Regulation of Cardiac Cell Fate by microRNAs: Implications for Heart Regeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulatory Programs Underlying Heart Development
2.1. Transcriptional Networks in Embryonic Heart Development
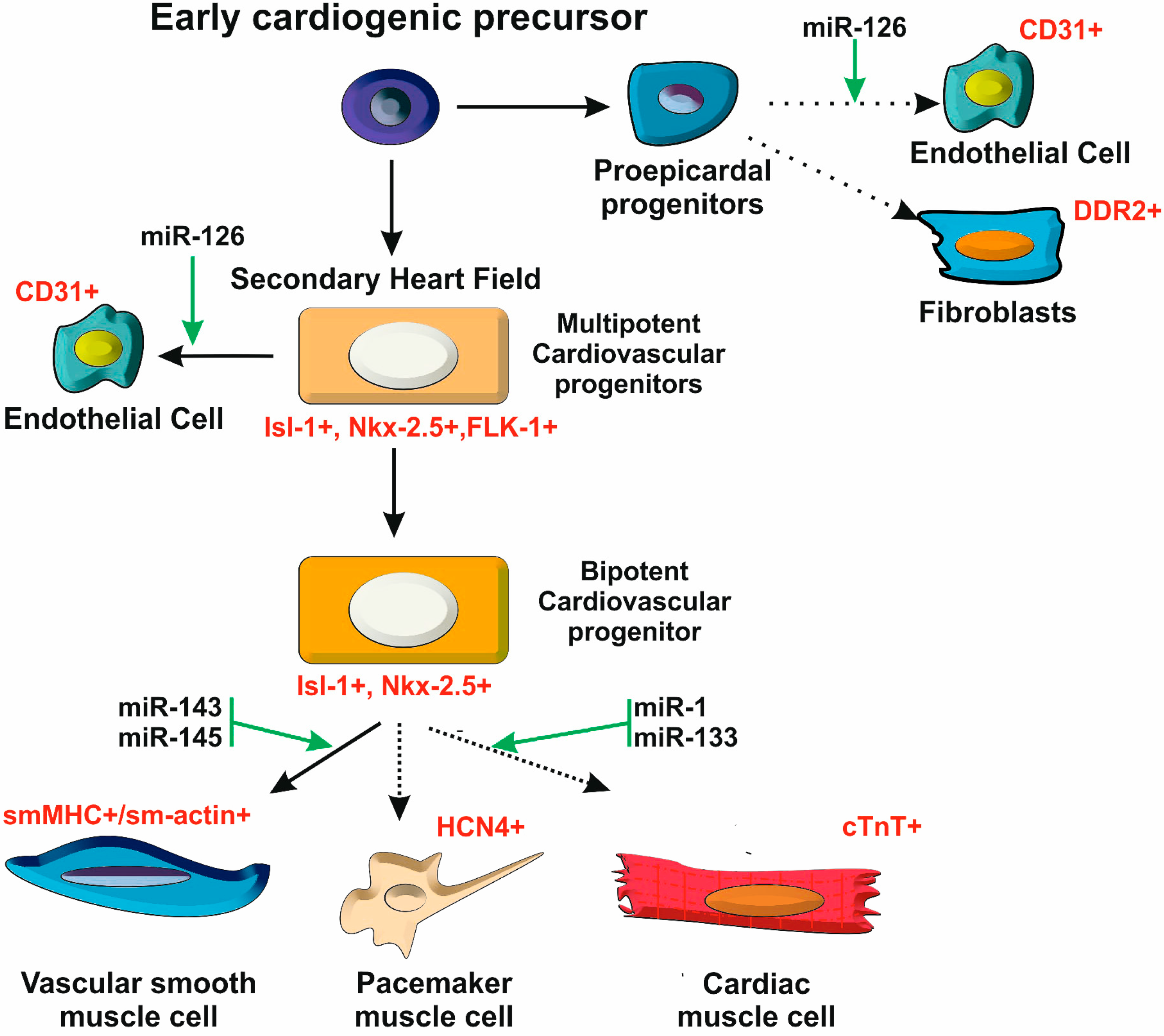
2.2. A Stem Cell Model for Heart Development

2.3. Postnatal Heart Development
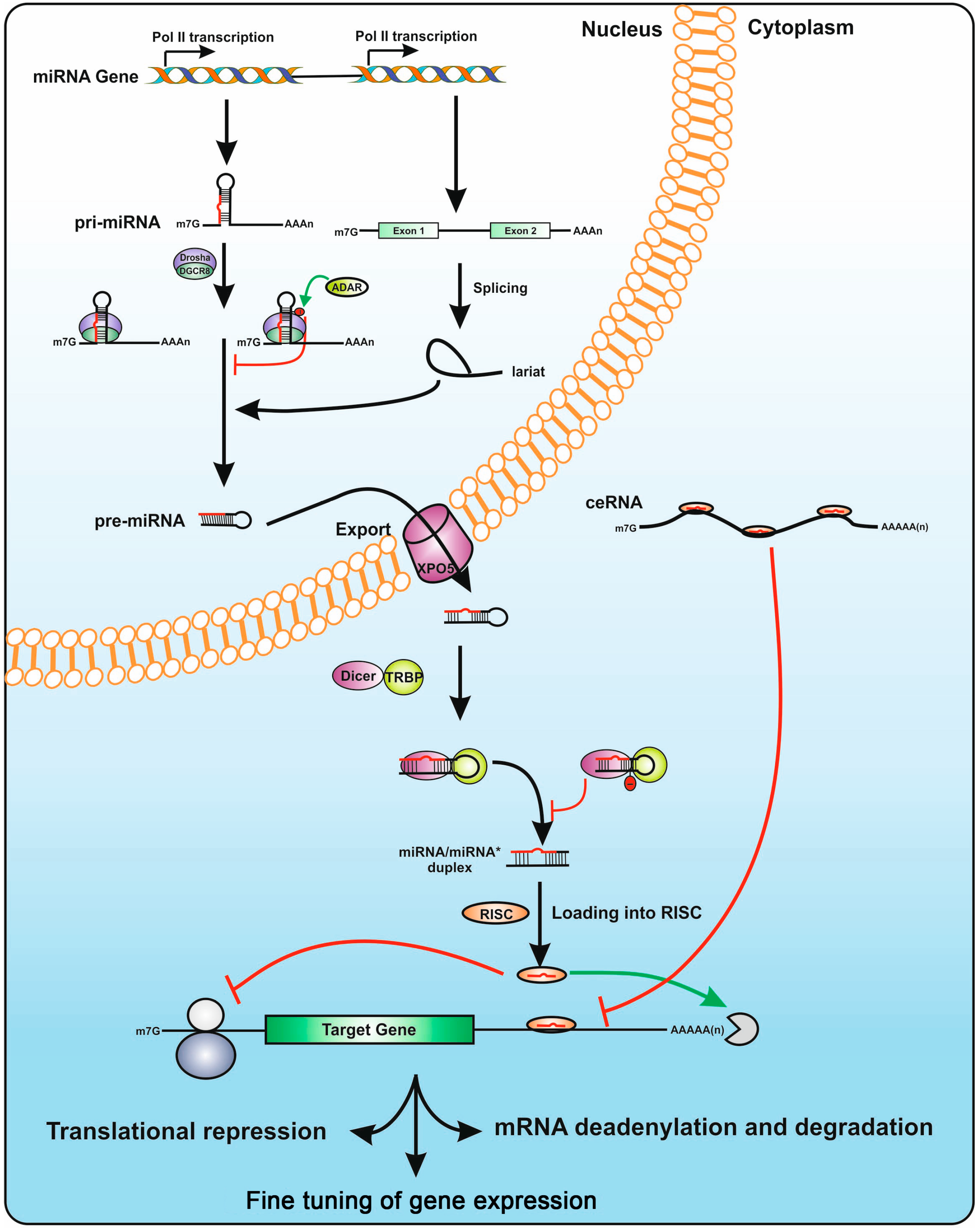
3. microRNAs: from Biogenesis to Post-Transcriptional Control of Gene Expression
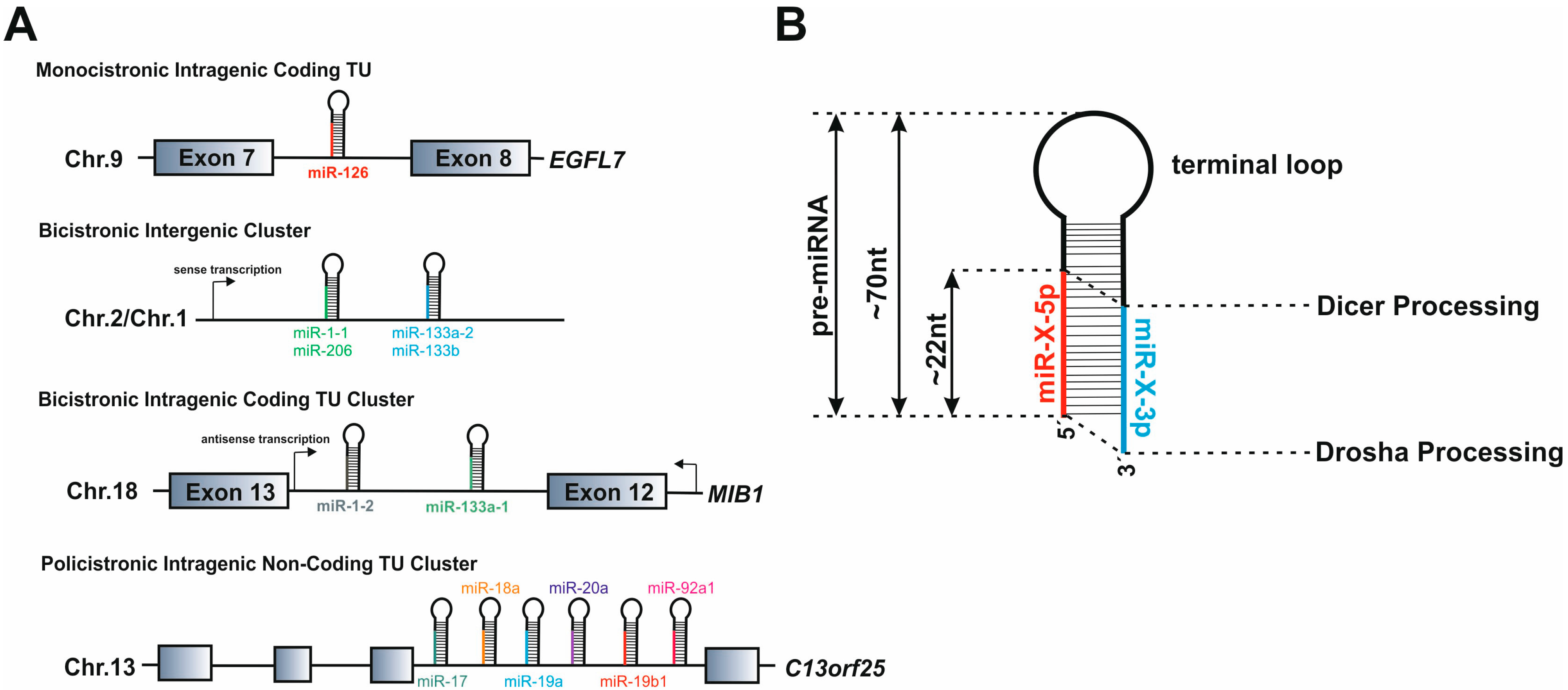
3.1. microRNA Gene Structure

3.2. The microRNA Biogenesis Machinery

3.3. microRNA Modes of Action
4. microRNAs’ Biological Functions: Getting to the Heart of the Matter
4.1. Regulation of Gene Expression Programs by microRNAs
4.2. Role of microRNAs in Cell Fate Decisions
4.3. miRNAs as Critical Regulators of Stem Cell Properties

4.4. Regulatory Feedback between miRNAs and Transcription Factors

5. microRNAs in Heart Development
5.1. Requirement of the microRNA Machinery for Heart Development
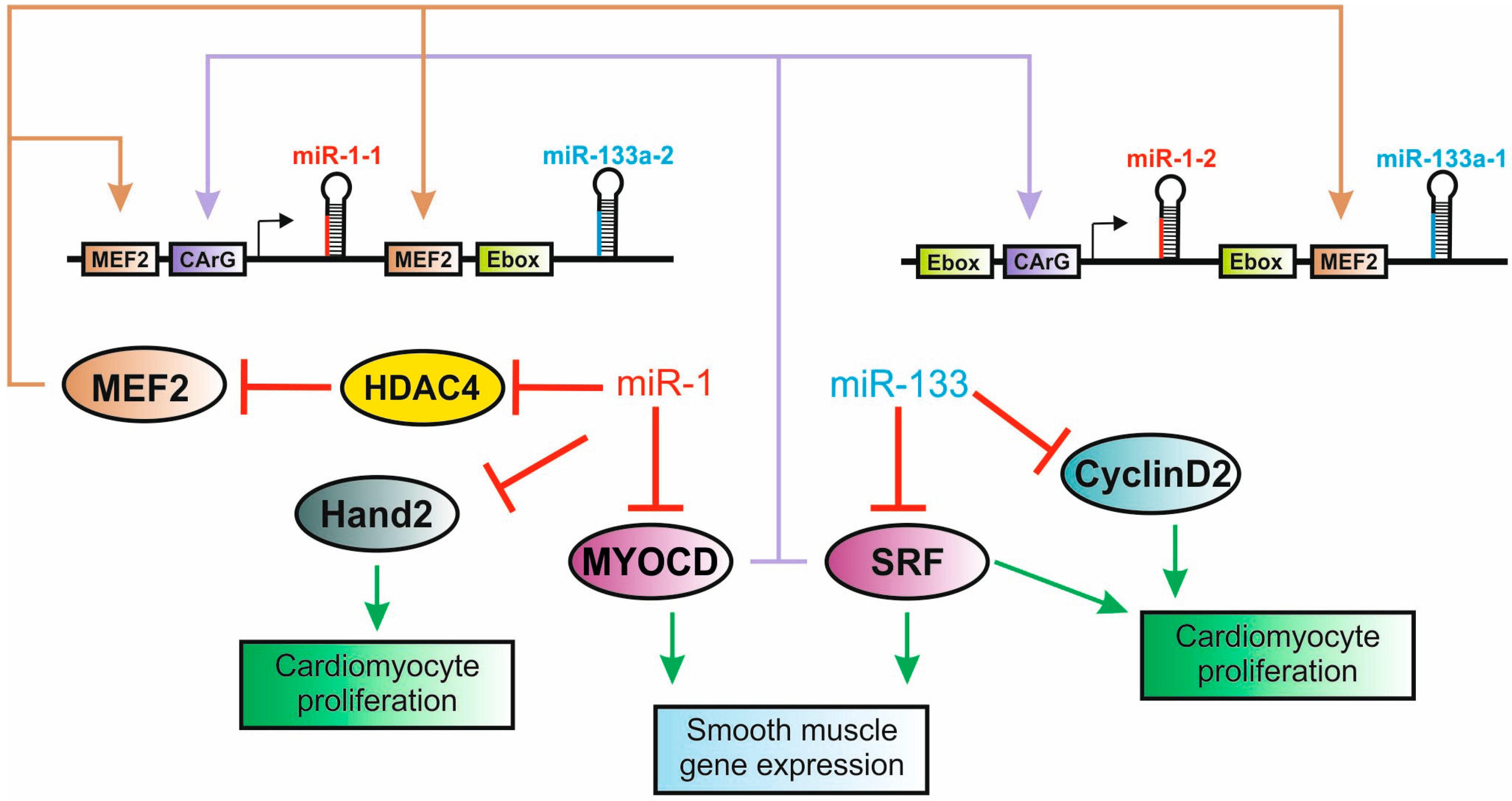
5.2. The miR-1/133 Family is a Critical Component of Cardiogenic Regulatory Networks
5.3. microRNAs Play a Critical Role in the Cardiac Fetal-to-Adult Switch
6. Heart Regeneration: A microRNA Connection to the Lost Link
6.1. Adult Cardiac Progenitor Cells under the Control of microRNAs Provide a Limited Source of Renewal

6.2. Persistence of Cardiomyocyte Division after Birth: Is the Key to Regeneration Locked away by microRNAs?
6.3. Reprograming of Cardiac Fibroblasts to Functional Cardiomyocytes
7. Conclusion and Future Perspectives
Acknowledgments
Author contributions
Conflicts of Interest
References
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar] [PubMed]
- Sempere, L.F.; Cole, C.N.; Mcpeek, M.A.; Peterson, K.J. The phylogenetic distribution of metazoan microRNAs: Insights into evolutionary complexity and constraint. J. Exp. Zool. Part B Mol. Dev. Evol. 2006, 306, 575–588. [Google Scholar] [CrossRef]
- Niwa, R.; Slack, F.J. The evolution of animal microRNA function. Curr. Opin. Genet. Dev. 2007, 17, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, F.; Raible, F.; Tomer, R.; Simakov, O.; Trachana, K.; Klaus, S.; Snyman, H.; Hannon, G.J.; Bork, P.; Arendt, D. Ancient animal microRNAs and the evolution of tissue identity. Nature 2010, 463, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Gregory, T.R. The case for junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.D.; Guerquin-Kern, J.L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Pedrazzini, T. Progenitor cell therapy for heart disease. Exp. Cell Res. 2009, 315, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Moradi, S.; Asgari, S.; Baharvand, H. Concise review: harmonies played by microRNAs in cell fate reprogramming. Stem Cells 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Gillis, W.Q.; St John, J.; Bowerman, B.; Schneider, S.Q. Whole genome duplications and expansion of the vertebrate GATA transcription factor gene family. BMC Evol. Biol. 2009, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E. K.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Domian, I.J.; Chien, K.R. Stem cell models of cardiac development and disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 667–687. [Google Scholar] [CrossRef] [PubMed]
- Sturzu, A.C.; Wu, S.M. Developmental and regenerative biology of multipotent cardiovascular progenitor cells. Circ. Res. 2011, 108, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, R. Molecular Control of Cardiac Fetal/Neonatal Remodeling. J. Cardiovasc. Dev. Dis. 2014, 1, 29–36. [Google Scholar] [CrossRef]
- Siedner, S.; Krüger, M.; Schroeter, M.; Metzler, D.; Roell, W.; Fleischmann, B.K.; Hescheler, J.; Pfitzer, G.; Stehle, R. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J. Physiol. 2003, 548, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Schanen, B.C.; Li, X. Transcriptional regulation of mammalian miRNA genes. Genomics 2011, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Siomi, M.C. Posttranscriptional Regulation of MicroRNA Biogenesis in Animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Canella, D.; Praz, V.; Reina, J.H.; Cousin, P.; Hernandez, N. Defining the RNA polymerase III transcriptome: Genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010, 20, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jiang, Q.; Wang, G.; Liu, B.; Teng, M.; Wang, Y. Chromatin structure characteristics of pre-miRNA genomic sequences. BMC Genomics 2011, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, D.L.; Pandit, K.V.; Gordon, B.; Bhattacharjee, A.; Kaminski, N.; Benos, P.V. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsico, A.; Huska, M.R.; Lasserre, J.; Hu, H.; Vucicevic, D.; Musahl, A.; Orom, U.A.; Vingron, M. PROmiRNA: a new miRNA promoter recognition method uncovers the complex regulation of intronic miRNAs. Genome Biol. 2013, 14, R84. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, V.N. Processing of intronic microRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. Rna 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cullen, B.R. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004, 32, 4776–4785. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Dahlberg, J.E. Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Suh, M.R.; Kim, V.N. The role of PACT in the RNA silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.H.; Ng, M.H.J.; Ching, Y.P.; Jin, D.Y. Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J. Biol. Chem. 2007, 282, 17649–17657. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Duelli, D.M.; Hastings, M.L. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. 2012, 40, 4626–4640. [Google Scholar] [CrossRef] [PubMed]
- Babiarz, J.E.; Ruby, J.G.; Wang, Y.; Bartel, D.P.; Blelloch, R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008, 22, 2773–2785. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W. S.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Ender, C.; Krek, A.; Friedländer, M.R.; Beitzinger, M.; Weinmann, L.; Chen, W.; Pfeffer, S.; Rajewsky, N.; Meister, G. A human snoRNA with microRNA-like functions. Mol. Cell 2008, 32, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Landweber, L.F. Hypothesis: RNA editing of microRNA target sites in humans? RNA 2007, 13, 463–467. [Google Scholar] [CrossRef]
- Blow, M.J.; Grocock, R.J.; van Dongen, S.; Enright, A.J.; Dicks, E.; Futreal, P.A.; Wooster, R.; Stratton, M.R. RNA editing of human microRNAs. Genome Biol. 2006, 7, R27. [Google Scholar] [CrossRef] [PubMed]
- Öhman, M. A-to-I editing challenger or ally to the microRNA process. Biochimie 2007, 89, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with dicer to promote MicroRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.; Nielsen, F.; Lund, A. MicroRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.C.; Wani, S.; Steptoe, A.L.; Krishnan, K.; Nones, K.; Nourbakhsh, E.; Vlassov, A.; Grimmond, S.M.; Cloonan, N. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014, 15, R51. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Fabian, M.R.; Sonenberg, N.; Bhattacharyya, S.N.; Filipowicz, W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res. 2012, 40, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; Agami, R. Interplay between microRNAs and RNA-binding proteins determines developmental processes. Cell Cycle 2008, 7, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Kedde, M.; Strasser, M.J.; Boldajipour, B.; Vrielink, J.A.O.; Slanchev, K.; le Sage, C.; Nagel, R.; Voorhoeve, P.M.; van Duijse, J.; Ørom, U.A.; et al. RNA-Binding Protein Dnd1 Inhibits MicroRNA Access to Target mRNA. Cell 2007, 131, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Matkovich, S.J. Menage a Trois: Intimate Relationship Among a MicroRNA, Long Noncoding RNA, and mRNA. Circ. Res. 2014, 114, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Flynt, A.S.; Lai, E.C. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat. Rev. Genet. 2008, 9, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Kizana, E.; Cingolani, E.; Marbán, E. Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther. 2009, 16, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. MicroRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Baek, M.; Gusev, Y.; Brackett, D.J.; Nuovo, G.J.; Schmittgen, T.D. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 2008, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Sokol, N.S.; Ambros, V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005, 19, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.-F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; Srivastava, D. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Srivastava, D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell 2010, 7, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.J.; Gregory, R.I. MicroRNA gene regulatory pathways in the establishment and maintenance of ESC identity. Cell Stem Cell 2010, 7, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, X.; Liao, B.; Benda, C.; Zhuang, Q.; Pei, D.; Qin, B.; Esteban, M.A. MicroRNAs in somatic cell reprogramming. Curr. Opin. Cell Biol. 2013, 25, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Yoo, A.S.; Sun, A.X.; Li, L.; Shcheglovitov, A.; Portmann, T.; Li, Y.; Lee-Messer, C.; Dolmetsch, R.E.; Tsien, R.W.; Crabtree, G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011, 476, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Zhang, W.; Liu, G.H.; Belmonte, J.C.I. Switching cell fate, ncRNAs coming to play. Cell Death Dis. 2013, 4, e464. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of Cardiac microRNA-Mediated Regulation Leads to Dilated Cardiomyopathy and Heart Failure. Circ. Res. 2009, 105, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA Regulatory Networks in Cardiovascular Development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20844–20849. [Google Scholar] [CrossRef] [PubMed]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Tabin, C.J. miRNA-processing enzyme Dicer is necessary for cardiac outflow tract alignment and chamber septation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Chapnik, E.; Sasson, V.; Blelloch, R.; Hornstein, E. Dgcr8 controls neural crest cells survival in cardiovascular development. Dev. Biol. 2012, 362, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.P.; Chen, J.F.; Regan, J.N.; Maguire, C.T.; Tang, R.H.; Dong, X.R.; Majesky, M.W.; Wang, D.Z. Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Shiohama, A.; Sasaki, T.; Noda, S.; Minoshima, S.; Shimizu, N. Molecular cloning and expression analysis of a novel gene DGCR8 located in the DiGeorge syndrome chromosomal region. Biochem. Biophys. Res. Commun. 2003, 304, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.; Han, Z.; Olson, E.N.; Srivastava, D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18986–18991. [Google Scholar] [CrossRef] [PubMed]
- Tani, S.; Kuraku, S.; Sakamoto, H.; Inoue, K.; Kusakabe, R. Developmental expression and evolution of muscle-specific microRNAs conserved in vertebrates. Evol. Dev. 2013, 15, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Choi, Y.S.; McManus, M.T. Analysis of microRNA knockouts in mice. Hum. Mol. Genet. 2010, 19, R169–R175. [Google Scholar] [CrossRef] [PubMed]
- Heidersbach, A.; Saxby, C.; Carver-Moore, K.; Huang, Y.; Ang, Y.S.; de Jong, P.J.; Ivey, K.N.; Srivastava, D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. Elife 2013, 2, e01323. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Peng, S.; Wu, M.; Sachidanandam, R.; Tu, Z.; Zhang, S.; Falce, C.; Sobie, E.A.; Lebeche, D.; Zhao, Y. Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell Res. 2014, 24, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Takahashi, S.; Rossant, J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood 2006, 107, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Wang, K.; Li, P.F.; Cooper, T.A. MicroRNAs coordinate an alternative splicing network during mouse postnatal heart development. Genes Dev. 2010, 24, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W.; van Rooij, E.; Olson, E.N. miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Da Costa Martins, P.A.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; De Windt, L.J. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; Gross, C.; Engelhardt, S.; Ertl, G.; Bauersachs, J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; Leri, A.; Kajstura, J.; Nadal-Ginard, B.; Anversa, P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; Kühn, B. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Sleep, E.; Raya, M.; Martí, M.; Raya, A.; Belmonte, J.C.I. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.R.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Urbanek, K.; Kajstura, J.; Yan, S.M.; Finato, N.; Bussani, R.; Nadal-Ginard, B.; Silvestri, F.; Leri, A.; Beltrami, C.A.; et al. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 2001, 344, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Quaini, F.; Urbanek, K.; Beltrami, A.P.; Finato, N.; Beltrami, C.A.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Chimerism of the transplanted heart. N. Engl. J. Med. 2002, 346, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hierlihy, A.M.; Seale, P.; Lobe, C.G.; Rudnicki, M.A.; Megeney, L.A. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002, 530, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.M.; Meeson, A.P.; Robertson, S.M.; Hawke, T.J.; Richardson, J.A.; Bates, S.; Goetsch, S.C.; Gallardo, T.D.; Garry, D.J. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev. Biol. 2004, 265, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12313–12318. [Google Scholar] [CrossRef] [PubMed]
- Laugwitz, K.L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.Z.; Cai, C.L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, T.; D’Amario, D.; Cabral-da-Silva, M.C.; Zheng, H.; Padin-Iruegas, M.E.; Ogórek, B.; Ferreira-Martins, J.; Yasuzawa-Amano, S.; Amano, K.; Ide-Iwata, N.; et al. Clonality of mouse and human cardiomyogenesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17169–17174. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.G.; van Mil, A.; van Vliet, P.; Metz, C.H.G.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Sirish, P.; López, J.E.; Li, N.; Wong, A.; Timofeyev, V.; Young, J.N.; Majdi, M.; Li, R.A.; Chen, H.S.V.; Chiamvimonvat, N. MicroRNA profiling predicts a variance in the proliferative potential of cardiac progenitor cells derived from neonatal and adult murine hearts. J. Mol. Cell. Cardiol. 2012, 52, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Bras-Rosario, L.; Matsuda, A.; Pinheiro, A.I.; Gardner, R.; Lopes, T.; Amaral, A.; Gama-Carvalho, M. Expression Profile of microRNAs Regulating Proliferation and Differentiation in Mouse Adult Cardiac Stem Cells. PLoS One 2013, 8, e63041. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Rota, M.; Nurzynska, D.; Misao, Y.; Tillmanns, J.; Ojaimi, C.; Padin-Iruegas, M.E.; Müller, P.; Esposito, G.; Bearzi, C.; et al. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ. Res. 2008, 102, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, T.; Zheng, H.; Cabral-da-Silva, M.; Sanada, F.; Ide-Iwata, N.; Ogórek, B.; Ferreira-Martins, J.; Arranto, C.; D’Amario, D.; del Monte, F.; et al. Human Cardiac Stem Cell Differentiation Is Regulated by a Mircrine Mechanism. Circulation 2011, 123, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.C.; Ganesalingam, S.; Jensen, C.H.; Sheikh, S.P. Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports 2014, 2, 406–413. [Google Scholar] [CrossRef]
- Kotlikoff, M.I.; Hesse, M.; Fleischmann, B.K. Comment on “Do Neonatal Mouse Hearts Regenerate following Heart Apex Resection”? Stem Cell Reports 2014, 3, 2. [Google Scholar] [CrossRef]
- Sadek, H.A.; Martin, J.F.; Takeuchi, J.K.; Leor, J.; Nei, Y.; Giacca, M.; Lee, R.T. Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Reports 2014, 3, 1. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA. 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Ausoni, S.; Sartore, S. From fish to amphibians to mammals: in search of novel strategies to optimize cardiac regeneration. J. Cell Biol. 2009, 184, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D. Getting to the heart of regeneration in zebrafish. Semin. Cell Dev. Biol. 2007, 18, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Hussain, S.R.A.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009, 82, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.M.; Montgomery, R.L.; Qi, X.; Obad, S.; Kauppinen, S.; Hill, J.A.; van Rooij, E.; Olson, E.N. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Invest. 2010, 120, 3912–3916. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yi, B.A.; Chien, K.R. In vivo reprogramming for heart disease. Cell Res. 2012, 22, 1521–1523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Han, P.; Yang, H.; Ouyang, K.; Lee, D.; Lin, Y.F.; Ocorr, K.; Kang, G.; Chen, J.; Stainier, D.Y.R.; et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 2013, 498, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gama-Carvalho, M.; Andrade, J.; Brás-Rosário, L. Regulation of Cardiac Cell Fate by microRNAs: Implications for Heart Regeneration. Cells 2014, 3, 996-1026. https://doi.org/10.3390/cells3040996
Gama-Carvalho M, Andrade J, Brás-Rosário L. Regulation of Cardiac Cell Fate by microRNAs: Implications for Heart Regeneration. Cells. 2014; 3(4):996-1026. https://doi.org/10.3390/cells3040996
Chicago/Turabian StyleGama-Carvalho, Margarida, Jorge Andrade, and Luis Brás-Rosário. 2014. "Regulation of Cardiac Cell Fate by microRNAs: Implications for Heart Regeneration" Cells 3, no. 4: 996-1026. https://doi.org/10.3390/cells3040996
APA StyleGama-Carvalho, M., Andrade, J., & Brás-Rosário, L. (2014). Regulation of Cardiac Cell Fate by microRNAs: Implications for Heart Regeneration. Cells, 3(4), 996-1026. https://doi.org/10.3390/cells3040996