
Specialized Cilia in Mammalian Sensory Systems


Abstract
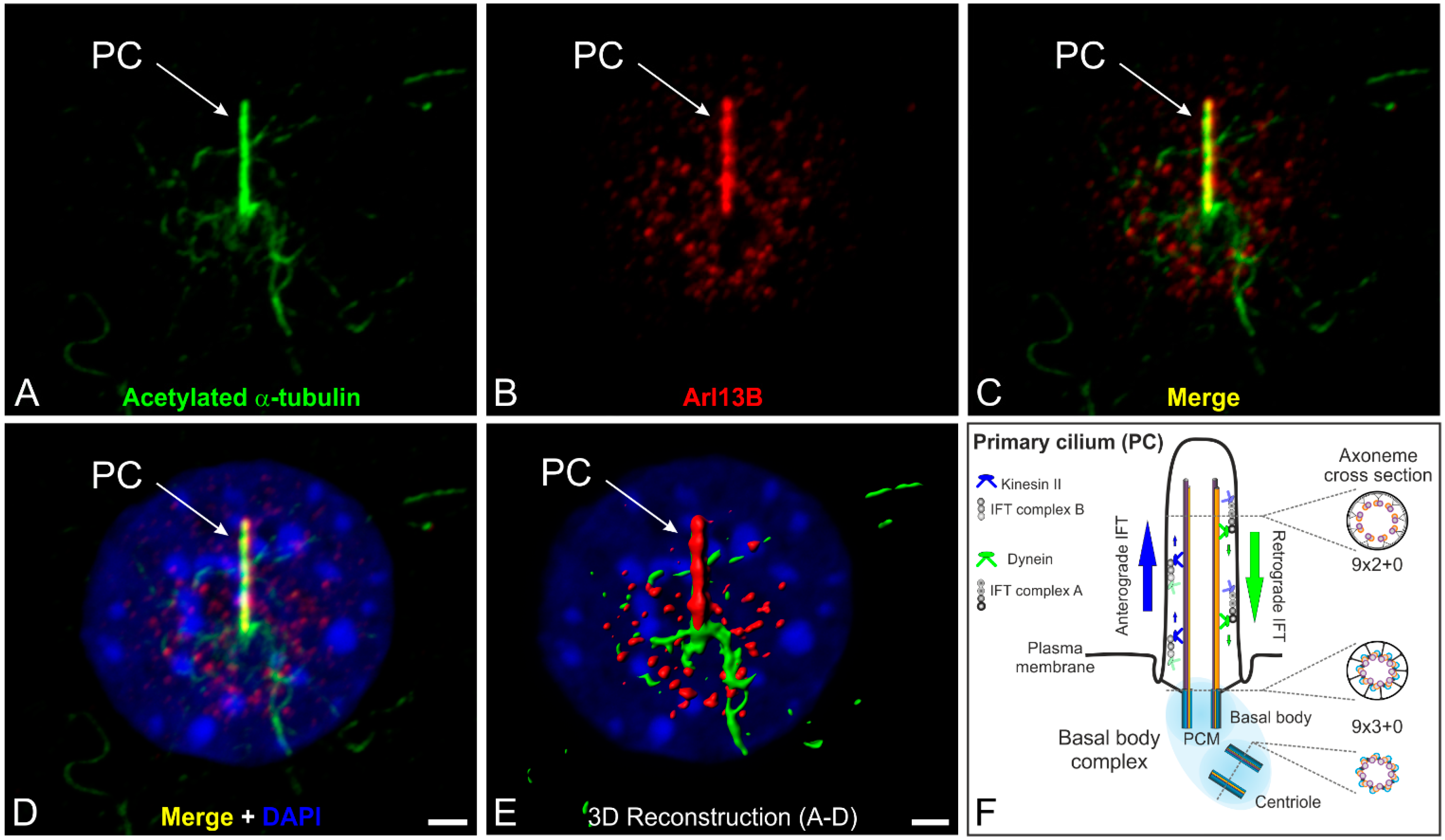
:1. Introduction
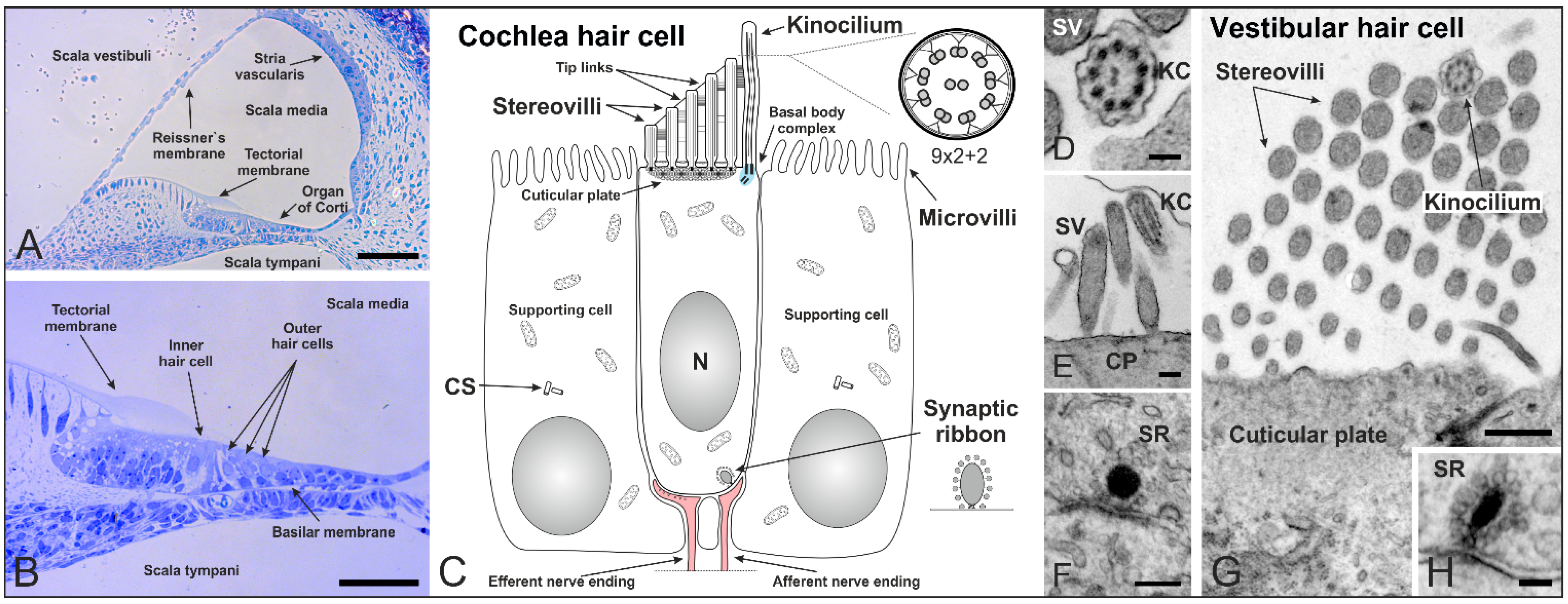
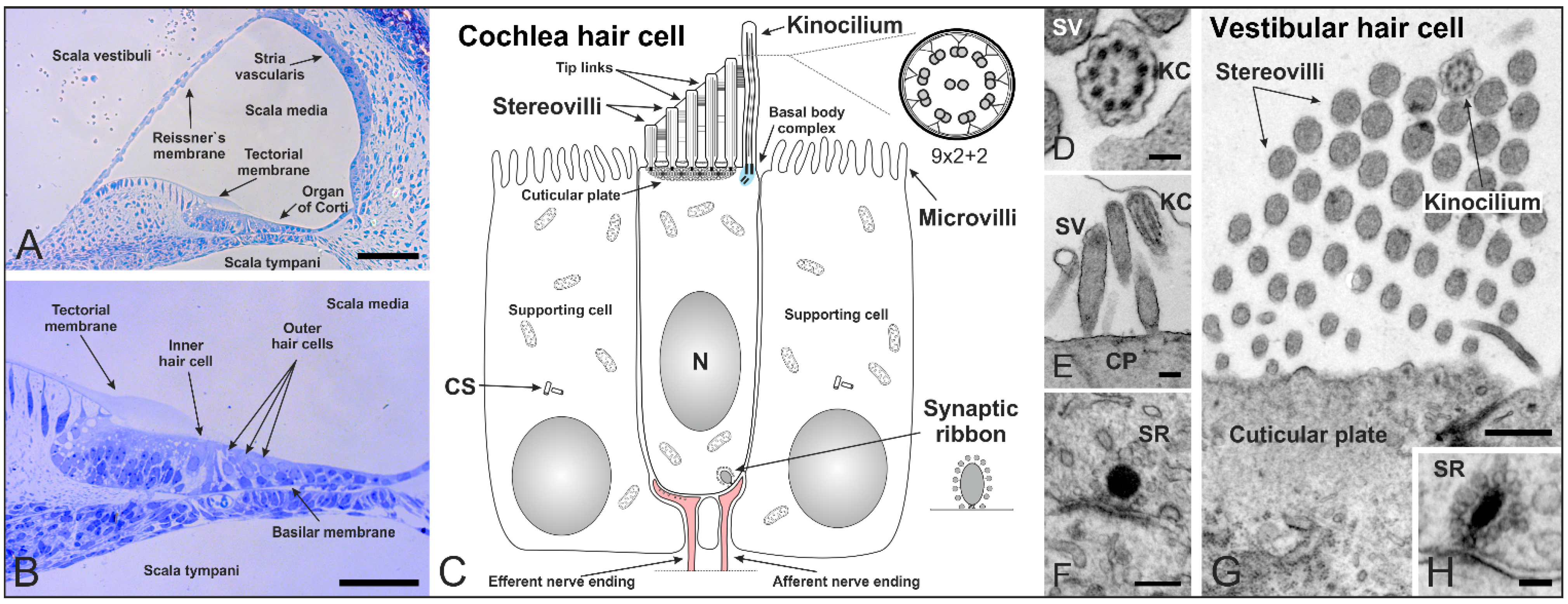
2. Cilia in the Inner Ear of Mammals
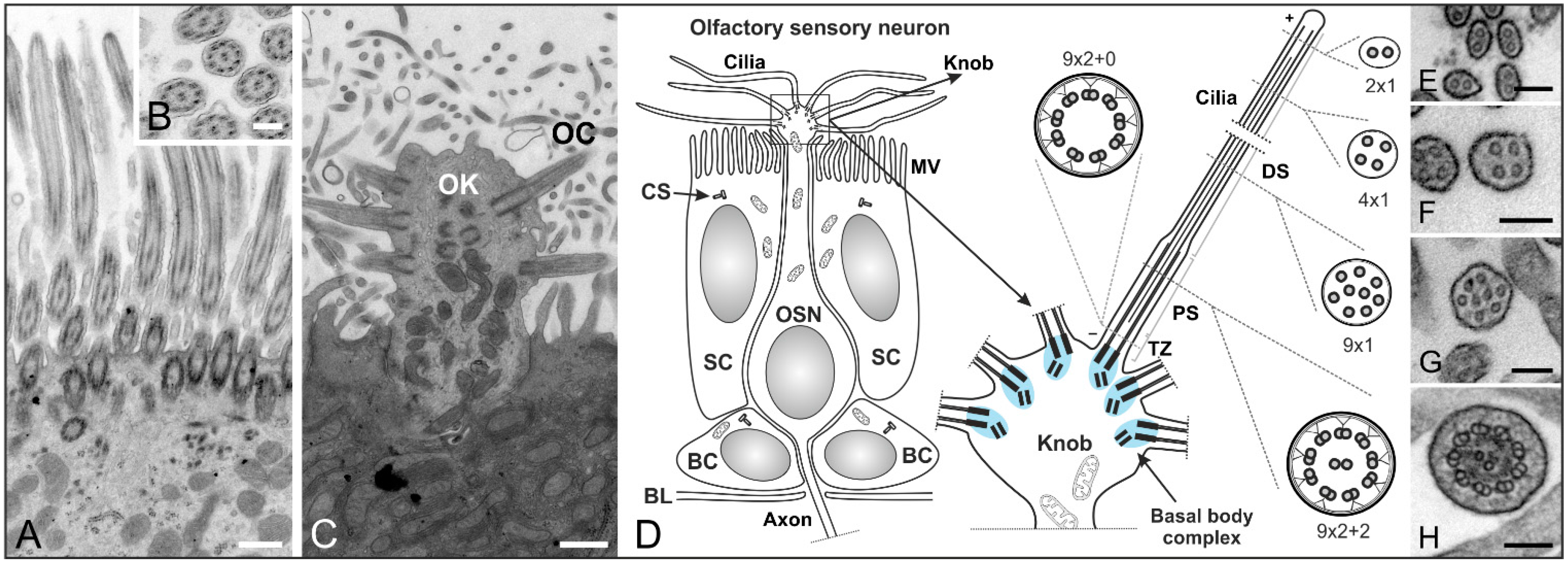
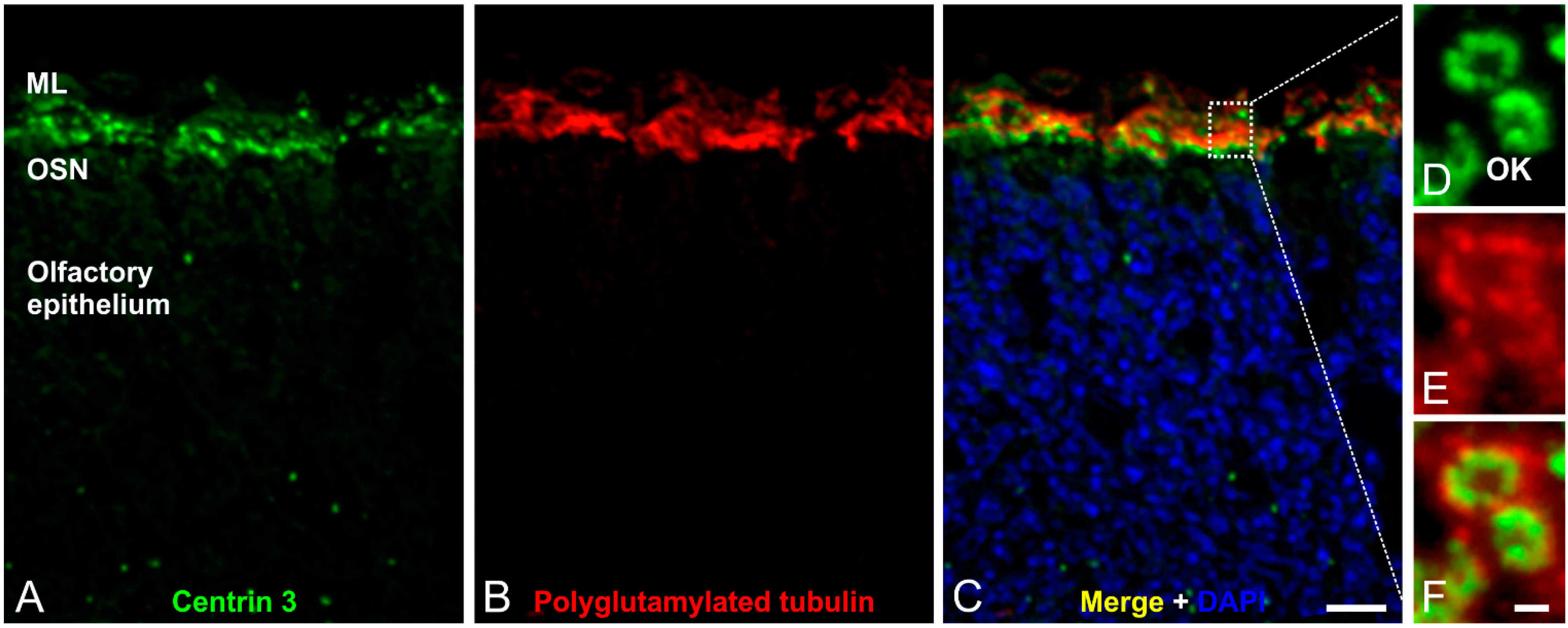
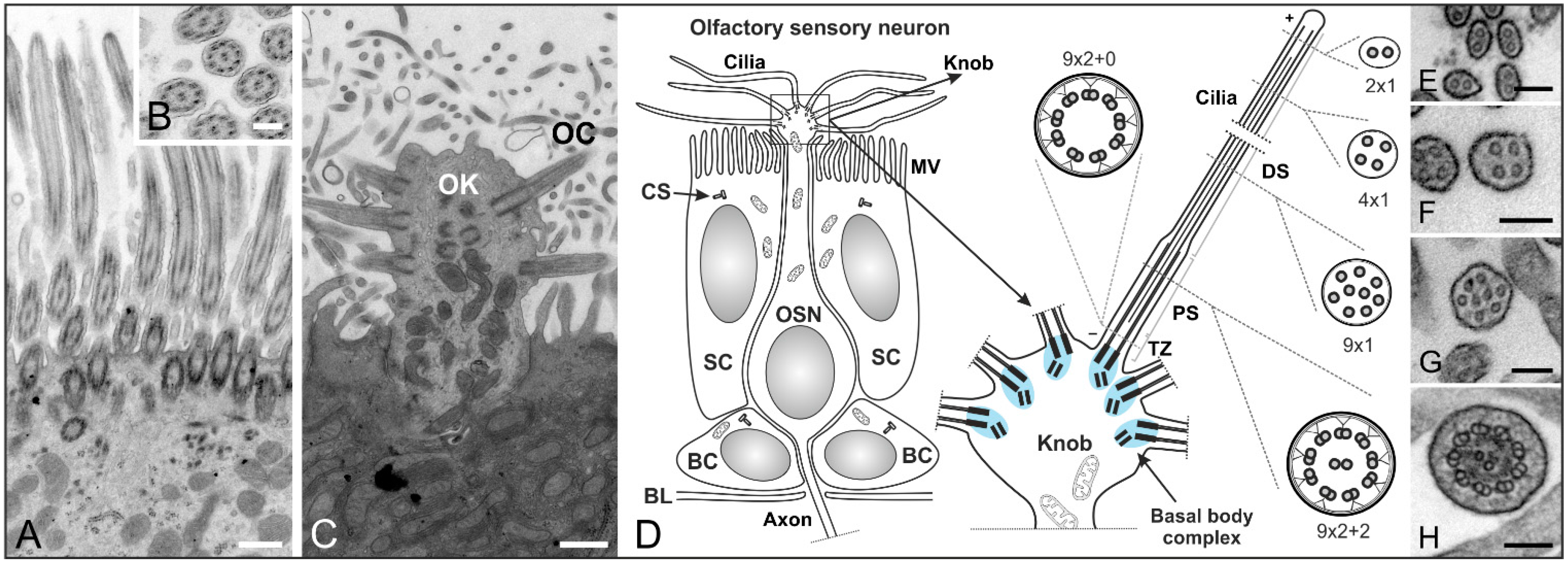
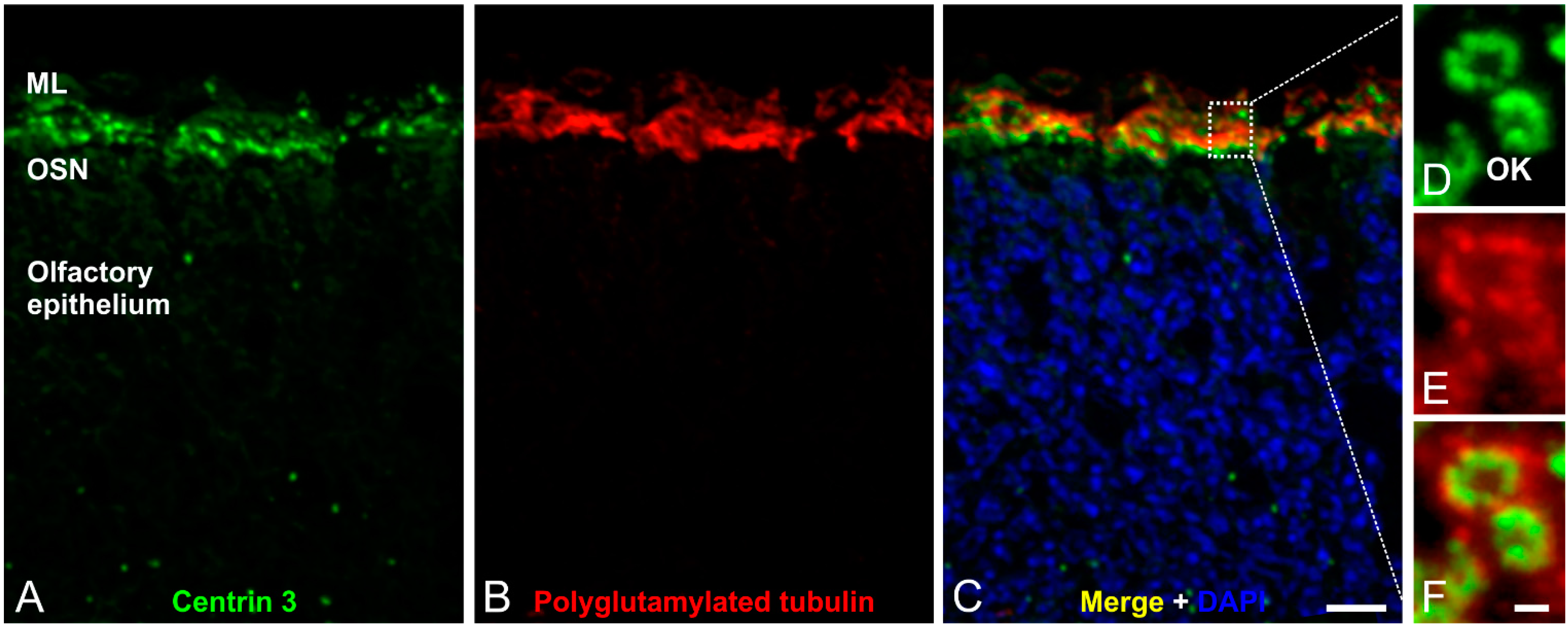
3. The Structure and Role of Cilia in Olfaction
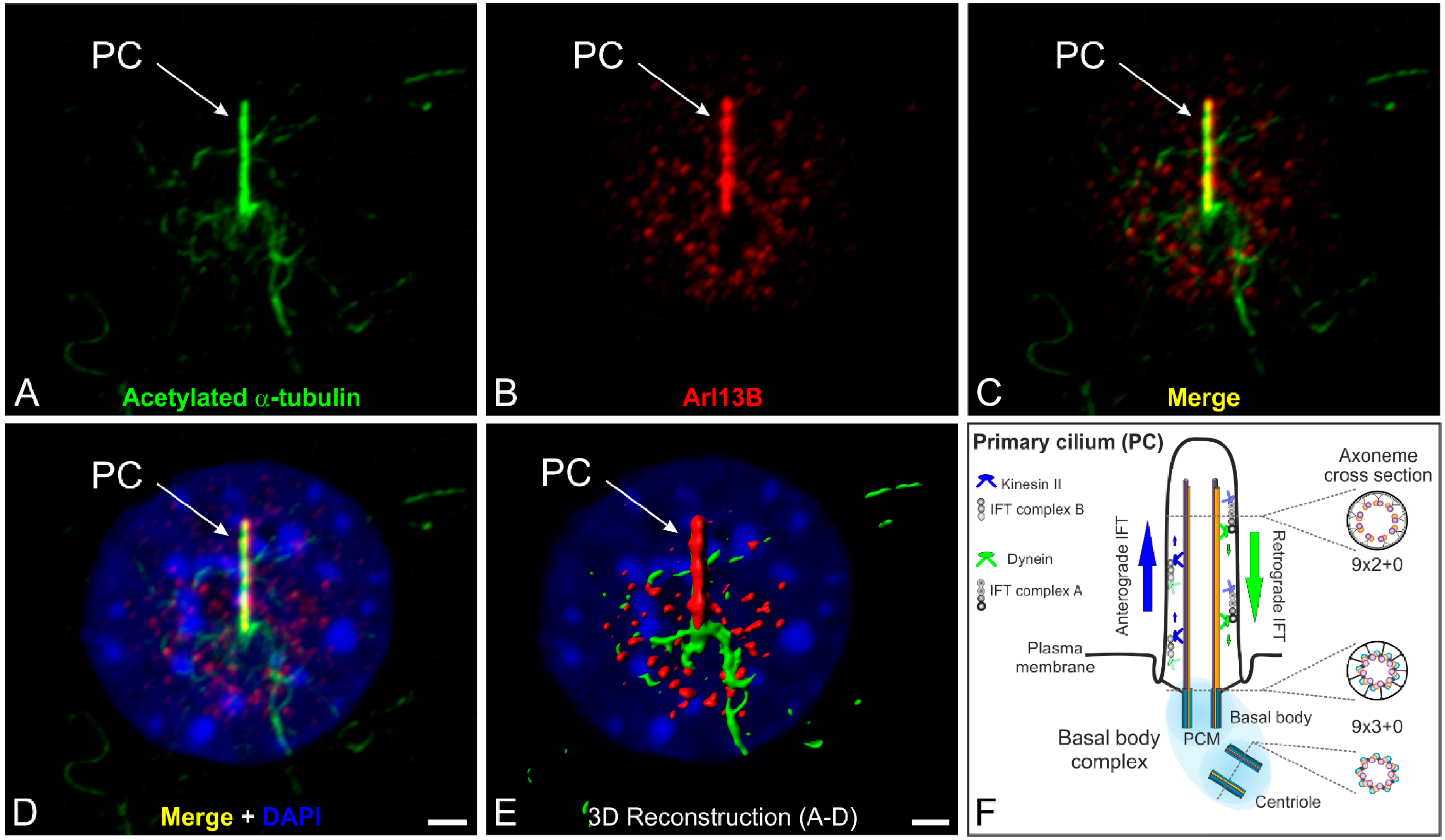
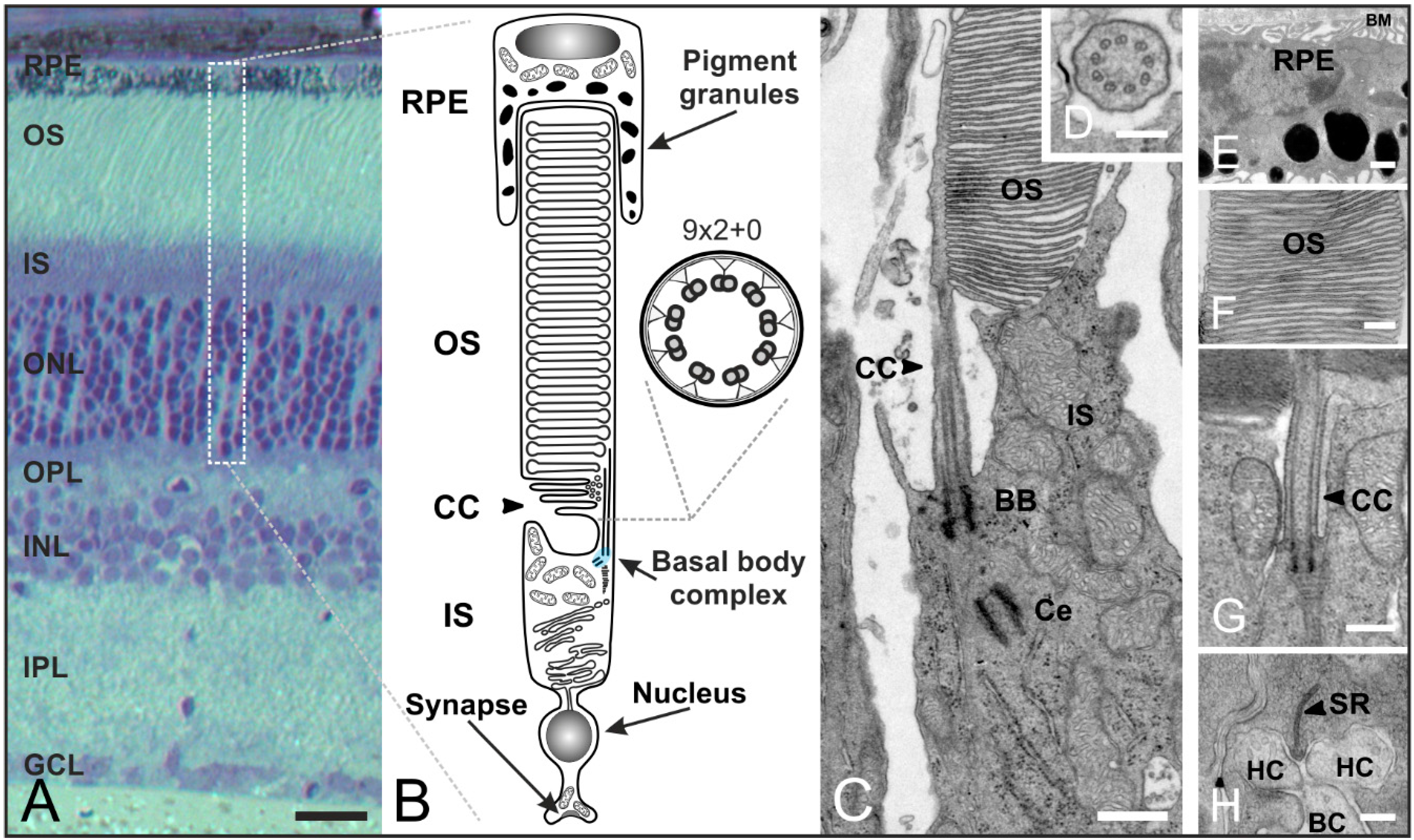
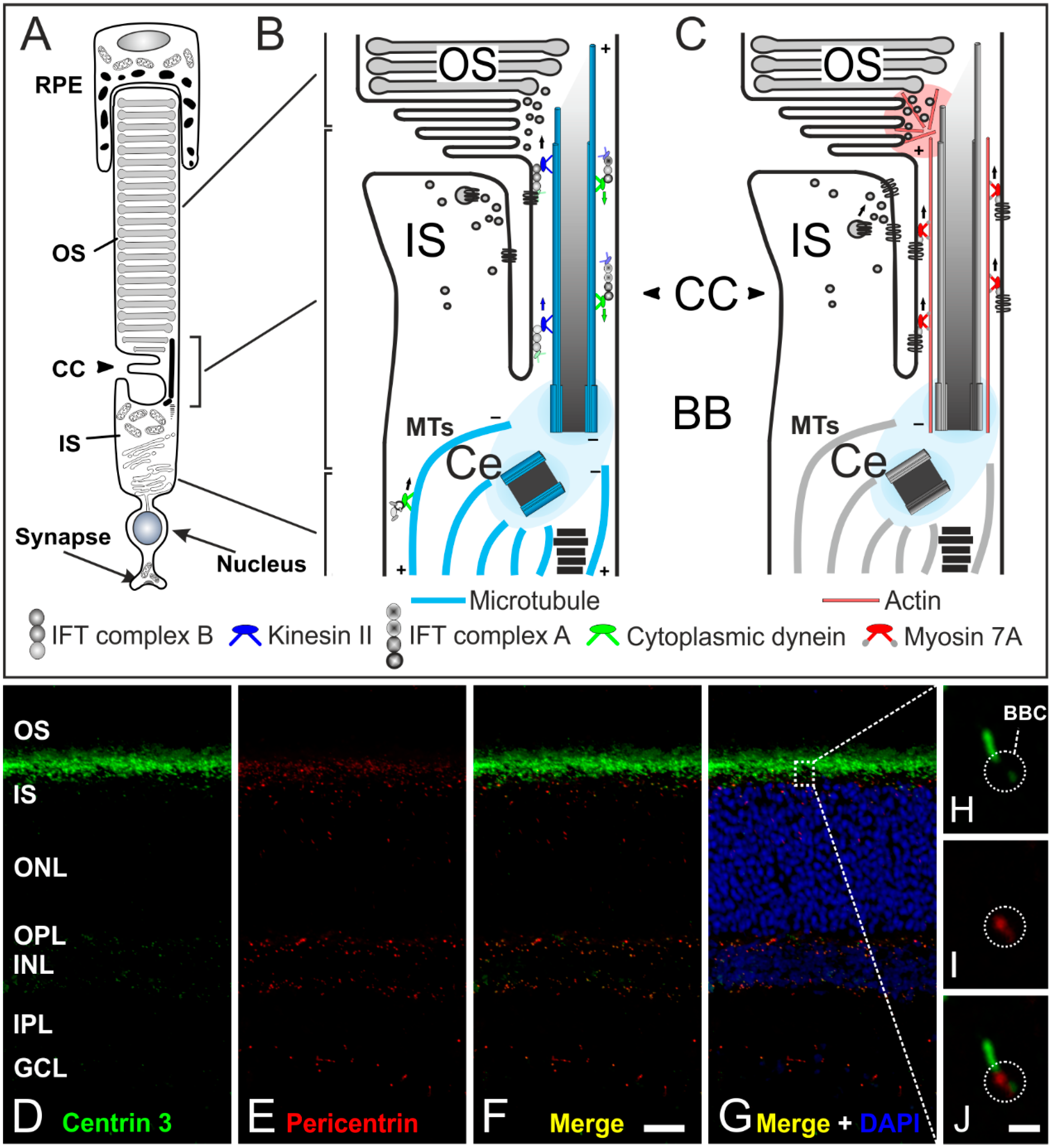
4. The Connecting Cilium as an Environmental Sensor in Light Detection
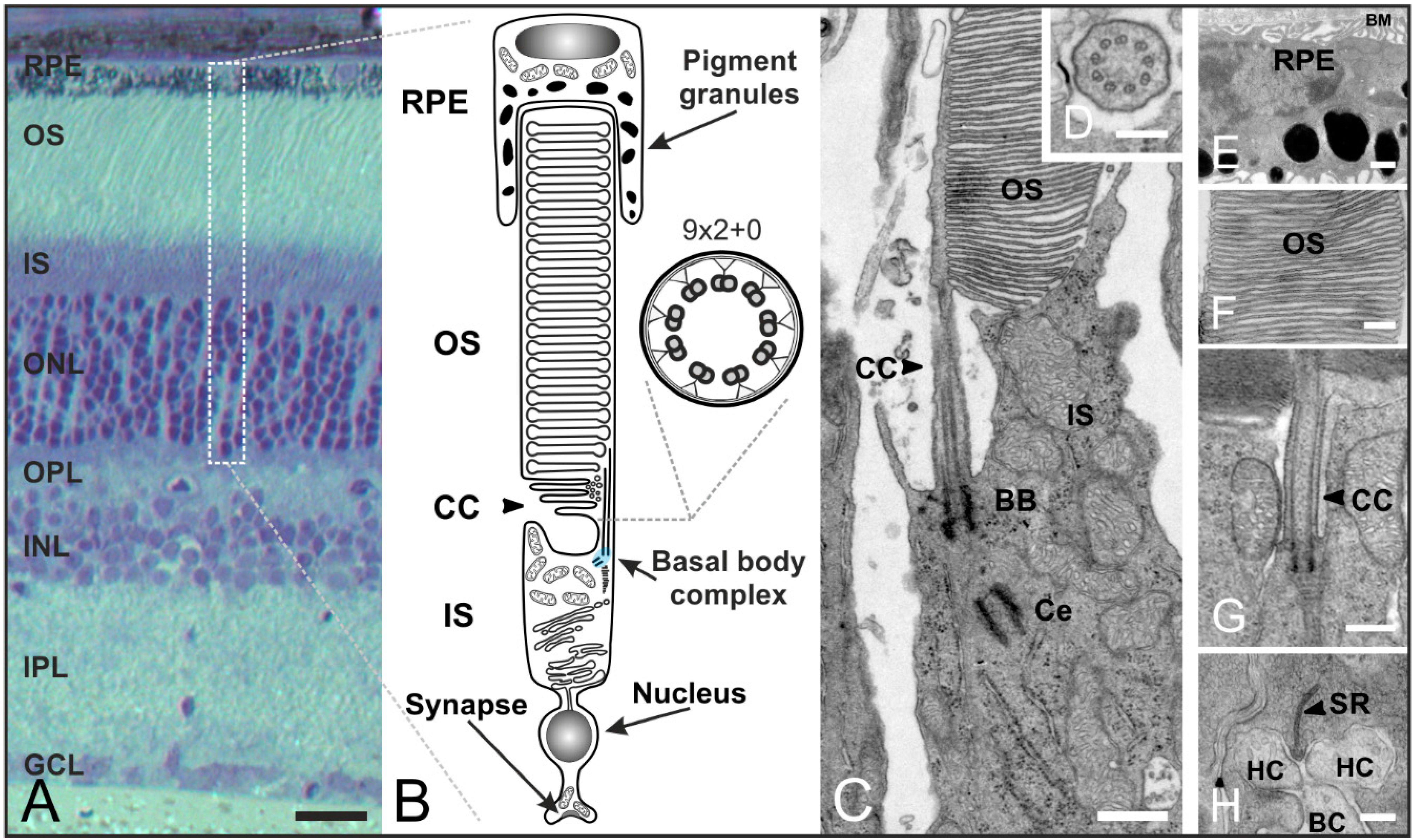
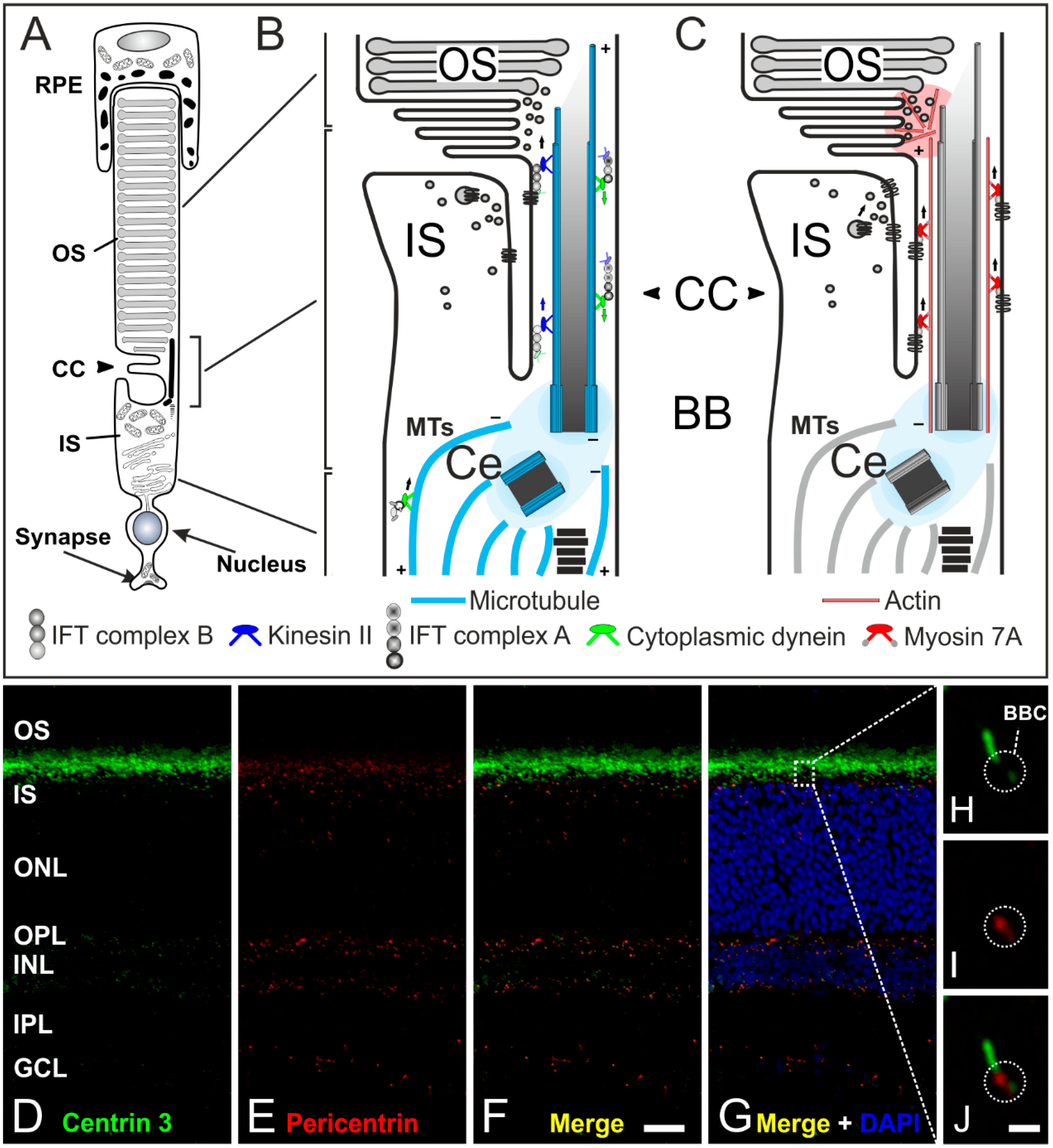
5. Concluding Remarks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ciliopathy or Related Disease | Associated Genes | Affected Tissues | Symptoms in Mouse and Human | Representative Mouse Mutants | Key References |
|---|---|---|---|---|---|
| Alström syndrome | ALMS1 | Eyes, inner ear, adipose tissue | Blindness, hearing loss, obesity | Alms1−/− | [92,93] |
| Bardet-Biedl syndrome | BBS1-BBS16 | Eyes, olfactory system, kidneys, bone, central nervous system, adipose tissue | Blindness, anosmia, renal dysfunction, polydactyly, obesity, mental retardation | Bbs1−/− | [24,94,95] |
| Bbs3−/− | |||||
| Bbs4−/− | |||||
| Joubert syndrome | JBTS1-JBTS15 | Brain, eyes, olfactory system, muscle, kidney, liver, bone | Mental retardation, blindness, anosmia, renal and liver dysfunction, uncoordinated movements | Cep290rd16 | [96,97] |
| Arl13bhnn | |||||
| Mainzer-Saldino syndrome | IFT140 | Eyes, bone, kidney | Blindness, phalangeal cone-shaped epiphyses, abnormality of the proximal femur, renal dysfunction | Ift140cauli/cauli | [98,99,100] |
| MOPD II and Seckel syndrome | PCNT POC1A | Olfactory system, internal organs | Anosmia, developmental defects, dwarfism | Pcntocd | [101,102,103] |
| Pcnt−/− | |||||
| Poc1aem1J | |||||
| Senior-Loken syndrome | NPHP1 NPHP4 NPHP6 | Eyes, internal organs, reproductive system | Blindness, male infertility heterotaxy | Cep290rd16 | [96] |
| Usher syndrome | USH1A-G USH2A-C USH3A-B | Inner ear, eyes, reproductive system | Hearing loss, blindness, infertility, movement anomalies | Ush1c−/− | [104,105,106] |
| Waltzer2J |
Acknowledgments
Conflicts of Interest
Ethics Statement
References
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell's antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Scholey, J.M. Intraflagellar transport motors in cilia: Moving along the cell’s antenna. J. Cell Biol. 2008, 180, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.G. The intraflagellar transport machinery of chlamydomonas reinhardtii. Traffic 2003, 4, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.A.; Leroux, M.R. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009, 19, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.A.; Huang, X.; Behal, R.H.; Cole, D.G.; Qin, H. The RABL5 homolog IFT22 regulates the cellular pool size and the amount of IFT particles partitioned to the flagellar compartment in chlamydomonas reinhardtii. Cytoskelet. (Hoboken NJ) 2012, 69, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Huet, D.; Blisnick, T.; Perrot, S.; Bastin, P. The gtpase IFT27 is involved in both anterograde and retrograde intraflagellar transport. eLife 2014, 3, e02419. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Vogan, K.J.; Wagner, M.K.; Tabin, C.J.; Yost, H.J.; Brueckner, M. Conserved function for embryonic nodal cilia. Nature 2002, 418, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Essner, J.J.; Amack, J.D.; Nyholm, M.K.; Harris, E.B.; Yost, H.J. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 2005, 132, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Zucker, A.G.; Olale, F.; Haycraft, C.J.; Yoder, B.K.; Schier, A.F.; Drummond, I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and kupffer’s vesicle is required for normal organogenesis. Development 2005, 132, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Schweickert, A.; Vick, P.; Wright, C.V.; Danilchik, M.V. Symmetry breakage in the vertebrate embryo: When does it happen and how does it work? Dev. Biol 2014, 393, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Conwell, M.D.; Chen, X.; Kettenhofen, C.I.; Westlake, C.J.; Cantor, L.B.; Wells, C.D.; Weinreb, R.N.; Corson, T.W.; Spandau, D.F.; et al. Primary cilia signaling mediates intraocular pressure sensation. Proc. Natl. Acad. Sci. USA 2014, 111, 12871–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahnleiter, D.; Hauer, N.N.; Kessler, K.; Uebe, S.; Sugano, Y.; Neuhauss, S.C.; Gießl, A.; Ekici, A.B.; Blessing, H.; Sticht, H.; et al. Map4-dependent regulation of microtubule formation affects centrosome, cilia, and golgi architecture as a central mechanism in growth regulation. Hum. Mutat. 2015, 36, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kessler, K.; Wunderlich, I.; Uebe, S.; Falk, N.S.; Gießl, A.; Helmut Brandstatter, J.; Popp, B.; Klinger, P.; Ekici, A.B.; Sticht, H.; et al. Dync2li1 mutations broaden the clinical spectrum of dynein-2 defects. Sci. Rep. 2015, 5, 11649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An sirna-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, D.; Lillo, C.; Roberts, E.A.; Goldstein, L.S.; Williams, D.S. Kinesin-2 and photoreceptor cell death: Requirement of motor subunits. Exp. Eye Res. 2006, 82, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Grati, M.; Chakchouk, I.; Ma, Q.; Bensaid, M.; Desmidt, A.; Turki, N.; Yan, D.; Baanannou, A.; Mittal, R.; Driss, N.; et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum. Mol. Genet. 2015, 24, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- McIntire, W.E.; Dingus, J.; Schey, K.L.; Hildebrandt, J.D. Characterization of the major bovine brain go alpha isoforms. Mapping the structural differences between the alpha subunit isoforms identifies a variable region of the protein involved in receptor interactions. J. Biol. Chem. 1998, 273, 33135–33141. [Google Scholar] [CrossRef] [PubMed]
- Kulaga, H.M.; Leitch, C.C.; Eichers, E.R.; Badano, J.L.; Lesemann, A.; Hoskins, B.E.; Lupski, J.R.; Beales, P.L.; Reed, R.R.; Katsanis, N. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 2004, 36, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, S.R.; Raghupathy, R.K.; Zhang, X.; Mansfield, D.; Shu, X. The role of RPGR and its interacting proteins in ciliopathies. J. Ophthalmol. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Gakovic, M.; Shu, X.; Kasioulis, I.; Carpanini, S.; Moraga, I.; Wright, A.F. The role of RPGR in cilia formation and actin stability. Hum. Mol. Genet. 2011, 20, 4840–4850. [Google Scholar] [CrossRef] [PubMed]
- Murga-Zamalloa, C.A.; Atkins, S.J.; Peranen, J.; Swaroop, A.; Khanna, H. Interaction of retinitis pigmentosa gtpase regulator (rpgr) with RAB8A gtpase: Implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet. 2010, 19, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; May-Simera, H.; Eichers, E.R.; Kai, M.; Hill, J.; Jagger, D.J.; Leitch, C.C.; Chapple, J.P.; Munro, P.M.; Fisher, S.; et al. Disruption of bardet-biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005, 37, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- M'Hamdi, O.; Ouertani, I.; Chaabouni-Bouhamed, H. Update on the genetics of bardet-biedl syndrome. Mol. Syndromol. 2014, 5, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Kremer, H.; van Wijk, E.; Marker, T.; Wolfrum, U.; Roepman, R. Usher syndrome: Molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006, 15, R262–R270. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.W.; Grillet, N.; Andrade, L.R.; Xiong, W.; Swarthout, L.; Della Santina, C.C.; Kachar, B.; Muller, U. Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 2011, 138, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Flock, A.; Duvall, A.J., III. The ultrastructure of the kinocilium of the sensory cells in the inner ear and lateral line organs. J. Cell Biol. 1965, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.D. Basal bodies, kinocilia and planar cell polarity. Nat. Genet. 2008, 40, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Chen, P. Primary cilia in planar cell polarity regulation of the inner ear. Curr. Top. Dev. Biol. 2008, 85, 197–224. [Google Scholar] [PubMed]
- Fukuda, T.; Kominami, K.; Wang, S.; Togashi, H.; Hirata, K.; Mizoguchi, A.; Rikitake, Y.; Takai, Y. Aberrant cochlear hair cell attachments caused by nectin-3 deficiency result in hair bundle. Development 2014, 141, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Roper, V.C.; Foucher, I.; Qian, D.; Banizs, B.; Petit, C.; Yoder, B.K.; Chen, P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat. Genet. 2008, 40, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Leibovici, M.; Verpy, E.; Goodyear, R.J.; Zwaenepoel, I.; Blanchard, S.; Laine, S.; Richardson, G.P.; Petit, C. Initial characterization of kinocilin, a protein of the hair cell kinocilium. Hear. Res. 2005, 203, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Jagger, D.; Collin, G.; Kelly, J.; Towers, E.; Nevill, G.; Longo-Guess, C.; Benson, J.; Halsey, K.; Dolan, D.; Marshall, J.; et al. Alstrom syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum. Mol. Genet. 2011, 20, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Xue, J.; Peterson, E.H. Architecture of the mouse utricle: Macular organization and hair bundle heights. J. Neurophysiol. 2008, 99, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Caggiano, M.; Kauer, J.S.; Hunter, D.D. Globose basal cells are neuronal progenitors in the olfactory epithelium: A lineage analysis using a replication-incompetent retrovirus. Neuron 1994, 13, 339–352. [Google Scholar] [CrossRef]
- Suzuki, J.; Sakurai, K.; Yamazaki, M.; Abe, M.; Inada, H.; Sakimura, K.; Katori, Y.; Osumi, N. Horizontal basal cell-specific deletion of PAX6 impedes recovery of the olfactory neuroepithelium following severe injury. Stem Cells Dev. 2015, 24, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Moran, D.T.; Rowley, J.C., III; Jafek, B.W. Electron microscopy of human olfactory epithelium reveals a new cell type: The microvillar cell. Brain Res. 1982, 253, 39–46. [Google Scholar] [CrossRef]
- Menco, B.P. Ciliated and microvillous structures of rat olfactory and nasal respiratory epithelia. A study using ultra-rapid cryo-fixation followed by freeze-substitution or freeze-etching. Cell Tissue Res. 1984, 235, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; McIntyre, J.C.; Norris, S.R.; Jenkins, P.M.; Zhang, L.; Pei, Q.; Verhey, K.; Martens, J.R. Direct evidence for bbsome-associated intraflagellar transport reveals distinct properties of native mammalian cilia. Nat. Commun. 2014, 5, 5813. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.A.; Cole, D.G.; Seeley, E.S.; Diener, D.R.; Rosenbaum, J.L. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 2001, 11, 1586–1590. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, Q.; Zhang, Y.; Li, Y.; Zhang, Q.; Hu, Z.; Harris, P.C.; Torres, V.E.; Ling, K.; Hu, J. Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat. Commun. 2013, 4, 2750. [Google Scholar] [CrossRef] [PubMed]
- Menco, B.P. Ultrastructural aspects of olfactory signaling. Chem. Senses 1997, 22, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.M.; McEwen, D.P.; Martens, J.R. Olfactory cilia: Linking sensory cilia function and human disease. Chem. Senses 2009, 34, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, K.; Fleischer, J.; Breer, H. Formation and maturation of olfactory cilia monitored by odorant receptor-specific antibodies. Histochem. Cell Biol. 2005, 123, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.R. Ultrastructural studies of microtubules and microtubule organizing centers of the vertebrate olfactory neuron. Microsc. Res. Tech. 1992, 23, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Niimura, Y.; Nei, M. Evolution of olfactory receptor genes in the human genome. Proc. Natl. Acad. Sci. USA 2003, 100, 12235–12240. [Google Scholar] [CrossRef] [PubMed]
- Barish, S.; Volkan, P.C. Mechanisms of olfactory receptor neuron specification in drosophila. Wiley Interdiscip. Rev. Dev. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Buck, L.B. Olfactory receptors and odor coding in mammals. Nutr. Rev. 2004, 62, S184–S188. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Lancet, D. Population differences in the human functional olfactory repertoire. Mol. Biol. Evol. 2003, 20, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Malnic, B.; Hirono, J.; Sato, T.; Buck, L.B. Combinatorial receptor codes for odors. Cell 1999, 96, 713–723. [Google Scholar] [CrossRef]
- Firestein, S. How the olfactory system makes sense of scents. Nature 2001, 413, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Ronnett, G.V. Molecular neurobiology of olfactory transducin. In Handbook on Olfaction and Gustation; Doty, R.L., Ed.; Information Health: New York, NY, USA, 2003; pp. 75–91. [Google Scholar]
- Berbari, N.F.; O’Connor, A.K.; Haycraft, C.J.; Yoder, B.K. The primary cilium as a complex signaling center. Curr. Biol. 2009, 19, R526–R535. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Pinto, J.M. Olfaction: Anatomy, physiology, and disease. Clin. Anat. (N. Y.) 2014, 27, 54–60. [Google Scholar] [CrossRef] [PubMed]
- McEwen, D.P.; Jenkins, P.M.; Martens, J.R. Olfactory cilia: Our direct neuronal connection to the external world. Curr. Top. Dev. Biol. 2008, 85, 333–370. [Google Scholar] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Rohatgi, R. G-protein-coupled receptors, hedgehog signaling and primary cilia. Semin. Cell Dev. Biol. 2014, 33, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Kefalov, V.J. The cone-specific visual cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Besharse, J.C.; Horst, C.J.; Bloodgood, R.A. The photoreceptor connecting cilium—A model for the transition zone. In Ciliary and Flagellar Membranes; Plenum: New York, NY, USA, 1990; pp. 389–417. [Google Scholar]
- Roepman, R.; Wolfrum, U. Protein networks and complexes in photoreceptor cilia. Subcell. Biochem. 2007, 43, 209–235. [Google Scholar] [PubMed]
- Pazour, G.J.; Bloodgood, R.A. Targeting proteins to the ciliary membrane. Curr. Top. Dev. Biol. 2008, 85, 115–149. [Google Scholar] [PubMed]
- Wang, J.; Deretic, D. Molecular complexes that direct rhodopsin transport to primary cilia. Prog. Retin. Eye Res. 2014, 38, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, O.; Khanna, H. Ciliary signaling cascades in photoreceptors. Vis. Res. 2012, 75, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Besharse, J.C.; Forestner, D.M.; Defoe, D.M. Membrane assembly in retinal photoreceptors. III. Distinct membrane domains of the connecting cilium of developing rods. J. Neurosci. 1985, 5, 1035–1048. [Google Scholar] [PubMed]
- Arshavsky, V.Y.; Lamb, T.D.; Pugh, E.N., Jr. G-proteins and phototransduction. Annu. Rev. Physiol. 2002, 64, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.E.; Arshavsky, V.Y. Beyond counting photons: Trials and trends in vertebrate visual transduction. Neuron 2005, 48, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Wensel, T.G. Signal transducing membrane complexes of photoreceptor outer segments. Vis. Res. 2008, 48, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Hardie, R.C. Phototransduction motifs and variations. Cell 2009, 139, 246–264. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.E. Deactivation mechanisms of rod phototransduction: The cogan lecture. Investig. Ophthalmol. Vis. Sci 2010, 51, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky, V.Y.; Burns, M.E. Photoreceptor signaling: Supporting vision across a wide range of light intensities. J. Biol. Chem. 2012, 287, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K. Chemistry and biology of the initial steps in vision: The friedenwald lecture. Investig. Ophthalmol. Vis. Sci 2014, 55, 6651–6672. [Google Scholar] [CrossRef] [PubMed]
- Insinna, C.; Pathak, N.; Perkins, B.; Drummond, I.; Besharse, J.C. The homodimeric kinesin, KIF17, is essential for vertebrate photoreceptor sensory outer segment development. Dev. Biol. 2008, 316, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Sedmak, T.; Wolfrum, U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell 2011, 103, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Vansant, G.; Udovichenko, I.P.; Wolfrum, U.; Williams, D.S. Myosin viia, the product of the usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil. Cytoskelet. 1997, 37, 240–252. [Google Scholar] [CrossRef]
- Colella, P.; Sommella, A.; Marrocco, E.; Di Vicino, U.; Polishchuk, E.; Garrido, M.G.; Seeliger, M.W.; Polishchuk, R.; Auricchio, A. Myosin7a deficiency results in reduced retinal activity which is improved by gene therapy. PLoS ONE 2013, 8, e72027. [Google Scholar] [CrossRef] [PubMed]
- Sedmak, T.; Wolfrum, U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell Biol. 2010, 189, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Deretic, D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vis. Res. 2006, 46, 4427–4433. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Chuang, J.Z.; Bode, C.; Wolfrum, U.; Sung, C.H. Rhodopsin’s carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain tctex-1. Cell 1999, 97, 877–887. [Google Scholar] [CrossRef]
- Artemyev, N.O. Light-dependent compartmentalization of transdu cin in rod photoreceptors. Mol. Neurobiol. 2008, 37, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Slepak, V.Z.; Hurley, J.B. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: Interaction-restricted diffusion. IUBMB Life 2008, 60, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Lobanova, E.S.; Herrmann, R.; Finkelstein, S.; Reidel, B.; Skiba, N.P.; Deng, W.T.; Jo, R.; Weiss, E.R.; Hauswirth, W.W.; Arshavsky, V.Y. Mechanistic basis for the failure of cone transducin to translocate: Why cones are never blinded by light. J. Neurosci. 2010, 30, 6815–6824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Doan, T.; Rieke, F.; Detwiler, P.B.; Frederick, J.M.; Baehr, W. Deletion of prbp/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 2007, 104, 8857–8862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Constantine, R.; Vorobiev, S.; Chen, Y.; Seetharaman, J.; Huang, Y.J.; Xiao, R.; Montelione, G.T.; Gerstner, C.D.; Davis, M.W.; et al. UNC119 is required for g protein trafficking in sensory neurons. Nat. Neurosci. 2011, 14, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Gießl, A.; Regus-Leidig, H.; Brandstätter, J.H. Signal transduction and signal transmission: The two faces of a photoreceptor. e-Neuroforum 2010. [Google Scholar] [CrossRef]
- Norris, D.P.; Grimes, D.T. Mouse models of ciliopathies: The state of the art. Dis. Models Mech. 2012, 5, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Arsov, T.; Larter, C.Z.; Nolan, C.J.; Petrovsky, N.; Goodnow, C.C.; Teoh, N.C.; Yeh, M.M.; Farrell, G.C. Adaptive failure to high-fat diet characterizes steatohepatitis in ALMS1 mutant mice. Biochem. Biophys. Res. Commun. 2006, 342, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. ALMS1-disrupted mice recapitulate human alstrom syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.; et al. BBS2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Nishimura, D.; Seo, S.; Vogel, T.; Morgan, D.A.; Searby, C.; Bugge, K.; Stone, E.M.; Rahmouni, K.; Sheffield, V.C. Bardet-biedl syndrome 3 (BBS3) knockout mouse model reveals common BBS-associated phenotypes and bbs3 unique phenotypes. Proc. Natl. Acad. Sci. USA 2011, 108, 20678–20683. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Khanna, H.; Hawes, N.; Jimeno, D.; He, S.; Lillo, C.; Parapuram, S.K.; Cheng, H.; Scott, A.; Hurd, R.E.; et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the RD16 mouse. Hum. Mol. Genet. 2006, 15, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The graded response to sonic hedgehog depends on cilia architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Saunier, S.; Hanein, S.; Filhol, E.; Bizet, A.A.; Collins, F.; Salih, M.A.; Gerber, S.; Delphin, N.; Bigot, K.; et al. Mainzer-saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012, 90, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Ah-Cann, C.J.; Welfare, M.F.; Tan, T.Y.; Pope, K.; Caruana, G.; Freckmann, M.L.; Savarirayan, R.; Bertram, J.F.; Dobbie, M.S.; et al. Cauli: A mouse strain with an IFT140 mutation that results in a skeletal ciliopathy modelling jeune syndrome. PLoS Genet. 2013, 9, e1003746. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M.; Frank, V.; Eisenberger, T.; Al Turki, S.; Bizet, A.A.; Antony, D.; Rix, S.; Decker, C.; Bachmann, N.; Bald, M.; et al. Combined ngs approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum. Mutat. 2013, 34, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Kasahara, K.; Miyazaki, I.; Shimizu, S.; Taniguchi, M.; Matsuzaki, S.; Tohyama, M.; Asanuma, M. Pericentrin, a centrosomal protein related to microcephalic primordial dwarfism, is required for olfactory cilia assembly in mice. FASEB J. 2009, 23, 3289–3297. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Karkar, K.M.; May, S.R.; Cobos, I.; Thwin, M.T.; Long, J.E.; Ashique, A.M.; Zarbalis, K.; Rubenstein, J.L.; Peterson, A.S. A mutation in the pericentrin gene causes abnormal interneuron migration to the olfactory bulb in mice. Dev. Biol. 2010, 340, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Faqeih, E.; Shamseldin, H.E.; Noche, R.R.; Sunker, A.; Alshammari, M.J.; Al-Sheddi, T.; Adly, N.; Al-Dosari, M.S.; Megason, S.G.; et al. Poc1a truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am. J. Hum. Genet. 2012, 91, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, F.; Holme, R.H.; Bryda, E.C.; Belyantseva, I.A.; Pellegrino, R.; Kachar, B.; Steel, K.P.; Noben-Trauth, K. Mutations in CDH23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for usher syndrome type 1D. Nat. Genet. 2001, 27, 103–107. [Google Scholar] [PubMed]
- Johnson, K.R.; Gagnon, L.H.; Webb, L.S.; Peters, L.L.; Hawes, N.L.; Chang, B.; Zheng, Q.Y. Mouse models of USH1C and DFNB18: Phenotypic and molecular analyses of two new spontaneous mutations of the USH1C gene. Hum. Mol. Genet. 2003, 12, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Kamiya, K.; Ouyang, X.M.; Liu, X.Z. Analysis of subcellular localization of MYO7A, PCDH15 and sans in USH1C knockout mice. Int. J. Exp. Pathol. 2011, 92, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Mühlhans, J.; Brandstätter, J.H.; Gießl, A. The centrosomal protein pericentrin identified at the basal body complex of the connecting cilium in mouse photoreceptors. PLoS ONE 2011, 6, e26496. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Kessler, K.; Gießl, A.; Dimmler, A.; Shalev, S.A.; von der Haar, S.; Zenker, M.; Zahnleiter, D.; Stoss, H.; Beinder, E.; et al. Nek1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011, 88, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamurthy, V.; Jolicoeur, C.; Koutroumbas, D.; Mühlhans, J.; Le, Y.Z.; Hauswirth, W.W.; Gießl, A.; Cayouette, M. Numb regulates the polarized delivery of cyclic nucleotide-gated ion channels in rod photoreceptor cilia. J. Neurosci. 2014, 34, 13976–13987. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falk, N.; Lösl, M.; Schröder, N.; Gießl, A. Specialized Cilia in Mammalian Sensory Systems. Cells 2015, 4, 500-519. https://doi.org/10.3390/cells4030500
Falk N, Lösl M, Schröder N, Gießl A. Specialized Cilia in Mammalian Sensory Systems. Cells. 2015; 4(3):500-519. https://doi.org/10.3390/cells4030500
Chicago/Turabian StyleFalk, Nathalie, Marlene Lösl, Nadja Schröder, and Andreas Gießl. 2015. "Specialized Cilia in Mammalian Sensory Systems" Cells 4, no. 3: 500-519. https://doi.org/10.3390/cells4030500
APA StyleFalk, N., Lösl, M., Schröder, N., & Gießl, A. (2015). Specialized Cilia in Mammalian Sensory Systems. Cells, 4(3), 500-519. https://doi.org/10.3390/cells4030500