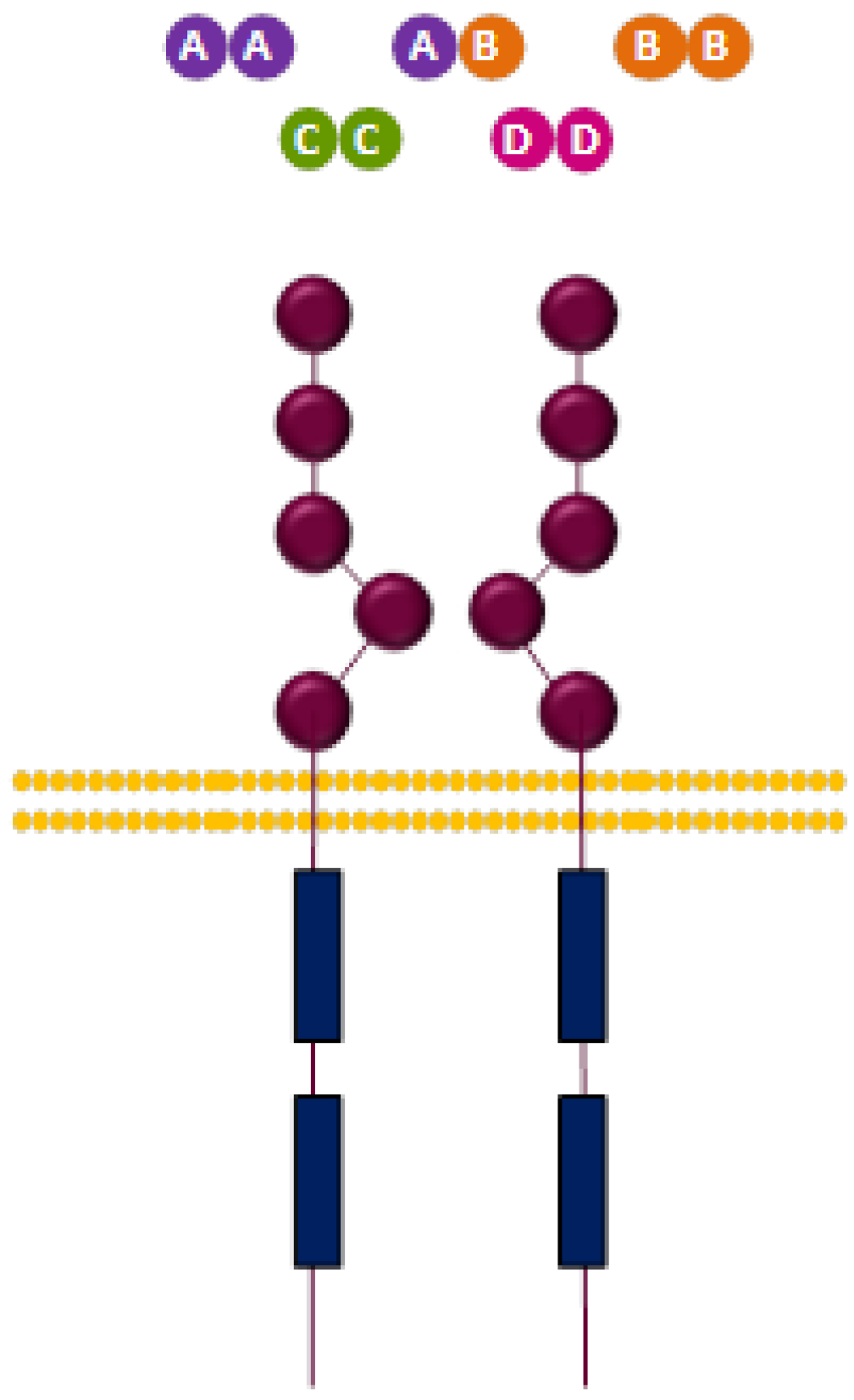
3.2.2. PDGFR Ligands
| Ligands | Dimers |
|---|---|
| AA, BB, CC, AB | PDGFR-α/α |
| BB, DD | PDGFR-β/β |
| BB, CC, AB | PDGFR-α/β |
3.2.3. PDGFR and Cancer
3.3. Insulin-Like Growth Factor 1 Receptor (IGF-1R)
3.3.1. Structure and Activation Mechanism

3.3.2. IGF-1R Ligands
| RECEPTOR | Locus | Structure | Kinase activity | Ligands |
|---|---|---|---|---|
| IR-A | 19p13.2 | dimer | yes | Insulin, IGF-I, IGF-II. |
| IR-B | 19p13.2 | dimer | yes | Insulin, IGF-I |
| IGF-1R | 15q26 | dimer | yes | IGF-I, IGF-II, Insulin |
| IGF-2R | 6q26-27 | monomer | no | IGF-II, lyosomal enzymes |
3.3.3. IGF-1R and Cancer
4. Current Targeted Therapies
4.1. EGFR
4.2. PDGFR
4.3. IGF-1R
4.4. MicroRNAs in Glioblastoma
4.5. Failure of Current Therapies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il'yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef]
- Ohgaki, H. Epidemiology of brain tumors. Methods Mol. Biol. 2009, 472, 323–342. [Google Scholar] [CrossRef]
- Gurney, J.G.; Kadan-Lottick, N. Brain and other central nervous system tumors: Rates, trends, and epidemiology. Curr. Opin. Oncol. 2001, 13, 160–166. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Van den Bent, M.J.; Mason, W.P.; Weller, M.; Mirimanoff, R.O.; Cairncross, J.G. Changing paradigms--an update on the multidisciplinary management of malignant glioma. Oncologist 2006, 11, 165–180. [Google Scholar] [CrossRef]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef]
- Koukourakis, G.V.; Kouloulias, V.; Zacharias, G.; Papadimitriou, C.; Pantelakos, P.; Maravelis, G.; Fotineas, A.; Beli, I.; Chaldeopoulos, D.; Kouvaris, J. Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules 2009, 14, 1561–1577. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Baeza, N.; Weller, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003, 106, 479–485. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Pages, G.; Lenormand, P.; L'Allemain, G.; Chambard, J.C.; Meloche, S.; Pouyssegur, J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. USA 1993, 90, 8319–8323. [Google Scholar] [CrossRef]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; de Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef]
- Lord, J.D.; McIntosh, B.C.; Greenberg, P.D.; Nelson, B.H. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J. Immunol. 2000, 164, 2533–2541. [Google Scholar]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef]
- Akiyama, T.; Sudo, C.; Ogawara, H.; Toyoshima, K.; Yamamoto, T. The product of the human c-erbB-2 gene: A 185-kilodalton glycoprotein with tyrosine kinase activity. Science 1986, 232, 1644–1646. [Google Scholar]
- Kraus, M.H.; Issing, W.; Miki, T.; Popescu, N.C.; Aaronson, S.A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. USA 1989, 86, 9193–9197. [Google Scholar] [CrossRef]
- Plowman, G.D.; Culouscou, J.M.; Whitney, G.S.; Green, J.M.; Carlton, G.W.; Foy, L.; Neubauer, M.G.; Shoyab, M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA 1993, 90, 1746–1750. [Google Scholar] [CrossRef]
- Carpenter, G.; Cohen, S. Human epidermal growth factor and the proliferation of human fibroblasts. J. Cell Physiol. 1976, 88, 227–237. [Google Scholar] [CrossRef]
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar]
- Chinkers, M.; Cohen, S. Purified EGF receptor-kinase interacts specifically with antibodies to Rous sarcoma virus transforming protein. Nature 1981, 290, 516–519. [Google Scholar] [CrossRef]
- Ushiro, H.; Cohen, S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 1980, 255, 8363–8365. [Google Scholar]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef]
- Lax, I.; Bellot, F.; Howk, R.; Ullrich, A.; Givol, D.; Schlessinger, J. Functional analysis of the ligand binding site of EGF-receptor utilizing chimeric chicken/human receptor molecules. EMBO J. 1989, 8, 421–427. [Google Scholar]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Bu, Z.; Ladbury, J.E.; Zhou, M.; Pinchasi, D.; Lax, I.; Engelman, D.M.; Schlessinger, J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997, 16, 281–294. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem. Sci. 1994, 19, 459–463. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Wiley, H.S.; Herbst, J.J.; Walsh, B.J.; Lauffenburger, D.A.; Rosenfeld, M.G.; Gill, G.N. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 1991, 266, 11083–11094. [Google Scholar]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef]
- Guy, P.M.; Platko, J.V.; Cantley, L.C.; Cerione, R.A.; Carraway, K.L., III. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8132–8136. [Google Scholar]
- Pinkas-Kramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M.; et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467. [Google Scholar]
- Lee, D.C.; Rose, T.M.; Webb, N.R.; Todaro, G.J. Cloning and sequence analysis of a cDNA for rat transforming growth factor-alpha. Nature 1985, 313, 489–491. [Google Scholar] [CrossRef]
- Pandiella, A.; Massague, J. Cleavage of the membrane precursor for transforming growth factor alpha is a regulated process. Proc. Natl. Acad. Sci. USA 1991, 88, 1726–1730. [Google Scholar] [CrossRef]
- Herrlich, A.; Klinman, E.; Fu, J.; Sadegh, C.; Lodish, H. Ectodomain cleavage of the EGF ligands HB-EGF, neuregulin1-beta, and TGF-alpha is specifically triggered by different stimuli and involves different PKC isoenzymes. FASEB J. 2008, 22, 4281–4295. [Google Scholar] [CrossRef]
- Anklesaria, P.; Teixido, J.; Laiho, M.; Pierce, J.H.; Greenberger, J.S.; Massague, J. Cell-cell adhesion mediated by binding of membrane-anchored transforming growth factor alpha to epidermal growth factor receptors promotes cell proliferation. Proc. Natl. Acad. Sci. USA 1990, 87, 3289–3293. [Google Scholar] [CrossRef]
- Riese, D.J.; Bermingham, Y.; van Raaij, T.M.; Buckley, S.; Plowman, G.D.; Stern, D.F. Betacellulin activates the epidermal growth factor receptor and erbB-4, and induces cellular response patterns distinct from those stimulated by epidermal growth factor or neuregulin-beta. Oncogene 1996, 12, 345–353. [Google Scholar]
- Carraway, K.L., III; Weber, J.L.; Unger, M.J.; Ledesma, J.; Yu, N.; Gassmann, M.; Lai, C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 1997, 387, 512–516. [Google Scholar] [CrossRef]
- Chang, H.; Riese, D.J.; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512. [Google Scholar] [CrossRef]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell. 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Earp, H.S.; Dawson, T.L.; Li, X.; Yu, H. Heterodimerization and functional interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research. Breast Cancer Res. Treat. 1995, 35, 115–132. [Google Scholar] [CrossRef]
- Riese, D.J.; Stern, D.F. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays 1998, 20, 41–48. [Google Scholar] [CrossRef]
- Gamett, D.C.; Pearson, G.; Cerione, R.A.; Friedberg, I. Secondary dimerization between members of the epidermal growth factor receptor family. J. Biol. Chem. 1997, 272, 12052–12056. [Google Scholar]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 1996, 16, 5276–5287. [Google Scholar]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Yeung, T.K.; Chen, X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell 1999, 10, 1621–1636. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Fedi, P.; Pierce, J.H.; di Fiore, P.P.; Kraus, M.H. Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase C gamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol. Cell Biol. 1994, 14, 492–500. [Google Scholar]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Watanabe, K.; Tachibana, O.; Sata, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223. [Google Scholar] [CrossRef]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat Cancer. 2001, 8, 83–96. [Google Scholar] [CrossRef]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Moscatello, D.K.; Holgado-Madruga, M.; Godwin, A.K.; Ramirez, G.; Gunn, G.; Zoltick, P.W.; Biegel, J.A.; Hayes, R.L.; Wong, A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995, 55, 5536–5539. [Google Scholar]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; Longo, N.; Hamid, M.L.; Olson, J.J.; Liu, L.; Collins, V.P.; James, C.D. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene 1994, 9, 2313–2320. [Google Scholar]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef]
- Malden, L.T.; Novak, U.; Kaye, A.H.; Burgess, A.W. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res. 1988, 48, 2711–2714. [Google Scholar]
- Panneerselvam, K.; Kanakaraj, P.; Raj, S.; Das, M.; Bishayee, S. Characterization of a novel epidermal-growth-factor-receptor-related 200-kDa tyrosine kinase in tumor cells. Eur. J. Biochem. 1995, 230, 951–957. [Google Scholar] [CrossRef]
- Steck, P.A.; Lee, P.; Hung, M.C.; Yung, W.K. Expression of an altered epidermal growth factor receptor by human glioblastoma cells. Cancer Res. 1988, 48, 5433–5439. [Google Scholar]
- Da Cunha, S.G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef]
- Seroogy, K.B.; Gall, C.M.; Lee, D.C.; Kornblum, H.I. Proliferative zones of postnatal rat brain express epidermal growth factor receptor mRNA. Brain Res. 1995, 670, 157–164. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Complex oncogenic signaling networks regulate brain tumor-initiating cells and their progenies: pivotal roles of wild-type EGFR, EGFRvIII mutant and hedgehog cascades and novel multitargeted therapies. Brain Pathol. 2011, 21, 479–500. [Google Scholar]
- Huang, H.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.D.; Huang, C.M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van, D.B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef]
- Ciesielski, M.J.; Fenstermaker, R.A. Oncogenic epidermal growth factor receptor mutants with tandem duplication: gene structure and effects on receptor function. Oncogene. 2000, 19, 810–820. [Google Scholar] [CrossRef]
- Lamszus, K.; Laterra, J.; Westphal, M.; Rosen, E.M. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int. J. Dev. Neurosci. 1999, 17, 517–530. [Google Scholar] [CrossRef]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; Vande Woude, G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar]
- Velpula, K.K.; Dasari, V.R.; Asuthkar, S.; Gorantla, B.; Tsung, A.J. EGFR and c-Met Cross Talk in Glioblastoma and Its Regulation by Human Cord Blood Stem Cells. Transl. Oncol. 2012, 5, 379–392. [Google Scholar] [CrossRef]
- Li, L.; Puliyappadamba, V.T.; Chakraborty, S.; Rehman, A.; Vemireddy, V.; Saha, D.; Souza, R.F.; Hatanpaa, K.J.; Koduru, P.; Burma, S.; et al. EGFR wild type antagonizes EGFRvIII-mediated activation of Met in glioblastoma. Oncogene 2013. [Google Scholar] [CrossRef]
- De, B.F.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D'Ambrosio, A.; Porrati, P.; et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. The radioprotector Bowman-Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiother. Oncol. 2008, 86, 375–382. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer 2008, 7. [Google Scholar] [CrossRef]
- Dittmann, K.H.; Mayer, C.; Ohneseit, P.A.; Raju, U.; Andratschke, N.H.; Milas, L.; Rodemann, H.P. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 203–212. [Google Scholar] [CrossRef]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef]
- Dasari, V.R.; Velpula, K.K.; Alapati, K.; Gujrati, M.; Tsung, A.J. Cord blood stem cells inhibit epidermal growth factor receptor translocation to mitochondria in glioblastoma. PLoS. One. 2012, 7, e31884. [Google Scholar]
- Gomes, D.A.; Rodrigues, M.A.; Leite, M.F.; Gomez, M.V.; Varnai, P.; Balla, T.; Bennett, A.M.; Nathanson, M.H. c-Met must translocate to the nucleus to initiate calcium signals. J. Biol. Chem. 2008, 283, 4344–4351. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Platelet-derived growth factor: Mechanism of action and possible in vivo function. Cell Regul. 1990, 1, 555–566. [Google Scholar]
- Matsui, T.; Heidaran, M.; Miki, T.; Popescu, N.; La Rochelle, W.; Kraus, M.; Pierce, J.; Aaronson, S. Isolation of a novel receptor cDNA establishes the existence of two PDGF receptor genes. Science 1989, 243, 800–804. [Google Scholar]
- Ferns, G.A.; Sprugel, K.H.; Seifert, R.A.; Bowen-Pope, D.F.; Kelly, J.D.; Murray, M.; Raines, E.W.; Ross, R. Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors. 1990, 3, 315–324. [Google Scholar] [CrossRef]
- Betsholtz, C.; Karlsson, L.; Lindahl, P. Developmental roles of platelet-derived growth factors. Bioessays. 2001, 23, 494–507. [Google Scholar] [CrossRef]
- Betsholtz, C.; Lindblom, P.; Bjarnegard, M.; Enge, M.; Gerhardt, H.; Lindahl, P. Role of platelet-derived growth factor in mesangium development and vasculopathies: lessons from platelet-derived growth factor and platelet-derived growth factor receptor mutations in mice. Curr. Opin. Nephrol. Hypertens. 2004, 13, 45–52. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Heidaran, M.A.; Pierce, J.H.; Jensen, R.A.; Matsui, T.; Aaronson, S.A. Chimeric alpha- and beta-platelet-derived growth factor (PDGF) receptors define three immunoglobulin-like domains of the alpha-PDGF receptor that determine PDGF-AA binding specificity. J. Biol. Chem. 1990, 265, 18741–18744. [Google Scholar]
- Kanakaraj, P.; Raj, S.; Khan, S.A.; Bishayee, S. Ligand-induced interaction between alpha- and beta-type platelet-derived growth factor (PDGF) receptors: Role of receptor heterodimers in kinase activation. Biochemistry. 1991, 30, 1761–1767. [Google Scholar] [CrossRef]
- Kelly, J.D.; Haldeman, B.A.; Grant, F.J.; Murray, M.J.; Seifert, R.A.; Bowen-Pope, D.F.; Cooper, J.A.; Kazlauskas, A. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 1991, 266, 8987–8992. [Google Scholar]
- Kumjian, D.A.; Wahl, M.I.; Rhee, S.G.; Daniel, T.O. Platelet-derived growth factor (PDGF) binding promotes physical association of PDGF receptor with phospholipase C. Proc. Natl. Acad. Sci. USA 1989, 86, 8232–8236. [Google Scholar] [CrossRef]
- Tallquist, M.; Kazlauskas, A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004, 15, 205–213. [Google Scholar] [CrossRef]
- Escobedo, J.A.; Kaplan, D.R.; Kavanaugh, W.M.; Turck, C.W.; Williams, L.T. A phosphatidylinositol-3 kinase binds to platelet-derived growth factor receptors through a specific receptor sequence containing phosphotyrosine. Mol. Cell Biol. 1991, 11, 1125–1132. [Google Scholar]
- Ronnstrand, L.; Mori, S.; Arridsson, A.K.; Eriksson, A.; Wernstedt, C.; Hellman, U.; Claesson-Welsh, L.; Heldin, C.H. Identification of two C-terminal autophosphorylation sites in the PDGF beta-receptor: involvement in the interaction with phospholipase C-gamma. EMBO J. 1992, 11, 3911–3919. [Google Scholar]
- Marra, F.; Pinzani, M.; DeFranco, R.; Laffi, G.; Gentilini, P. Involvement of phosphatidylinositol 3-kinase in the activation of extracellular signal-regulated kinase by PDGF in hepatic stellate cells. FEBS Lett. 1995, 376, 141–145. [Google Scholar] [CrossRef]
- Heidaran, M.A.; Beeler, J.F.; Yu, J.C.; Ishibashi, T.; LaRochelle, W.J.; Pierce, J.H.; Aaronson, S.A. Differences in substrate specificities of alpha and beta platelet-derived growth factor (PDGF) receptors. Correlation with their ability to mediate PDGF transforming functions. J. Biol. Chem. 1993, 268, 9287–9295. [Google Scholar]
- Diliberto, P.A.; Gordon, G.W.; Yu, C.L.; Earp, H.S.; Herman, B. Platelet-derived growth factor (PDGF) alpha receptor activation modulates the calcium mobilizing activity of the PDGF beta receptor in Balb/c3T3 fibroblasts. J. Biol. Chem. 1992, 267, 11888–11897. [Google Scholar]
- Klinghoffer, R.A.; Mueting-Nelsen, P.F.; Faerman, A.; Shani, M.; Soriano, P. The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol. Cell 2001, 7, 343–354. [Google Scholar] [CrossRef]
- Antoniades, H.N.; Scher, C.D.; Stiles, C.D. Purification of human platelet-derived growth factor. Proc. Natl. Acad. Sci. USA 1979, 76, 1809–1813. [Google Scholar] [CrossRef]
- Li, X.; Ponten, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Backstrom, G.; Hellstrom, M.; Bostrom, H.; Li, H.; et al. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999, 79, 1283–1316. [Google Scholar]
- Eriksson, A.; Rorsman, C.; Ernlund, A.; Claesson-Welsh, L.; Heldin, C.H. Ligand-induced homo- and hetero-dimerization of platelet-derived growth factor alpha- and beta-receptors in intact cells. Growth Factors. 1992, 6, 1–14. [Google Scholar] [CrossRef]
- Dai, C.; Celestino, J.C.; Okada, Y.; Louis, D.N.; Fuller, G.N.; Holland, E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001, 15, 1913–1925. [Google Scholar] [CrossRef]
- Uhrbom, L.; Hesselager, G.; Nister, M.; Westermark, B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998, 58, 5275–5279. [Google Scholar]
- Schiffer, D.; Annovazzi, L.; Caldera, V.; Mellai, M. On the origin and growth of gliomas. Anticancer Res. 2010, 30, 1977–1998. [Google Scholar]
- Jackson, E.L.; Garcia-Verdugo, J.M.; Gil-Perotin, S.; Roy, M.; Quinones-Hinojosa, A.; VandenBerg, S.; Alvarez-Buylla, A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 2006, 51, 187–199. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Jeske, I.; Muller, D.; Buslei, R.; Coras, R.; Hahnen, E.; Huttner, H.B.; Corbeil, D.; Kaesbauer, J.; Appl, T.; et al. Spontaneous in vitro transformation of adult neural precursors into stem-like cancer cells. Brain Pathol. 2009, 19, 399–408. [Google Scholar] [CrossRef]
- Antoniades, H.N.; Galanopoulos, T.; Neville-Golden, J.; O'Hara, C.J. Malignant epithelial cells in primary human lung carcinomas coexpress in vivo platelet-derived growth factor (PDGF) and PDGF receptor mRNAs and their protein products. Proc. Natl. Acad. Sci. USA 1992, 89, 3942–3946. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Koopmann, J.; Maintz, D.; Waha, A.; Westermark, B.; Heldin, C.H.; Wiestler, O.D.; Louis, D.N.; von Deimling, A.; et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor alpha receptor expression in human malignant gliomas. Cancer Res. 1996, 56, 164–171. [Google Scholar]
- Seymour, L.; Dajee, D.; Bezwoda, W.R. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res. Treat. 1993, 26, 247–252. [Google Scholar] [CrossRef]
- Paulsson, J.; Sjoblom, T.; Micke, P.; Ponten, F.; Landberg, G.; Heldin, C.H.; Bergh, J.; Brennan, D.J.; Jirstrom, K.; Ostman, A. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am. J. Pathol. 2009, 175, 334–341. [Google Scholar] [CrossRef]
- Dolloff, N.G.; Shulby, S.S.; Nelson, A.V.; Stearns, M.E.; Johannes, G.J.; Thomas, J.D.; Meucci, O.; Fatatis, A. Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor. Oncogene 2005, 24, 6848–6854. [Google Scholar] [CrossRef]
- Kong, D.; Wang, Z.; Sarkar, S.H.; Li, Y.; Banerjee, S.; Saliganan, A.; Kim, H.R.; Cher, M.L.; Sarkar, F.H. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells 2008, 26, 1425–1435. [Google Scholar] [CrossRef]
- Nister, M.; Libermann, T.A.; Betsholtz, C.; Pettersson, M.; Claesson-Welsh, L.; Heldin, C.H.; Schlessinger, J.; Westermark, B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988, 48, 3910–3918. [Google Scholar]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grunert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Ho, C.L.; Hsu, L.F.; Phyliky, R.L.; Li, C.Y. Autocrine expression of platelet-derived growth factor B in B cell chronic lymphocytic leukemia. Acta Haematol. 2005, 114, 133–140. [Google Scholar] [CrossRef]
- Furuhashi, M.; Sjoblom, T.; Abramsson, A.; Ellingsen, J.; Micke, P.; Li, H.; Bergsten-Folestad, E.; Eriksson, U.; Heuchel, R.; Betsholtz, C.; et al. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004, 64, 2725–2733. [Google Scholar] [CrossRef]
- Clarke, I.D.; Dirks, P.B. A human brain tumor-derived PDGFR-alpha deletion mutant is transforming. Oncogene 2003, 22, 722–733. [Google Scholar] [CrossRef]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010, 24, 2205–2218. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von, M.M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Kumabe, T.; Sohma, Y.; Kayama, T.; Yoshimoto, T.; Yamamoto, T. Amplification of alpha-platelet-derived growth factor receptor gene lacking an exon coding for a portion of the extracellular region in a primary brain tumor of glial origin. Oncogene 1992, 7, 627–633. [Google Scholar]
- Cross, N.C.; Reiter, A. Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia 2002, 16, 1207–1212. [Google Scholar] [CrossRef]
- Baxter, E.J.; Kulkarni, S.; Vizmanos, J.L.; Jaju, R.; Martinelli, G.; Testoni, N.; Hughes, G.; Salamanchuk, Z.; Calasanz, M.J.; Lahortiga, I.; et al. Novel translocations that disrupt the platelet-derived growth factor receptor beta (PDGFRB) gene in BCR-ABL-negative chronic myeloproliferative disorders. Br. J. Haematol. 2003, 120, 251–256. [Google Scholar] [CrossRef]
- Gotlib, J.; Cools, J.; Malone, J.M., III; Schrier, S.L.; Gilliland, D.G.; Coutre, S.E. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: Implications for diagnosis, classification, and management. Blood 2004, 103, 2879–2891. [Google Scholar]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar]
- Baserga, R.; Hongo, A.; Rubini, M.; Prisco, M.; Valentinis, B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta 1997, 1332, F105–F126. [Google Scholar]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar]
- De la Monte, S.M.; Tong, M.; Bowling, N.; Moskal, P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: relevance to fetal alcohol spectrum disorder. Mol. Brain 2011, 4, 13. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, C.; Blakesley, V.; Kalebic, T.; Helman, L.; LeRoith, D. The role of the tyrosine kinase domain of the insulin-like growth factor-I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J. Biol. Chem. 1995, 270, 29176–29181. [Google Scholar] [CrossRef]
- Hubbard, S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 464–471. [Google Scholar] [CrossRef]
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8, 1058–1063. [Google Scholar] [CrossRef]
- Kato, H.; Faria, T.N.; Stannard, B.; Roberts, C.T., Jr.; LeRoith, D. Essential role of tyrosine residues 1131, 1135, and 1136 of the insulin-like growth factor-I (IGF-I) receptor in IGF-I action. Mol. Endocrinol. 1994, 8, 40–50. [Google Scholar]
- Furlanetto, R.W. Receptor-mediated endocytosis and lysosomal processing of insulin-like growth factor I by mitogenically responsive cells. Endocrinology 1988, 122, 2044–2053. [Google Scholar] [CrossRef]
- Lou, M.; Garrett, T.P.; McKern, N.M.; Hoyne, P.A.; Epa, V.C.; Bentley, J.D.; Lovrecz, G.O.; Cosgrove, L.J.; Frenkel, M.J.; Ward, C.W. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 12429–12434. [Google Scholar] [CrossRef]
- Lawrence, M.C.; McKern, N.M.; Ward, C.W. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 2007, 17, 699–705. [Google Scholar] [CrossRef]
- Seino, S.; Bell, G.I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 1989, 159, 312–316. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Pavelic, K.; Bukovic, D.; Pavelic, J. The role of insulin-like growth factor 2 and its receptors in human tumors. Mol. Med. 2002, 8, 771–780. [Google Scholar]
- Ludwig, T.; Munier-Lehmann, H.; Bauer, U.; Hollinshead, M.; Ovitt, C.; Lobel, P.; Hoflack, B. Differential sorting of lysosomal enzymes in mannose 6-phosphate receptor-deficient fibroblasts. EMBO J. 1994, 13, 3430–3437. [Google Scholar]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- Moxham, C.P.; Duronio, V.; Jacobs, S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J. Biol. Chem. 1989, 264, 13238–13244. [Google Scholar]
- Soos, M.A.; Siddle, K. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem. J. 1989, 263, 553–563. [Google Scholar]
- Treadway, J.L.; Morrison, B.D.; Goldfine, I.D.; Pessin, J.E. Assembly of insulin/insulin-like growth factor-1 hybrid receptors in vitro. J. Biol. Chem. 1989, 264, 21450–21453. [Google Scholar]
- Baxter, R.C. Insulin-like growth factor (IGF)-binding proteins: Interactions with IGFs and intrinsic bioactivities. Am. J. Physiol Endocrinol. Metab 2000, 278, E967–E976. [Google Scholar]
- Werner, H.; Shalita-Chesner, M.; Abramovitch, S.; Idelman, G.; Shaharabani-Gargir, L.; Glaser, T. Regulation of the insulin-like growth factor-I receptor gene by oncogenes and antioncogenes: implications in human cancer. Mol. Genet. Metab 2000, 71, 315–320. [Google Scholar] [CrossRef]
- Sell, C.; Rubini, M.; Rubin, R.; Liu, J.P.; Efstratiadis, A.; Baserga, R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11217–11221. [Google Scholar]
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell Biol. 1994, 14, 3604–3612. [Google Scholar]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef]
- Carboni, J.M.; Lee, A.V.; Hadsell, D.L.; Rowley, B.R.; Lee, F.Y.; Bol, D.K.; Camuso, A.E.; Gottardis, M.; Greer, A.F.; Ho, C.P.; et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005, 65, 3781–3787. [Google Scholar] [CrossRef]
- Linnerth, N.M.; Siwicky, M.D.; Campbell, C.I.; Watson, K.L.; Petrik, J.J.; Whitsett, J.A.; Moorehead, R.A. Type I insulin-like growth factor receptor induces pulmonary tumorigenesis. Neoplasia 2009, 11, 672–682. [Google Scholar]
- All-Ericsson, C.; Girnita, L.; Seregard, S.; Bartolazzi, A.; Jager, M.J.; Larsson, O. Insulin-like growth factor-1 receptor in uveal melanoma: A predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol. Vis. Sci. 2002, 43, 1–8. [Google Scholar]
- Hellawell, G.O.; Turner, G.D.; Davies, D.R.; Poulsom, R.; Brewster, S.F.; Macaulay, V.M. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002, 62, 2942–2950. [Google Scholar]
- Weber, M.M.; Fottner, C.; Liu, S.B.; Jung, M.C.; Engelhardt, D.; Baretton, G.B. Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer 2002, 95, 2086–2095. [Google Scholar] [CrossRef]
- Bergmann, U.; Funatomi, H.; Yokoyama, M.; Beger, H.G.; Korc, M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995, 55, 2007–2011. [Google Scholar]
- Sichani, M.M.; Yazdi, F.S.; Moghaddam, N.A.; Chehrei, A.; Kabiri, M.; Naeimi, A.; Taheri, D. Prognostic value of insulin- like growth factor-I receptor expression in renal cell carcinoma. Saudi. J. Kidney Dis. Transpl. 2010, 21, 69–74. [Google Scholar]
- Resnicoff, M.; Sell, C.; Rubini, M.; Coppola, D.; Ambrose, D.; Baserga, R.; Rubin, R. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994, 54, 2218–2222. [Google Scholar]
- Resnicoff, M.; Li, W.; Basak, S.; Herlyn, D.; Baserga, R.; Rubin, R. Inhibition of rat C6 glioblastoma tumor growth by expression of insulin-like growth factor I receptor antisense mRNA. Cancer Immunol. Immunother. 1996, 42, 64–68. [Google Scholar] [CrossRef]
- Rininsland, F.; Johnson, T.R.; Chernicky, C.L.; Schulze, E.; Burfeind, P.; Ilan, J. Suppression of insulin-like growth factor type I receptor by a triple-helix strategy inhibits IGF-I transcription and tumorigenic potential of rat C6 glioblastoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 5854–5859. [Google Scholar] [CrossRef]
- Resnicoff, M.; Tjuvajev, J.; Rotman, H.L.; Abraham, D.; Curtis, M.; Aiken, R.; Baserga, R. Regression of C6 rat brain tumors by cells expressing an antisense insulin-like growth factor I receptor RNA. J. Exp. Ther. Oncol. 1996, 1, 385–389. [Google Scholar]
- Berns, E.M.; Klijn, J.G.; van Staveren, I.L.; Portengen, H.; Foekens, J.A. Sporadic amplification of the insulin-like growth factor 1 receptor gene in human breast tumors. Cancer Res. 1992, 52, 1036–1039. [Google Scholar]
- Sekyi-Otu, A.; Bell, R.S.; Ohashi, C.; Pollak, M.; Andrulis, I.L. Insulin-like growth factor 1 (IGF-1) receptors, IGF-1, and IGF-2 are expressed in primary human sarcomas. Cancer Res. 1995, 55, 129–134. [Google Scholar]
- DiGiovanni, J.; Bol, D.K.; Wilker, E.; Beltran, L.; Carbajal, S.; Moats, S.; Ramirez, A.; Jorcano, J.; Kiguchi, K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000, 60, 1561–1570. [Google Scholar]
- DiGiovanni, J.; Kiguchi, K.; Frijhoff, A.; Wilker, E.; Bol, D.K.; Beltran, L.; Moats, S.; Ramirez, A.; Jorcano, J.; Conti, C. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 3455–3460. [Google Scholar] [CrossRef]
- Kiess, W.; Lee, L.; Graham, D.E.; Greenstein, L.; Tseng, L.Y.; Rechler, M.M.; Nissley, S.P. Rat C6 glial cells synthesize insulin-like growth factor I (IGF-I) and express IGF-I receptors and IGF-II/mannose 6-phosphate receptors. Endocrinology 1989, 124, 1727–1736. [Google Scholar] [CrossRef]
- Trojan, J.; Blossey, B.K.; Johnson, T.R.; Rudin, S.D.; Tykocinski, M.; Ilan, J.; Ilan, J. Loss of tumorigenicity of rat glioblastoma directed by episome-based antisense cDNA transcription of insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1992, 89, 4874–4878. [Google Scholar] [CrossRef]
- Schlenska-Lange, A.; Knupfer, H.; Lange, T.J.; Kiess, W.; Knupfer, M. Cell proliferation and migration in glioblastoma multiforme cell lines are influenced by insulin-like growth factor I in vitro. Anticancer Res. 2008, 28, 1055–1060. [Google Scholar]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Rosenzweig, S.A.; Atreya, H.S. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem. Pharmacol. 2010, 80, 1115–1124. [Google Scholar] [CrossRef]
- Goya, M.; Miyamoto, S.; Nagai, K.; Ohki, Y.; Nakamura, K.; Shitara, K.; Maeda, H.; Sangai, T.; Kodama, K.; Endoh, Y.; et al. Growth inhibition of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice by a ligand-specific antibody to human insulin-like growth factors. Cancer Res. 2004, 64, 6252–6258. [Google Scholar] [CrossRef]
- Kimura, T.; Kuwata, T.; Ashimine, S.; Yamazaki, M.; Yamauchi, C.; Nagai, K.; Ikehara, A.; Feng, Y.; Dimitrov, D.S.; Saito, S.; et al. Targeting of bone-derived insulin-like growth factor-II by a human neutralizing antibody suppresses the growth of prostate cancer cells in a human bone environment. Clin. Cancer Res. 2010, 16, 121–129. [Google Scholar] [CrossRef]
- Gucev, Z.S.; Oh, Y.; Kelley, K.M.; Rosenfeld, R.G. Insulin-like growth factor binding protein 3 mediates retinoic acid- and transforming growth factor beta2-induced growth inhibition in human breast cancer cells. Cancer Res. 1996, 56, 1545–1550. [Google Scholar]
- Higo, H.; Duan, C.; Clemmons, D.R.; Herman, B. Retinoic acid inhibits cell growth in HPV negative cervical carcinoma cells by induction of insulin-like growth factor binding protein-5 (IGFBP-5) secretion. Biochem. Biophys. Res. Commun. 1997, 239, 706–709. [Google Scholar] [CrossRef]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocme, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar]
- Schechter, Y.; Hernaez, L.; Schlessinger, J.; Cuatrecasas, P. Local aggregation of hormone-receptor complexes is required for activation by epidermal growth factor. Nature 1979, 278, 835–838. [Google Scholar] [CrossRef]
- Carraway, K.L., III; Koland, J.G.; Cerione, R.A. Visualization of epidermal growth factor (EGF) receptor aggregation in plasma membranes by fluorescence resonance energy transfer. Correlation of receptor activation with aggregation. J. Biol. Chem. 1989, 264, 8699–8707. [Google Scholar]
- Gadella, T.W., Jr.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef]
- Van den Eynde, M.; Baurain, J.F.; Mazzeo, F.; Machiels, J.P. Epidermal growth factor receptor targeted therapies for solid tumours. Acta Clin. Belg. 2011, 66, 10–17. [Google Scholar] [CrossRef]
- Krex, D.; Klink, B.; Hartmann, C.; von, D.A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. Long-term survival with glioblastoma multiforme. Brain 2007, 130, 2596–2606. [Google Scholar] [CrossRef]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Quan, A.L.; Barnett, G.H.; Lee, S.Y.; Vogelbaum, M.A.; Toms, S.A.; Staugaitis, S.M.; Prayson, R.A.; Peereboom, D.M.; Stevens, G.H.; Cohen, B.H.; et al. Epidermal growth factor receptor amplification does not have prognostic significance in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 695–703. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van, M.G.; de, T.N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Halatsch, M.E.; Schmidt, U.; Behnke-Mursch, J.; Unterberg, A.; Wirtz, C.R. Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat. Rev. 2006, 32, 74–89. [Google Scholar] [CrossRef]
- Haas-Kogan, D.A.; Prados, M.D.; Tihan, T.; Eberhard, D.A.; Jelluma, N.; Arvold, N.D.; Baumber, R.; Lamborn, K.R.; Kapadia, A.; Malec, M.; et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005, 97, 880–887. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Saceda, M.; Grasso, S.; Rocamora-Reverte, L.; Conde, M.; Gomez-Martinez, A.; Garcia-Morales, P.; Ferragut, J.A.; Martinez-Lacaci, I. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp. Cell Res. 2011, 317, 1476–1489. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available online: http://www.clinicaltrials.gov/ct2/results?term=EGFR%2C+glioblastoma&Search=Search (accessed on 12 March 2014).
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar]
- Learn, C.A.; Hartzell, T.L.; Wikstrand, C.J.; Archer, G.E.; Rich, J.N.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin. Cancer Res. 2004, 10, 3216–3224. [Google Scholar] [CrossRef]
- Voelzke, W.R.; Petty, W.J.; Lesser, G.J. Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr. Treat. Options. Oncol. 2008, 9, 23–31. [Google Scholar] [CrossRef]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nister, M. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Gross, D.; Bernhardt, G.; Buschauer, A. Platelet-derived growth factor receptor independent proliferation of human glioblastoma cells: Selective tyrosine kinase inhibitors lack antiproliferative activity. J. Cancer Res. Clin. Oncol. 2006, 132, 589–599. [Google Scholar] [CrossRef]
- Vassbotn, F.S.; Ostman, A.; Langeland, N.; Holmsen, H.; Westermark, B.; Heldin, C.H.; Nister, M. Activated platelet-derived growth factor autocrine pathway drives the transformed phenotype of a human glioblastoma cell line. J. Cell Physiol. 1994, 158, 381–389. [Google Scholar] [CrossRef]
- Ranza, E.; Mazzini, G.; Facoetti, A.; Nano, R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J. Neurooncol. 2010, 96, 349–357. [Google Scholar] [CrossRef]
- Ren, H.; Tan, X.; Dong, Y.; Giese, A.; Chou, T.C.; Rainov, N.; Yang, B. Differential effect of imatinib and synergism of combination treatment with chemotherapeutic agents in malignant glioma cells. Basic Clin. Pharmacol. Toxicol. 2009, 104, 241–252. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Chaskis, C.; Dujardin, M.; Everaert, H.; Lv, S.; Duerinck, J.; Tynninen, O.; Nupponen, N.; Michotte, A.; et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J. Neurooncol. 2011, 103, 491–501. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available online: http://www.clinicaltrials.gov/ct2/results?term=PDGFR%2C+glioma&Search=Search (accessed on 12 March 2014).
- Hewish, M.; Chau, I.; Cunningham, D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Pat. Anticancer Drug Discov. 2009, 4, 54–72. [Google Scholar] [CrossRef]
- Casa, A.J.; Dearth, R.K.; Litzenburger, B.C.; Lee, A.V.; Cui, X. The type I insulin-like growth factor receptor pathway: a key player in cancer therapeutic resistance. Front Biosci. 2008, 13, 3273–3287. [Google Scholar]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar]
- Giovannone, B.; Scaldaferri, M.L.; Federici, M.; Porzio, O.; Lauro, D.; Fusco, A.; Sbraccia, P.; Borboni, P.; Lauro, R.; Sesti, G. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res. Rev. 2000, 16, 434–441. [Google Scholar] [CrossRef]
- Mukohara, T.; Shimada, H.; Ogasawara, N.; Wanikawa, R.; Shimomura, M.; Nakatsura, T.; Ishii, G.; Park, J.O.; Janne, P.A.; Saijo, N.; et al. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009, 282, 14–24. [Google Scholar] [CrossRef]
- Werner, H.; Bruchim, I. The insulin-like growth factor-I receptor as an oncogene. Arch. Physiol. Biochem. 2009, 115, 58–71. [Google Scholar] [CrossRef]
- Oh, Y. IGF-independent regulation of breast cancer growth by IGF binding proteins. Breast Cancer Res. Treat. 1998, 47, 283–293. [Google Scholar] [CrossRef]
- Girnita, A.; Girnita, L.; del Prete, F.; Bartolazzi, A.; Larsson, O.; Axelson, M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004, 64, 236–242. [Google Scholar] [CrossRef]
- Stromberg, T.; Ekman, S.; Girnita, L.; Dimberg, L.Y.; Larsson, O.; Axelson, M.; Lennartsson, J.; Hellman, U.; Carlson, K.; Osterborg, A.; et al. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood 2006, 107, 669–678. [Google Scholar] [CrossRef]
- Yin, S.; Girnita, A.; Stromberg, T.; Khan, Z.; Andersson, S.; Zheng, H.; Ericsson, C.; Axelson, M.; Nister, M.; Larsson, O.; et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro. Oncol. 2010, 12, 19–27. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A service of the U.S. National Institutes of Health. Available online: http://www.clinicaltrials.gov/ct2/results?term=IGF1R%2C+glioma&Search=Search (accessed on 12 March 2014).
- Moller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of microRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef]
- Hernando, E. microRNAs and cancer: Role in tumorigenesis, patient classification and therapy. Clin. Transl. Oncol. 2007, 9, 155–160. [Google Scholar] [CrossRef]
- Karsy, M.; Arslan, E.; Moy, F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer 2012, 3, 3–15. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Friedmann-Morvinski, D.; Neveu, P.; Dugas, J.C.; Gill, R.M.; Huillard, E.; Liu, C.; Zong, H.; Rowitch, D.H.; Barres, B.A.; et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene 2012, 31, 1884–1895. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Lian, H.; Liu, J.; Zhou, B.; Han, S.; Peng, B.; Yin, J.; Liu, W.; He, X. MicroRNA-503 acts as a tumor suppressor in glioblastoma for multiple antitumor effects by targeting IGF-1R. Oncol. Rep. 2014, 31, 1445–1452. [Google Scholar]
- Shao, M.; Rossi, S.; Chelladurai, B.; Shimizu, M.; Ntukogu, O.; Ivan, M.; Calin, G.A.; Matei, D. PDGF induced microRNA alterations in cancer cells. Nucleic Acids Res. 2011, 39, 4035–4047. [Google Scholar] [CrossRef]
- Katakowski, M.; Zheng, X.; Jiang, F.; Rogers, T.; Szalad, A.; Chopp, M. MiR-146b-5p suppresses EGFR expression and reduces in vitro migration and invasion of glioma. Cancer Invest 2010, 28, 1024–1030. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab Invest 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Jun, H.J.; Bronson, R.T.; Charest, A. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells 2014, 32, 338–348. [Google Scholar] [CrossRef]
- Martinez-Lacaci, I.; Garcia, M.P.; Soto, J.L.; Saceda, M. Tumour cells resistance in cancer therapy. Clin. Transl. Oncol. 2007, 9, 13–20. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Tan, B.; Piwnica-Worms, D.; Ratner, L. Multidrug resistance transporters and modulation. Curr. Opin. Oncol. 2000, 12, 450–458. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Smyth, M.J. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem. Sci. 2000, 25, 1–6. [Google Scholar] [CrossRef]
- Decleves, X.; Fajac, A.; Lehmann-Che, J.; Tardy, M.; Mercier, C.; Hurbain, I.; Laplanche, J.L.; Bernaudin, J.F.; Scherrmann, J.M. Molecular and functional MDR1-Pgp and MRPs expression in human glioblastoma multiforme cell lines. Int. J. Cancer 2002, 98, 173–180. [Google Scholar] [CrossRef]
- Beaulieu, E.; Demeule, M.; Ghitescu, L.; Beliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326, 539–544. [Google Scholar]
- Toth, K.; Vaughan, M.M.; Peress, N.S.; Slocum, H.K.; Rustum, Y.M. MDR1 P-glycoprotein is expressed by endothelial cells of newly formed capillaries in human gliomas but is not expressed in the neovasculature of other primary tumors. Am. J. Pathol. 1996, 149, 853–858. [Google Scholar]
- Nakagawa, T.; Ido, K.; Sakuma, T.; Takeuchi, H.; Sato, K.; Kubota, T. Prognostic significance of the immunohistochemical expression of O6-methylguanine-DNA methyltransferase, P-glycoprotein, and multidrug resistance protein-1 in glioblastomas. Neuropathology 2009, 29, 379–388. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009, 10, 709–717. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 2008, 118, 2609–2619. [Google Scholar]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef]
- Rexer, B.N.; Engelman, J.A.; Arteaga, C.L. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle 2009, 8, 18–22. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef]
- Ogino, A.; Kitao, H.; Hirano, S.; Uchida, A.; Ishiai, M.; Kozuki, T.; Takigawa, N.; Takata, M.; Kiura, K.; Tanimoto, M. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res. 2007, 67, 7807–7814. [Google Scholar] [CrossRef]
- Daub, H.; Specht, K.; Ullrich, A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat. Rev. Drug Discov. 2004, 3, 1001–1010. [Google Scholar] [CrossRef]
- Morgillo, F.; Lee, H.Y. Resistance to epidermal growth factor receptor-targeted therapy. Drug Resist. Updat. 2005, 8, 298–310. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carrasco-García, E.; Saceda, M.; Martínez-Lacaci, I. Role of Receptor Tyrosine Kinases and Their Ligands in Glioblastoma. Cells 2014, 3, 199-235. https://doi.org/10.3390/cells3020199
Carrasco-García E, Saceda M, Martínez-Lacaci I. Role of Receptor Tyrosine Kinases and Their Ligands in Glioblastoma. Cells. 2014; 3(2):199-235. https://doi.org/10.3390/cells3020199
Chicago/Turabian StyleCarrasco-García, Estefanía, Miguel Saceda, and Isabel Martínez-Lacaci. 2014. "Role of Receptor Tyrosine Kinases and Their Ligands in Glioblastoma" Cells 3, no. 2: 199-235. https://doi.org/10.3390/cells3020199
APA StyleCarrasco-García, E., Saceda, M., & Martínez-Lacaci, I. (2014). Role of Receptor Tyrosine Kinases and Their Ligands in Glioblastoma. Cells, 3(2), 199-235. https://doi.org/10.3390/cells3020199