3.1. Heh-2 Derived Reporter Proteins for Studying Import
In our previous work [
13], we have defined the minimal features of the yeast
S. cerevisiae INM protein Heh2 that govern its accumulation in the INM. In this process, Heh2 was truncated, removing the extralumenal LEM and MAN domains as well as its second transmembrane helix (TM) and lumenal domain, thereby changing it into a monotopic membrane protein. The LEM and MAN domains could cause retention in the nucleus. The resulting reporter is composed of GFP and Heh293-378 encoding the NLS, the ID linker and the first transmembrane helix, and is named G-NLS-L-TM (
Figure 1A). We have previously shown that this reporter still accumulates in the INM, despite the removal of over a half of the molecular weight of native Heh2; the presence of the NLS and linker regions is required and sufficient for INM accumulation. We also showed that the accumulation is reversible upon inhibition of active transport by depletion of cytosolic Kap95 [
15].
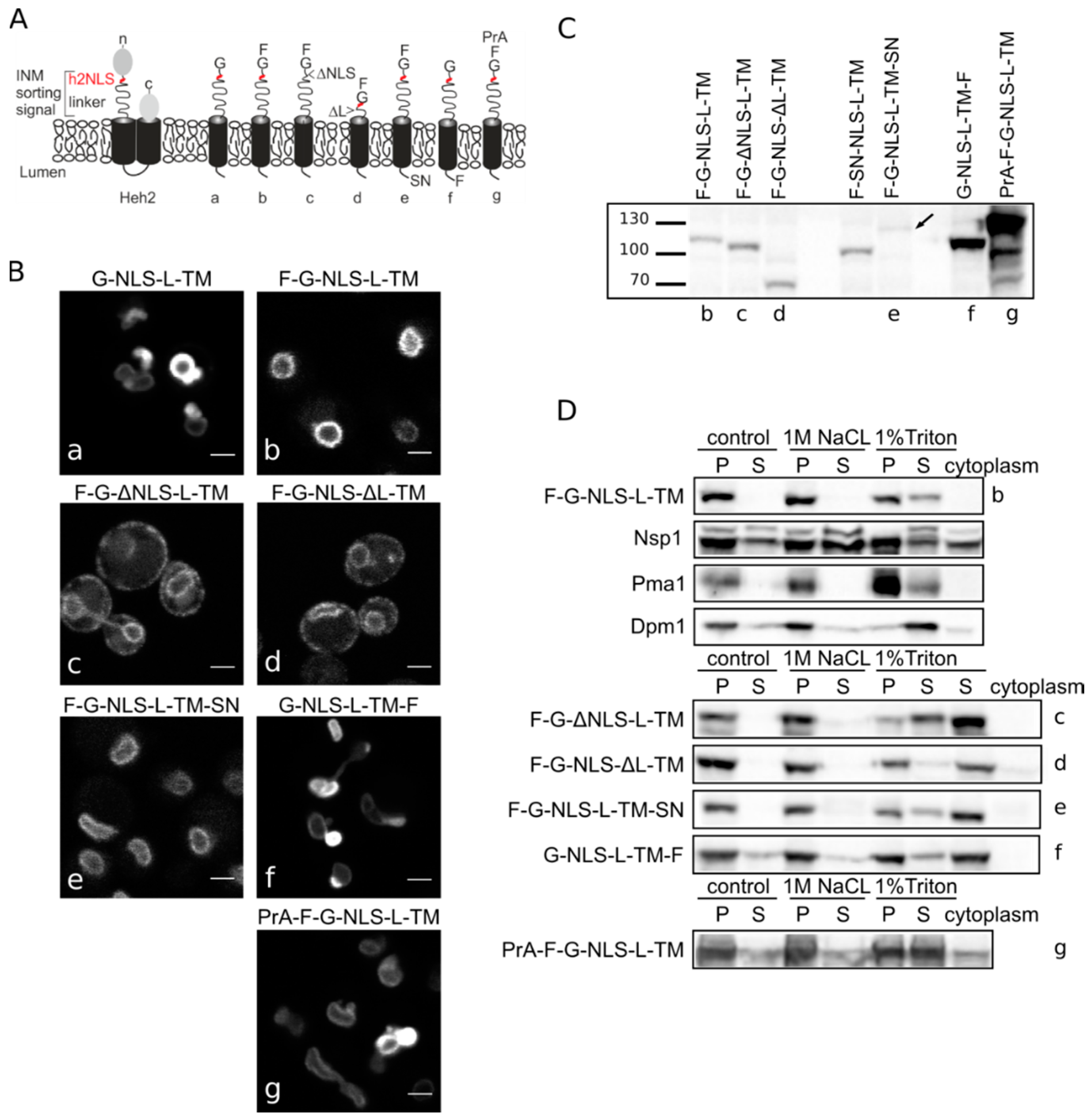
Figure 1.
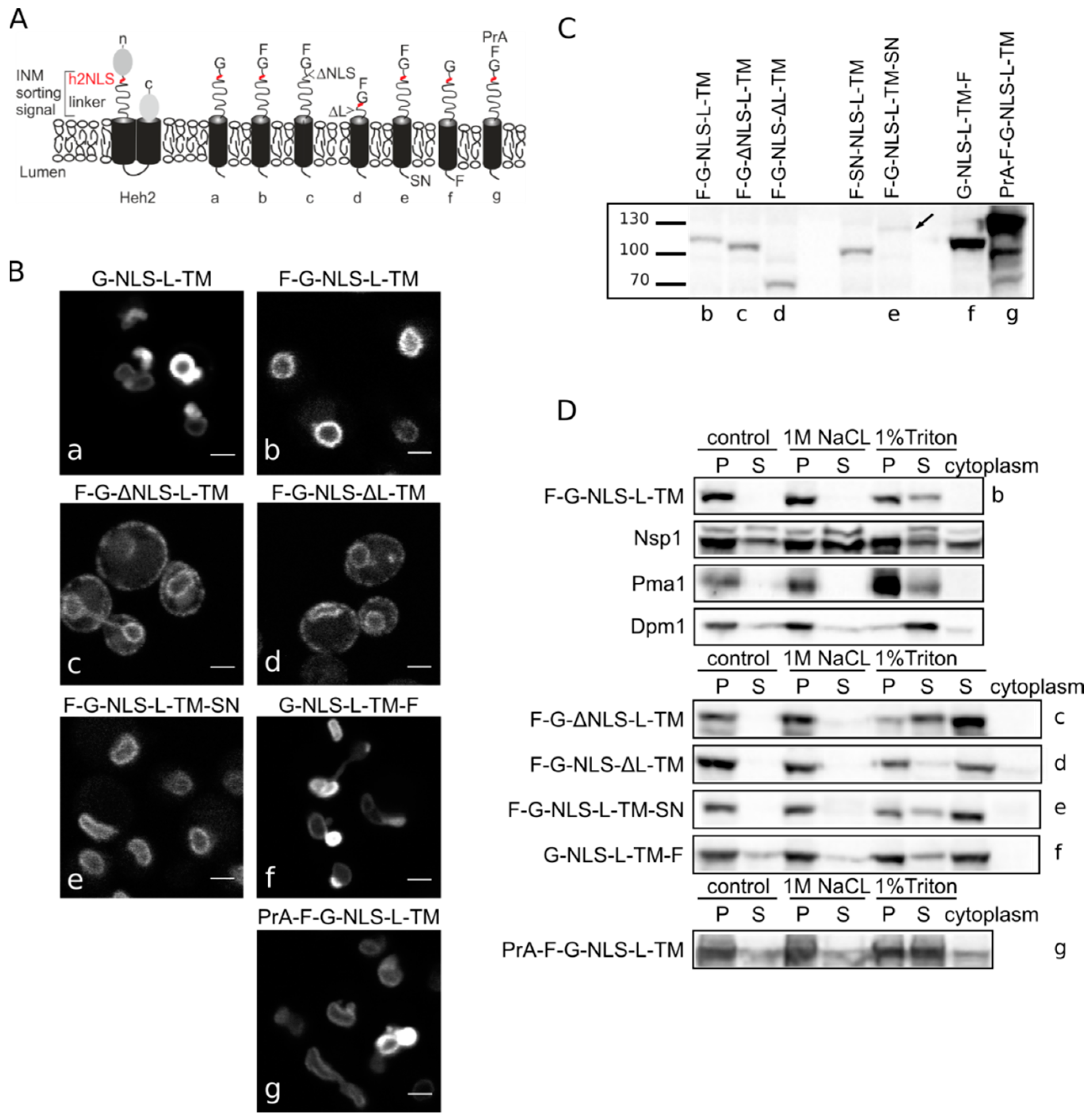
Heh2-derived reporter proteins localizing to the NE-ER network are membrane embedded. (A) Cartoons showing domain composition of native Heh2 and derived reporters. F: FKBP (FK506 binding protein); G: GFP; SN: SNAP-tag; PrA: ProteinA. (B) Confocal fluorescence microscopy images showing K14708 cells expressing the indicated Heh2-derived reporters. Scale bars: 2 μm. (C) Western blot (anti-FKBP) of whole cells extracts of cells expressing the indicated reporters; equal protein amounts were loaded. (D) Western Blots showing the results from salt and detergent extraction of crude yeast membranes fractions. Crude membranes are incubated in control buffer (20 mM Tris with 150 mM NaCl), or in buffer with 1 M NaCl or 1% Triton and ultracentrifuged, as described in Materials and Methods. The pellet (P) and supernatant (S) fractions were loaded onto the gel. The cytoplasm fraction represents the proteins present in the supernatant of the lysate after the first centrifugation step.
Figure 1.
Heh2-derived reporter proteins localizing to the NE-ER network are membrane embedded. (A) Cartoons showing domain composition of native Heh2 and derived reporters. F: FKBP (FK506 binding protein); G: GFP; SN: SNAP-tag; PrA: ProteinA. (B) Confocal fluorescence microscopy images showing K14708 cells expressing the indicated Heh2-derived reporters. Scale bars: 2 μm. (C) Western blot (anti-FKBP) of whole cells extracts of cells expressing the indicated reporters; equal protein amounts were loaded. (D) Western Blots showing the results from salt and detergent extraction of crude yeast membranes fractions. Crude membranes are incubated in control buffer (20 mM Tris with 150 mM NaCl), or in buffer with 1 M NaCl or 1% Triton and ultracentrifuged, as described in Materials and Methods. The pellet (P) and supernatant (S) fractions were loaded onto the gel. The cytoplasm fraction represents the proteins present in the supernatant of the lysate after the first centrifugation step.
In the here presented experiments, we use the above-mentioned reporter and a reporter with an
N-terminal FKBP tag, F-G-NLS-L-TM. Addition of the FKBP tag has a dual role—it is used in trapping experiments described later, but it also reduces the expression level of the entire protein and the NE deformation caused by high expression of INM proteins. Analysis of the microscopy data of the reporters (
Figure 1B) reinstated the necessity of both the NLS and the linker region for targeting the NE as removal of the NLS (F-G-ΔNLS-L-TM) or the linker domain (F-G-NLS-ΔL-TM) abolishes NE accumulation: reporters are evenly distributed between the NE and ER. Comparison of the expression levels of all the reporters is presented in
Figure 1C. They range from very low in the case of F-G-NLS-L-TM-SN (SN for SNAP tag), to very high in case of G-NLS-L-TM-F and PrA-F-G-NLS-L-TM. Higher expression levels of G-NLS-L-TM, or the reporters with C-terminal FKBP-tag (G-NLS-L-TM-F) or
N-terminal Protein A tag (PrA-F-G-NLS-L-TM) do not interfere with accumulation at the NE, but cells do display deformation of the NE.
3.2. Heh-2 Derived Reporters are Integral Membrane Proteins of the NE-ER
Next, we performed biochemical fractionation studies to confirm that the reporters are membrane embedded. We measured the steady state membrane integration of the reporters by salt and detergent extraction (as published in [
25]). In this method, a crude membrane fraction is isolated from exponentially growing yeast and subsequently incubated with buffers containing a high concentration of salt or detergent. After ultracentrifugation, the soluble and pellet fractions are analyzed. Proteins that solubilize with salt are only peripherally associated with the membrane, such as the nucleoporin Nsp1, which is used as a control protein. Transmembrane proteins, such as the ER protein Dpm1 with one transmembrane helix and the plasma membrane protein Pma1 with 10 transmembrane helices, require detergents to be extracted from the membrane. As shown on Western blots in
Figure 1D, the integral membrane proteins solubilize only (Pma1) or predominantly (Dpm1) in 1% Triton, while the peripheral protein Nsp1 is also found in the soluble fractions when membranes were incubated with the control buffer containing 150 mM NaCl or the buffer with 1 M NaCl. The F-G-NLS-L-TM reporter behaved similarly to Dpm1 and Pma1, and solubilized in buffer with 1% Triton only. We next checked if deletion of the NLS (F-G-ΔNLS-L-TM), deletion of the linker region (F-G-NLS-ΔL-TM) or expanding the size of the soluble domains (from the cytoplasmic side PrA-F-G-NLS-L-TM and from the lumenal side F-G-NLS-L-TM-SN or G-NLS-L-TM-F) changes the solubilization pattern of these reporters. All of them solubilized predominantly or exclusively when treated with 1% Triton (
Figure 1D). On the blots with G-NLS-L-TM-F and PrA-F-G-NLS-L-TM, which are the higher expressed proteins, a fraction of the protein appears in the soluble extract after incubation with the control buffer or with 1 M NaCl, similar to Dpm1. This suggests that a fraction of the reporter proteins is not well inserted in the membrane.
Altogether, the biochemical fractionation studies on whole cell lysates and the in vivo localization studies confirm that the majority of Heh2-derived reporter proteins are membrane embedded, and that the linker region and NLS are not critical for membrane insertion.
3.3. Evidence against Membrane Insertion Post Nuclear-Import
The biochemical assay presented above does not exclude if temporarily, especially during the transport via the NPC channel, a fraction of the proteins is not embedded in the membrane. This could be particularly relevant for proteins that are inserted in the membrane post-translationally and which exist shortly as a soluble, chaperoned protein. The possibility of insertion to the inner nuclear membrane after nuclear import has thus far not been tested. Many of our reporters have C-terminal transmembrane helices with a short C-terminal tail (38 residues for the G-NLS-L-TM reporter), and could thus classify as tail-anchored proteins. These types of proteins are inserted into the membrane environment post-translationally via the GET pathway [
20].
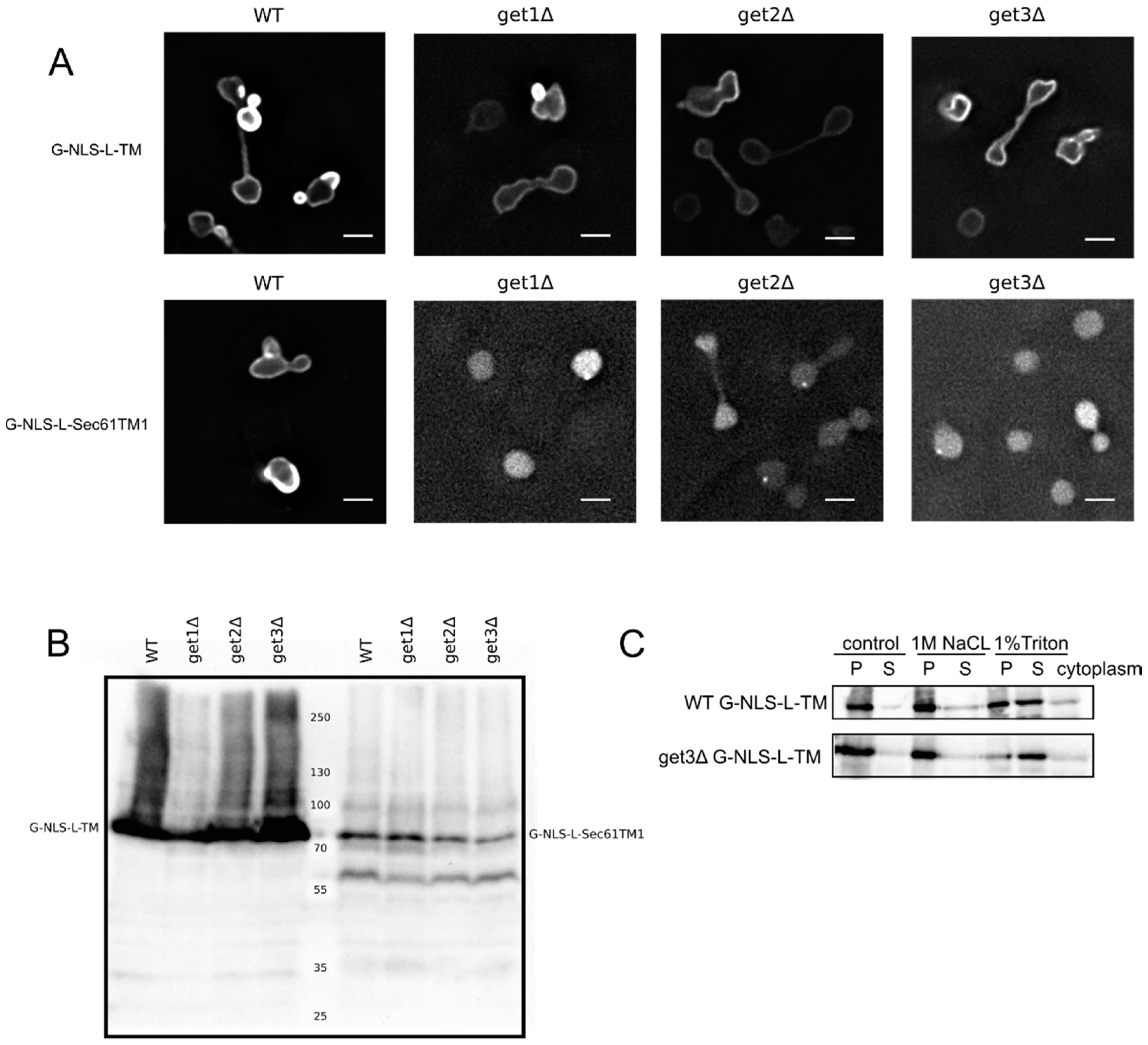
We first aimed to resolve the uncertainty if our reporters depend on the GET system for membrane insertion, and tested their localization in a series of GET deletion mutants. The microscopy images and the membrane extractions with salt and detergent presented in
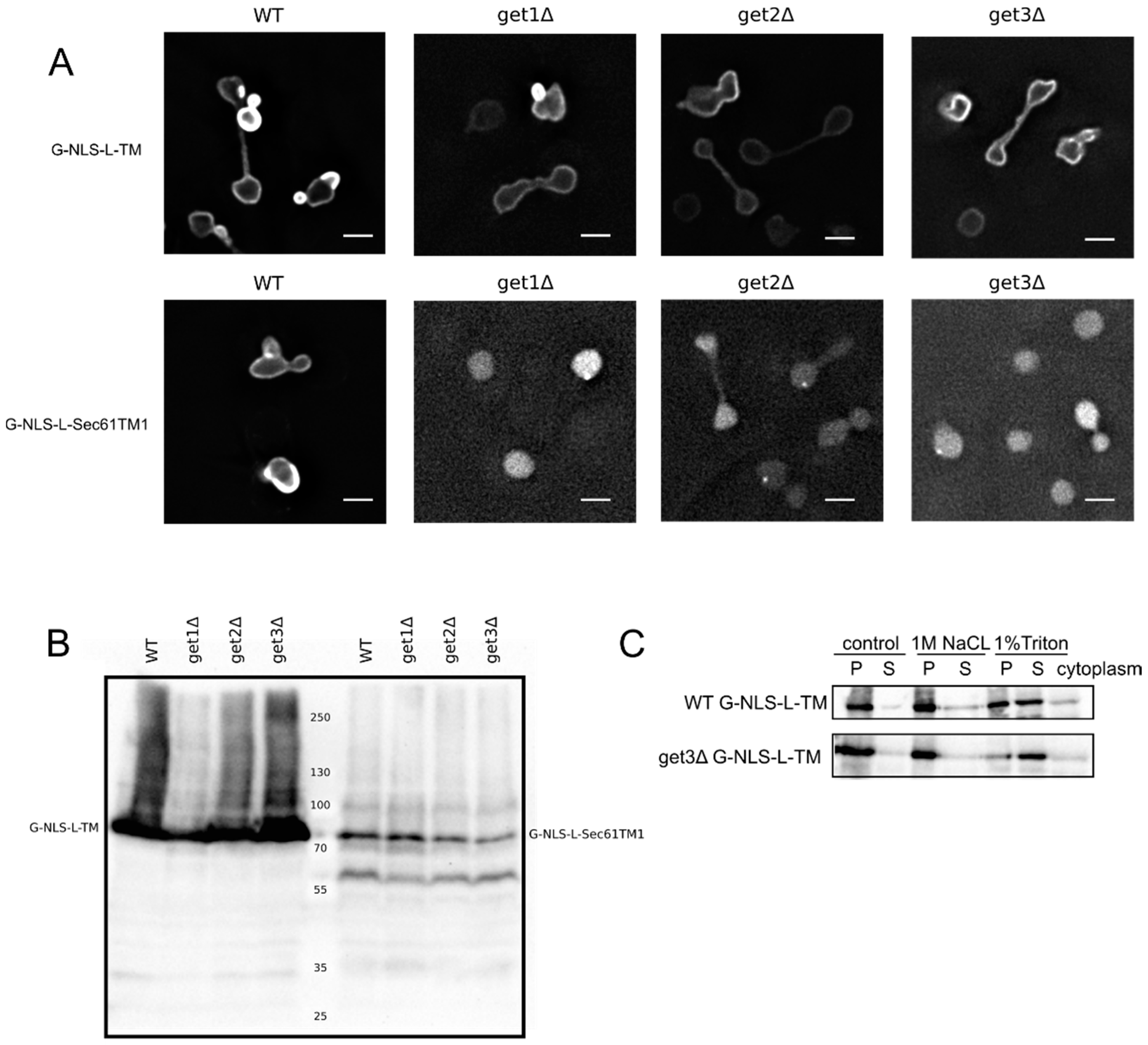
Figure 2A,C clearly show that the G-NLS-L-TM reporter is NE localized and membrane embedded, also when the GET system is nonfunctional,
i.e., in a get3Δ mutant, which lacks the protein that chaperones the newly synthesized tail-anchored protein and brings it to the membrane insertion machinery. Also in get1Δ and a get2Δ mutant, lacking the proteins that are responsible for membrane insertion step of tail-anchored proteins, the reporter protein is NE localized (
Figure 2A). As a control we expressed the G-NLS-L-Sec61TM1 reporter in the wildtype and GET deletion strains. In this reporter the transmembrane helix of Heh2 is replaced by the first transmembrane helix of the yeast membrane protein Sec61, and it contains a short C-terminal tail of only 10 amino acids [
13]. When expressed in the wildtype yeast BY4742 the G-NLS-L-Sec61TM1 reporter localizes to the NE, in GET deletion mutants, however, this reporter localizes to the nucleoplasm and is not present in the NE (
Figure 2A). Expression levels of the full length protein, and the degree of degradation of the protein, are very similar in WT and GET deletion strains (
Figure 2B), showing that in the case of the G-NLS-L-Sec61TM1 reporter, membrane insertion is GET dependent and that this tail-anchored protein can be imported to the nucleus as a soluble protein when the GET insertion machinery is not functional. As we see no difference in the localization of G-NLS-L-TM between the wild type and the GET mutants, we conclude that the insertion of these reporters is not strictly dependent on GET, which would point to either Sec61-dependent co-translational insertion or by another still unidentified system.
Although chaperoned soluble nuclear import is less likely for reporters that are co-translationally inserted by Sec61, instead of post-translationally via the GET system, we aimed to further experimentally test this possibility for two reasons. Firstly, since the h2NLS-linker motif is a potent signal for sorting to the nuclear envelope outcompeting other classical NLSs [
13,
15,
16], a competition between the membrane insertion and nuclear transport machineries might take place. Secondly, one report indicates the presence of the Sec61 machinery on the inner side of the nuclear envelope in yeast [
26]. We therefore developed a system to monitor if membrane proteins were membrane embedded when they are trapped in the NPC. Our trapping experiments are based on the Anchor Away system [
22], which utilizes rapamycin-dependent interaction between FRB and FKBP molecules. Previously, the FG-Nup Nsp1 had been used as the anchor in the central channel [
13]. However, as recent publications show, a non-NPC cytoplasmic pool of Nsp1 [
27,
28], in our new set up FRB was fused to the inner ring scaffold nucleoporin Nup170.
Figure 2.
Membrane insertion of the G-NLS-L-TM reporter is GET-independent. (
A) Fluorescence microscopy of wild type and get1Δ, get2Δ and get3Δ mutant yeasts expressing G-NLS-L-TM or G-NLS-L-Sec61TM1. Scale bars: 2 μm. (
B) Expression levels (anti-GFP Western blot) of G-NLS-L-TM and G-NLS-L-Sec61TM1 in WT and GET mutants. (
C) Western Blots (anti-GFP) showing the results form salt and detergent extraction assay on WT and get3Δ mutant expressing G-NLS-L-TM. Crude membrane preparations were treated as described in Material and Methods and in the legend to
Figure 1. P, pellet; S, supernatant.
Figure 2.
Membrane insertion of the G-NLS-L-TM reporter is GET-independent. (
A) Fluorescence microscopy of wild type and get1Δ, get2Δ and get3Δ mutant yeasts expressing G-NLS-L-TM or G-NLS-L-Sec61TM1. Scale bars: 2 μm. (
B) Expression levels (anti-GFP Western blot) of G-NLS-L-TM and G-NLS-L-Sec61TM1 in WT and GET mutants. (
C) Western Blots (anti-GFP) showing the results form salt and detergent extraction assay on WT and get3Δ mutant expressing G-NLS-L-TM. Crude membrane preparations were treated as described in Material and Methods and in the legend to
Figure 1. P, pellet; S, supernatant.
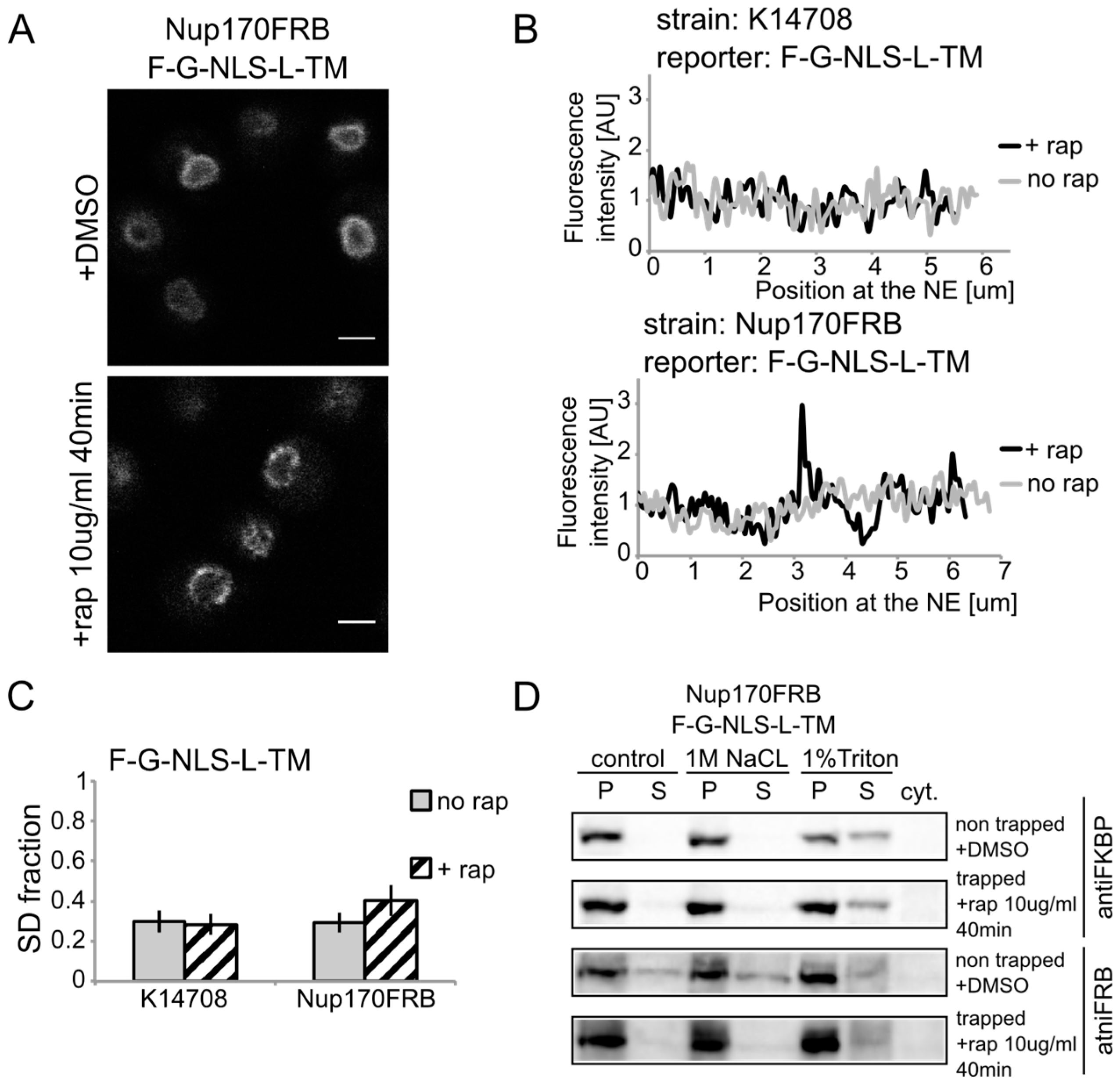
We show rapamycin-dependent trapping of the reporter with the
N-terminal FKBP tag, F-G-NLS-L-TM, at Nup170 (
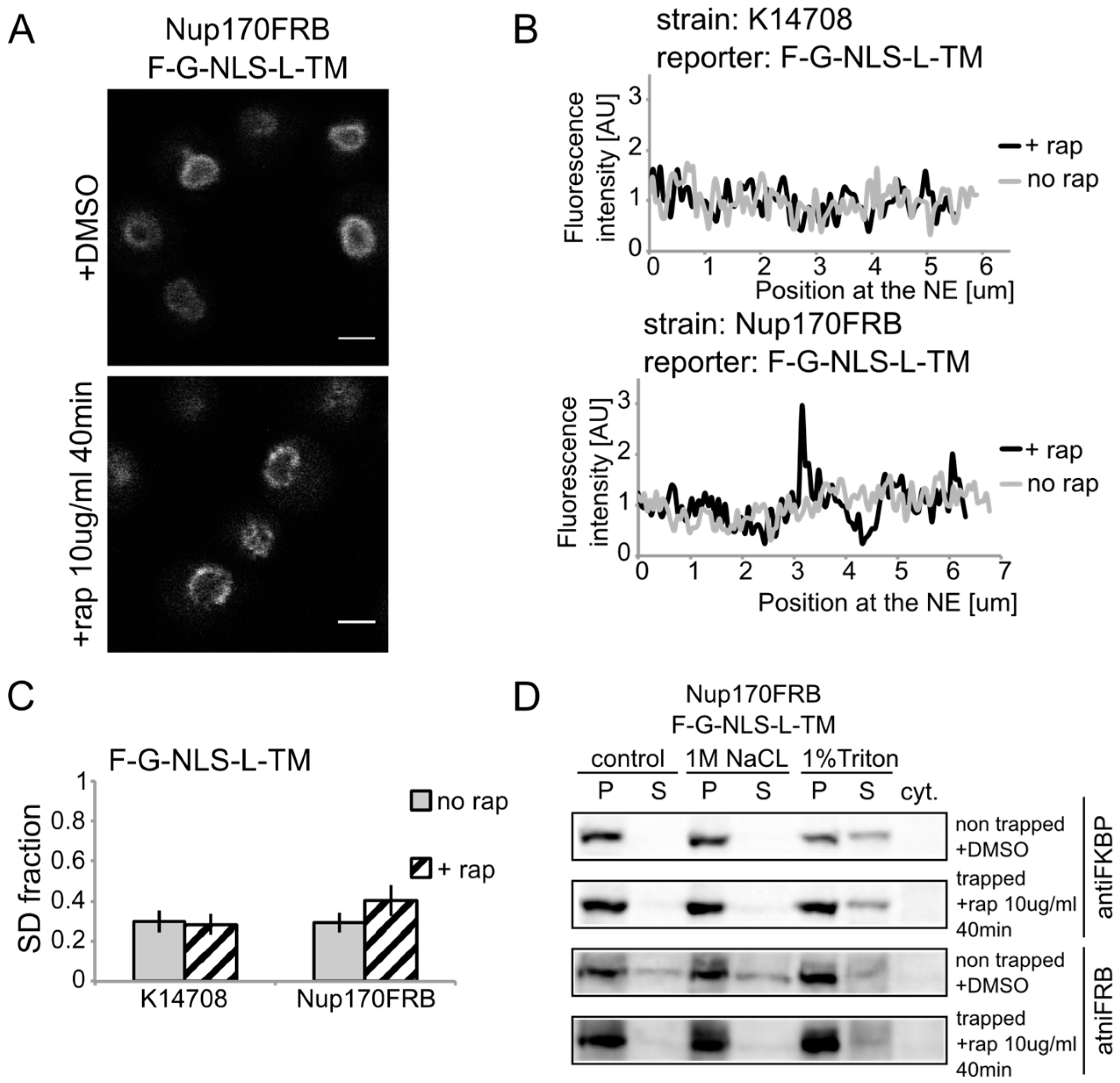
Figure 3A). After cells were exposed to rapamycin, the fluorescence signal in the NE was observed in an exclusive punctate pattern, similar to what is seen with fluorescently labeled nucleoporins. The change in fluorescence pattern is also apparent when measuring the fluorescence intensity in the NE (
Figure 3B), and calculating the SD fraction (as described in the Experimental section), which represents to which degree the fluorescence on a specific location in the NE deviates from the average fluorescence intensity. The average SD fraction over multiple cells is indicated in
Figure 3C. The average SD fraction increases significantly in conditions with rapamycin in the strain expressing Nup170FRB but not in the background strain (K14708), which does not have an FRB anchor at Nup170. Therefore, we conclude that a significant fraction (or possibly all) of the expressed reporter molecules were trapped at Nup170.
Figure 3.
Heh2-derived reporters are membrane-embedded while transiting the NPC. (A) Confocal fluorescence microscopy images of cells expressing FRB (FKBP12-rapamycin binding)-tagged Nup170 (Nup170FRB) and F-G-NLS-L-TM reporter after incubation with rapamycin (+rap 10 μg/mL) and in control conditions (+DMSO). The fluorescence patterns change from continues to punctate upon rapamycin treatment. Scale bars: 2 μm. (B) Line-scans of the fluorescence intensity in the NE of the representative cells expressing F-G-NLS-L-TM after incubation with rapamycin (black line, +rap) or in control conditions (grey line, no rap). Top panel: control K14708 strain. Bottom panel: Nup170FRB strain. (C) Comparison of the average SD fraction for each reporter in the Nup170FRB strain (n = 37 cells for both conditions) and in the wild type (K14708, n = 7 cells for no rap, n = 55 cells for +rap); standard deviation is indicated. The SD fraction is calculated from the standard deviation of the fluorescence intensity along the NE in a cell divided by the average fluorescence intensity at the NE in that cell. (D) Western Blots showing the results from salt and detergent extraction of crude yeast membranes fractions of cells expressing Nup170FRB and F-G-NLS-L-TM. Membrane extractions were performed in trapped (+rap) and in non-trapped (+DMSO) conditions. F-G-NLS-L-TM solubilizes with the buffer with 1% Triton both in trapped and non-trapped conditions. Nup170FRB is salt-soluble before trapping and becomes salt-resistant upon trapping as the bands of solubilized fractions treated with control buffer and 1 M NaCl disappear.
Figure 3.
Heh2-derived reporters are membrane-embedded while transiting the NPC. (A) Confocal fluorescence microscopy images of cells expressing FRB (FKBP12-rapamycin binding)-tagged Nup170 (Nup170FRB) and F-G-NLS-L-TM reporter after incubation with rapamycin (+rap 10 μg/mL) and in control conditions (+DMSO). The fluorescence patterns change from continues to punctate upon rapamycin treatment. Scale bars: 2 μm. (B) Line-scans of the fluorescence intensity in the NE of the representative cells expressing F-G-NLS-L-TM after incubation with rapamycin (black line, +rap) or in control conditions (grey line, no rap). Top panel: control K14708 strain. Bottom panel: Nup170FRB strain. (C) Comparison of the average SD fraction for each reporter in the Nup170FRB strain (n = 37 cells for both conditions) and in the wild type (K14708, n = 7 cells for no rap, n = 55 cells for +rap); standard deviation is indicated. The SD fraction is calculated from the standard deviation of the fluorescence intensity along the NE in a cell divided by the average fluorescence intensity at the NE in that cell. (D) Western Blots showing the results from salt and detergent extraction of crude yeast membranes fractions of cells expressing Nup170FRB and F-G-NLS-L-TM. Membrane extractions were performed in trapped (+rap) and in non-trapped (+DMSO) conditions. F-G-NLS-L-TM solubilizes with the buffer with 1% Triton both in trapped and non-trapped conditions. Nup170FRB is salt-soluble before trapping and becomes salt-resistant upon trapping as the bands of solubilized fractions treated with control buffer and 1 M NaCl disappear.
![]()
Using biochemical fractionation methods, we now addressed if the reporter molecules are membrane embedded when trapped in the NPC. If the reporter protein passes the NPC as a soluble protein, one expects a change in the membrane extraction characteristics (
Figure 3D). When trapped at Nup170, the reporter is detergent-soluble and therefore a true transmembrane protein after entry in the pore. Consistent with this, the tight interaction with the reporter causes a change in Nup170 solubility: in non-trapped conditions Nup170 is partially extracted with salt as expected for a non-transmembrane protein, while salt-extraction is clearly prevented after rapamycin-induced binding to the reporter.
Altogether, the experiments presented in
Figure 1,
Figure 2 and
Figure 3 show the transmembrane nature of our INM reporter proteins, also when trapped in the NPC. We find no support for a mechanism where membrane insertion occurs post nuclear import.
3.4. Active Transport Breaks Size Restrictions for Passive Leak through the NPC
We revisit our claim that in active import large extralumenal domains were imported; larger than what was estimated to pass the lateral channels passively. Firstly, the assays are now performed with reporter proteins with multi-pass transmembrane domain to validate that our previous studies were not biased from the use of monotopic membrane proteins. Secondly, we mapped the size limitations for passive entry of membrane proteins more precisely and established that membrane proteins with an extralumenal domain of 90 kDa still diffuse to the INM on a time-scale of an hour [
29]. This now allows us to better assess if active transport indeed breaks size restrictions for passive diffusion. Thirdly, we improved the assay to better account for the impact of protein synthesis in our assay.
The new reporters consisted of the transmembrane domain of Sec61 (10 transmembrane segments). However, previous publications have suggested that Sec61 may exist in the INM [
26] and thus Sec61 may have specific protein interaction partners in the INM. To exclude that the Sec61 transmembrane domain contributed to the sorting, we additionally repeated the INM targeting with reporters with a TM domain unrelated to yeast. We used the polytopic TM domain from RibU from the Gram-positive bacterium
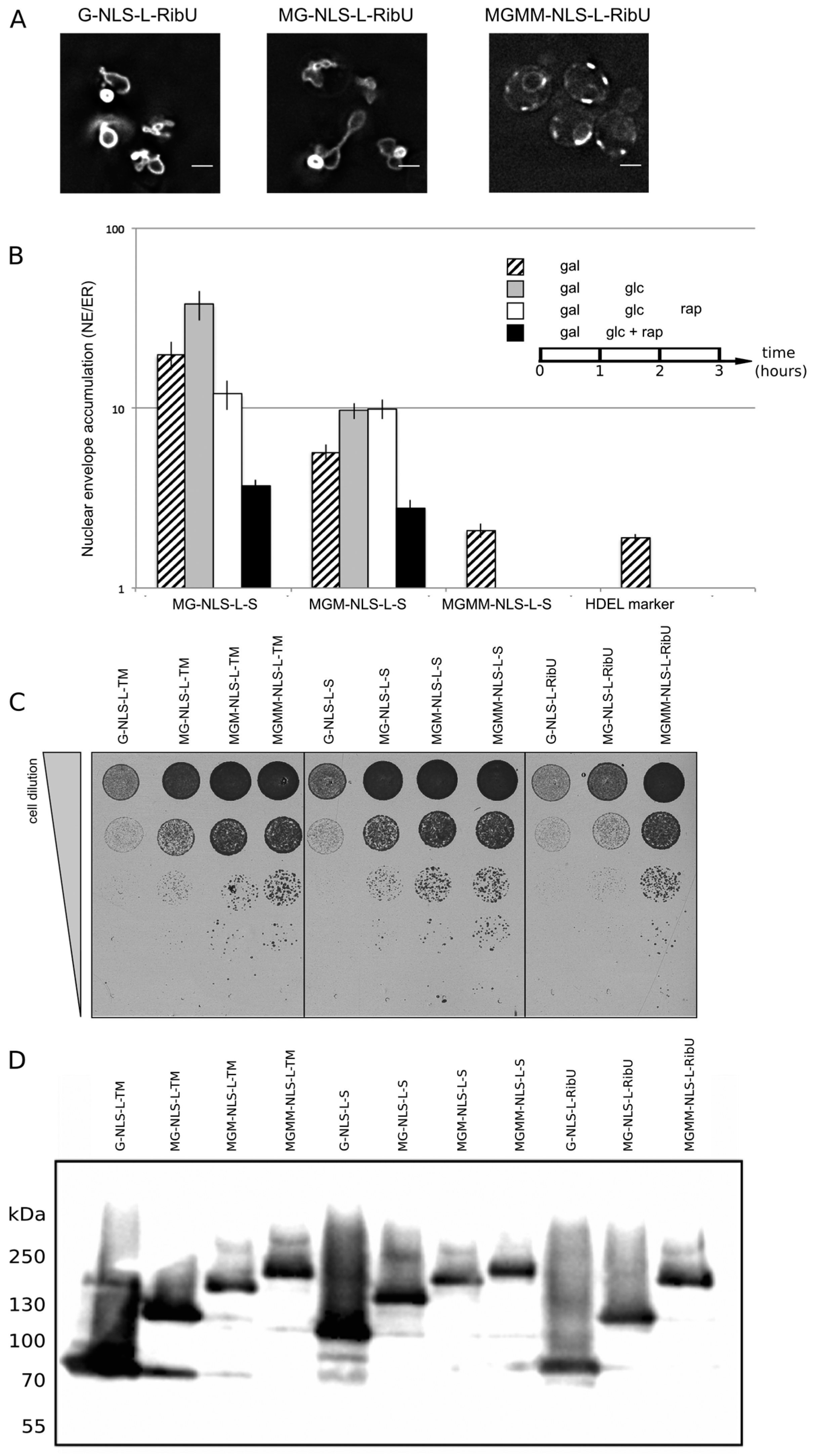
Lactococcus lactis, thereby excluding the chance of specific retention of the reporter in the INM via interaction with the other INM proteins. The fusion indeed sorts effectively to the INM (
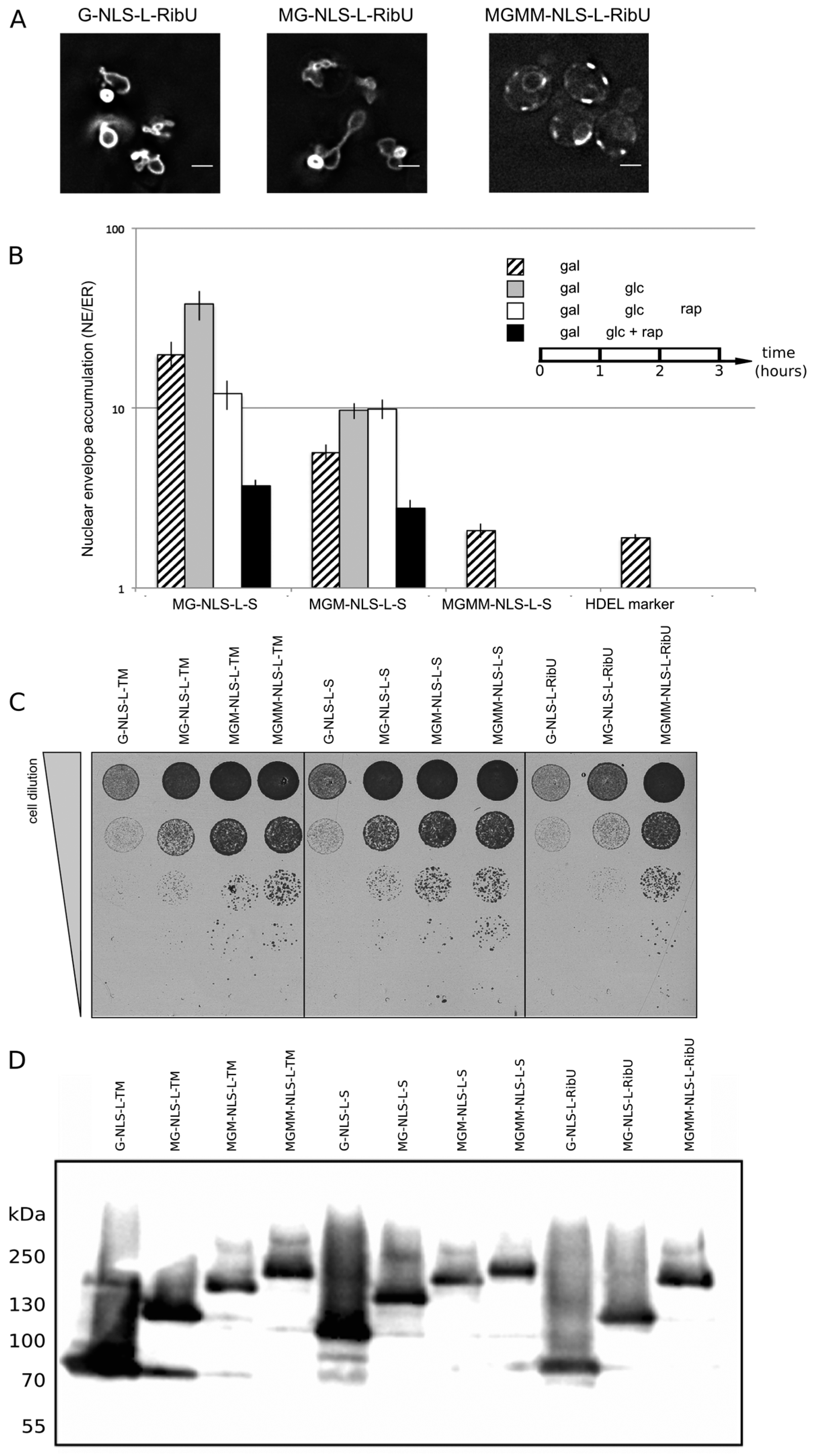
Figure 4A, G-NLS-L-RibU) confirming our previous claims that import is not dependent on the nature of the TM domain.
Next, a set of reporter membrane proteins with extralumenal domains of increasing size was constructed. Their extralumenal domains were composed of GFP, one, two or three copies of MBP together with the NLS-L motif, resulting in extralumenal domain size of 95 kDa (MG-NLS-L-S and MG-NLS-L-RibU); 136 kDa (MGM-NLS-L-S) and 176 kDa (MGMM-NLS-L-S and MGMM-NLS-L-RibU). From past analysis monitoring Heh2, Heh2ΔNLS [
14], GFP-h2NLS-L-TM and GFP-L-TM [
13] using immune electron microscopy, we know that the ratio of GFP-fluorescence in the NE and ER, (the NE/ER ratios), is a good readout of accumulation at the INM as compared to the ONM. In addition, for reporter proteins with Sec61 transmembrane domains we confirm that high NE/ER ratio’s report accumulation of the proteins at the INM as compared to the ONM (Popken
et al., unpublished). We thus assume that also for the here presented reporter proteins the NE/ER ratio reports localization at the INM. In the Kap95AA strain background, we find that a protein lacking complete sorting signals, such as G-ΔNLS-L-TM, gives an NE/ER ratio similar to the NE/ER ratio found for the ER-marker protein mCherry-HDEL (
Figure 4B, NE/ER ratio of 1.9+/−0.1). When expressed in yeast, the MG-NLS-L-S and MGM-NLS-L-S reporters enter the nucleus and accumulate at the INM (NE/ER ratios of 19.8 and 5.7, respectively), while the MGMM-NLS-L-S reporter does not accumulate and has a NE/ER ratio of 2.1, similar to mCherry-HDEL (
Figure 4B, striped bars).
To estimate how synthesis of new reporter proteins and import of already synthesized reporters affect the NE/ER ratios, we inhibited the expression of the reporters from the GAL promoter after one hour of induction, by adding glucose to the growth medium (
Figure 4B, grey bars). The cultures were imaged again after one hour: the total fluorescence levels measured in the NE and the ER increased, which indicates that some reporter protein has still been synthesized and/or matured. Importantly, the accumulation levels (NE/ER) increase for the reporters MG-NLS-L-S and MGM-NLS-L-S, consistent with continued import at a reduced synthesis rates after transcription repression (
Figure 4B, compare striped and grey bars). The NE/ER ratios of the MGMM-NLS-L-S and mCherry-HDEL reporters do not change in this timeframe.
Next, we determined the passive diffusion or leak of the reporters from the nucleus to the ER. For this purpose, the reporter proteins were expressed in the KAP95AA strain, which expresses Pma1-FKBP and Kap95-FRB, and in which the Kap95/Kap60-dependent active import can be conditionally blocked by the addition of rapamycin. The experiments were performed as follows: first protein expression was induced (1 h galactose), and then expression was repressed (1 h glucose), as described above. As we showed in
Figure 4B, grey bars, during this hour in glucose, translation of existing mRNAs, maturation of fluorophores and import continues. Finally, rapamycin was added which results in a fast block of nuclear import [
13,
15,
22]. The distribution of the MG-NLS-L-S and MGM-NLS-L-S reporter proteins over the NE-ER network was determined after one hour of incubation with rapamycin (
Figure 4B, white bars). Only the reporter protein with the smallest extralumenal domain, MG-NLS-L-S, leaks out of the nucleus when import is blocked: the NE/ER ratio drops from 37.9 to 12.0, 1 h after addition of rapamycin (
Figure 4B, compare grey and white bars). The accumulation levels of the MGM-NLS-L-S reporter do not decrease when the import is blocked, indicating that this reporter protein cannot or only very slowly passively diffuses out of the nucleus on these timescales, consistent with the size limits in [
29].
When active import and transcription were inhibited simultaneously,
i.e., by the addition of rapamycin and glucose simultaneously after 1 h of galactose-induced expression, the NE/ER ratios decrease for both MG-NLS-L-S (from 19.8 to 3.7) and MGM-NLS-L-S (from 5.7 to 2.8) (
Figure 4B, compare striped and black bars). This decrease in the NE/ER is the result of the reporter being synthesized, albeit at a reduced rate, while Kap-dependent INM import is blocked, resulting in increased fluorescence in the ER, while the NE fluorescence does not increase. The experimental scheme presented here is better than the one used previously [
13,
15], where we did not take into account the effect of protein synthesis in the ER.
Additional support that the type of transmembrane domain, monotopic or polytopic, is not critical for the targeting comes from viability tests. As shown in
Figure 2, INM accumulation of the G-NLS-L-TM reporter results in deformed nuclei. In
Figure 4 we show that the deformation correlates with the levels of INM accumulation, and also with the viability of the cells: bigger extralumenal domains result in lower accumulation (
Figure 4B) and increased viability (
Figure 4C). This is observed irrespective of the type of transmembrane domain, or the expression and degradation levels (
Figure 4D) of the reporter proteins.
Figure 4.
NE accumulation of membrane protein reporters with extralumenal domains of increasing size. (A) Fluorescence microscopy of cells expressing G-NLS-L-RibU, MG-NLS-L-RibU, MGM2-NLS-L-RibU. M: MBP. (B) Average accumulation of reporter proteins at the NE over the ER after different regimes of expression, and import inhibition. The reporter proteins were expressed for 1 h (striped bars); subsequently expression was inhibited by glucose for 1 h (grey bars) and finally import was blocked by rapamycin for 1 h (white bars). Alternatively, transcription and import were inhibited simultaneously (black bars). Average of 20 cells; SEM are indicated. (C) Viability of cells expressing membrane proteins with different transmembrane domains and differently-sized extralumenal domains. (D) Western blot (anti-GFP) showing expression of the membrane protein transporters with different transmembrane domains and differently-sized extralumenal domains used in this figure.
Figure 4.
NE accumulation of membrane protein reporters with extralumenal domains of increasing size. (A) Fluorescence microscopy of cells expressing G-NLS-L-RibU, MG-NLS-L-RibU, MGM2-NLS-L-RibU. M: MBP. (B) Average accumulation of reporter proteins at the NE over the ER after different regimes of expression, and import inhibition. The reporter proteins were expressed for 1 h (striped bars); subsequently expression was inhibited by glucose for 1 h (grey bars) and finally import was blocked by rapamycin for 1 h (white bars). Alternatively, transcription and import were inhibited simultaneously (black bars). Average of 20 cells; SEM are indicated. (C) Viability of cells expressing membrane proteins with different transmembrane domains and differently-sized extralumenal domains. (D) Western blot (anti-GFP) showing expression of the membrane protein transporters with different transmembrane domains and differently-sized extralumenal domains used in this figure.
In conclusion, our findings in
Figure 4 confirm that the NLS-L motif enables the import of extralumenal domains bigger than by passive diffusion. This is best illustrated by the reporter MGM-NLS-L-S; on the time scale of an hour this molecule is small enough to be accumulated at the INM by active import but it is too large to passively efflux from the NPC on this timescale.
3.5. Transport Route through the NPC
Previously we have shown that we could trap the same reporter as used here (F-G-NLS-L-TM) at Nsp1-FRB and concluded that the linker can span the distance between the pore membrane and the central channel. Accounting for this finding, plus the FG-Nup dependence and the large sizes of extralumenal domains that can be imported, we proposed that the extralumenal domains would travel through the center of the NPC [
13]. However, recent publications show a non-NPC cytoplasmic pool of Nsp1 [
27,
28] that may have affected our measurements. We thus constructed new strains targeting the scaffold nucleoporin Nup170 (
Figure 3), and the FG-Nups Nup53 and Nup59 as trapping sites for our reporters and tested the trapping of membrane proteins with and without the NLS-L sorting signals at these positions. All three nucleoporins are located relatively close to the pore membrane [
30,
31,
32,
33,
34].
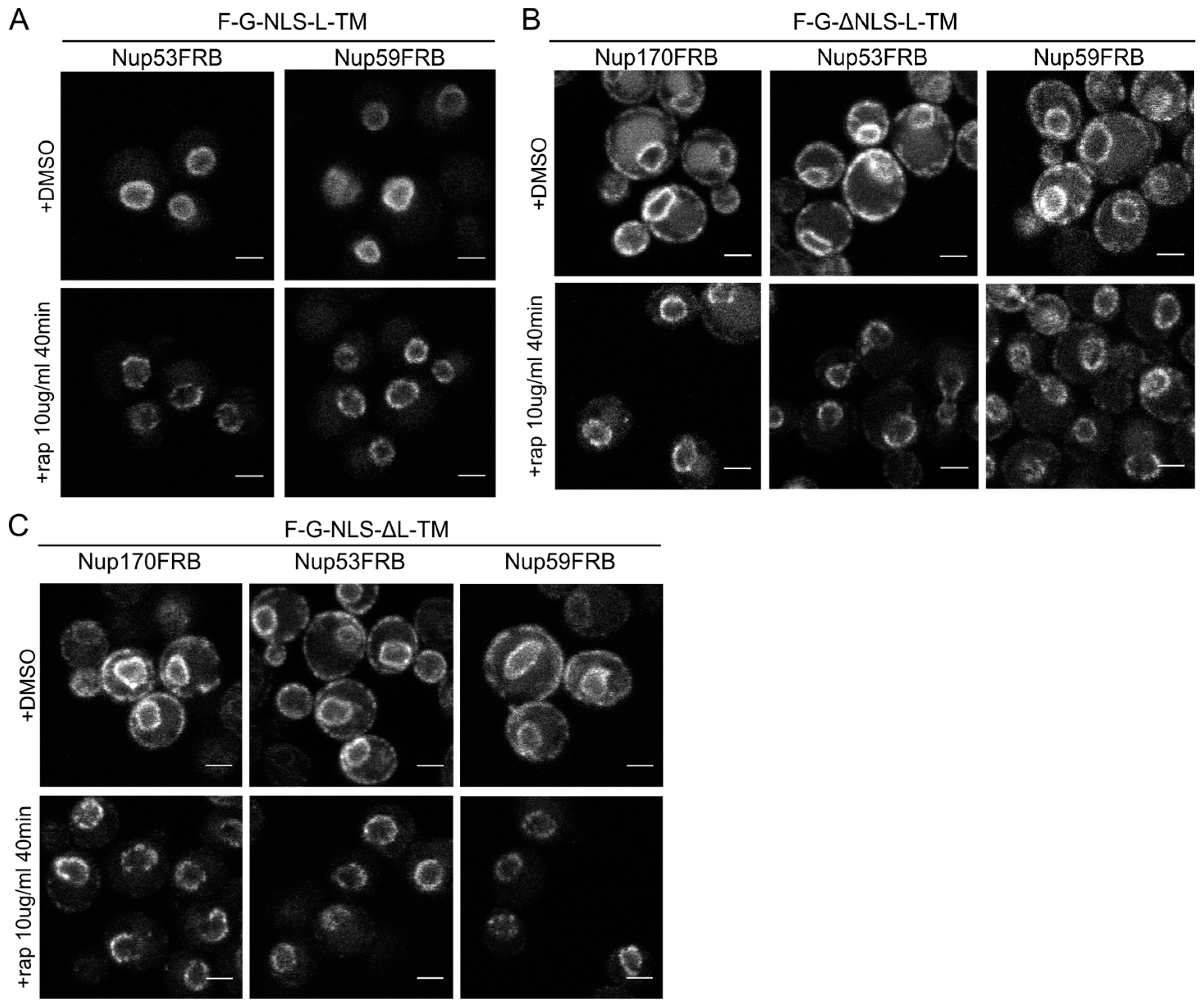
Figure 5.
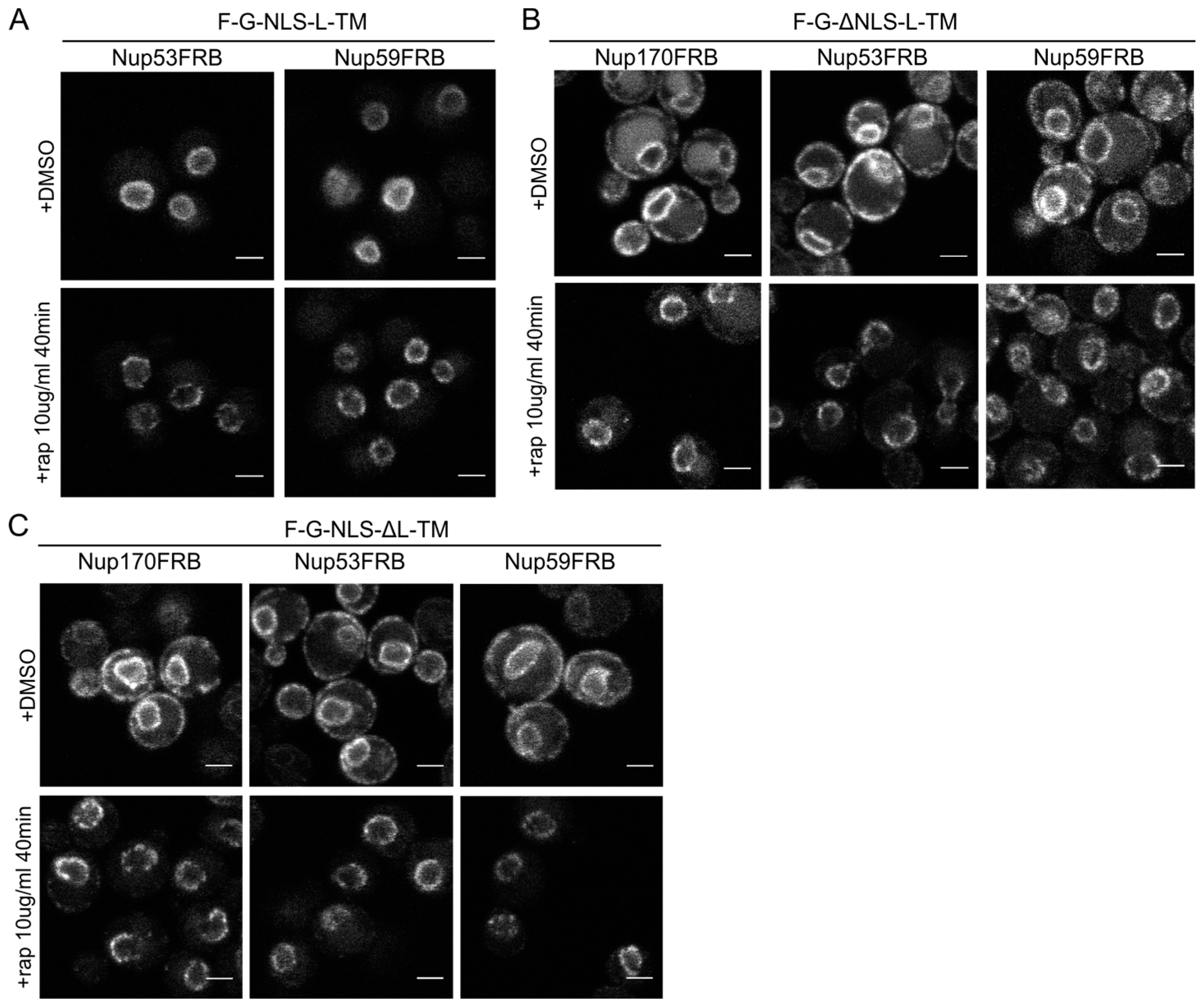
The reporters without the NLS or the linker domain can enter the NPC. (A) Confocal fluorescence microscopy images showing cells expressing F-G-NLS-L-TM in Nup53FRB and Nup59FRB strains background after incubation with rapamycin (+rap 10 μg/mL) and in control conditions (+DMSO). The fluorescence patterns change from continues to punctate upon rapamycin treatment (B,C). Same as (A), but with cells expressing F-G-ΔNLS-L-TM (B) or F-G-NLS-ΔL-TM (C) in Nup170FRB, Nup53FRB and Nup59FRB strain background. The fluorescence signal disappears from the ER upon rapamycin treatment. Scale bars: 2 μm.
Figure 5.
The reporters without the NLS or the linker domain can enter the NPC. (A) Confocal fluorescence microscopy images showing cells expressing F-G-NLS-L-TM in Nup53FRB and Nup59FRB strains background after incubation with rapamycin (+rap 10 μg/mL) and in control conditions (+DMSO). The fluorescence patterns change from continues to punctate upon rapamycin treatment (B,C). Same as (A), but with cells expressing F-G-ΔNLS-L-TM (B) or F-G-NLS-ΔL-TM (C) in Nup170FRB, Nup53FRB and Nup59FRB strain background. The fluorescence signal disappears from the ER upon rapamycin treatment. Scale bars: 2 μm.
Upon incubation with rapamycin, we saw the characteristic punctate localization pattern of the F-G-NLS-L-TM reporter in all tested Nup-FRB strains (
Figure 5A). The two reporters that fail to accumulate in the nuclear envelope (F-G-ΔNLS-L-TM and F-G-NLS-ΔL-TM) can also be trapped at Nup170, Nup53 and Nup59. In these cases, rapamycin does not only cause clustering of the fluorescence signal in distinct points of the nuclear envelope, but also an almost complete shift of the ER-localized molecules to the NE (
Figure 5B,C). This data reinforces the mobility of our reporters between ER and the pore membrane, and also confirms that the tested reporters are not restricted from entering the NPC in the absence of a NLS or a linker region. Unfortunately, as trapping also occurs in the absence of the sorting signals, we cannot interpret the trapping event as one that uniquely reflects active import.
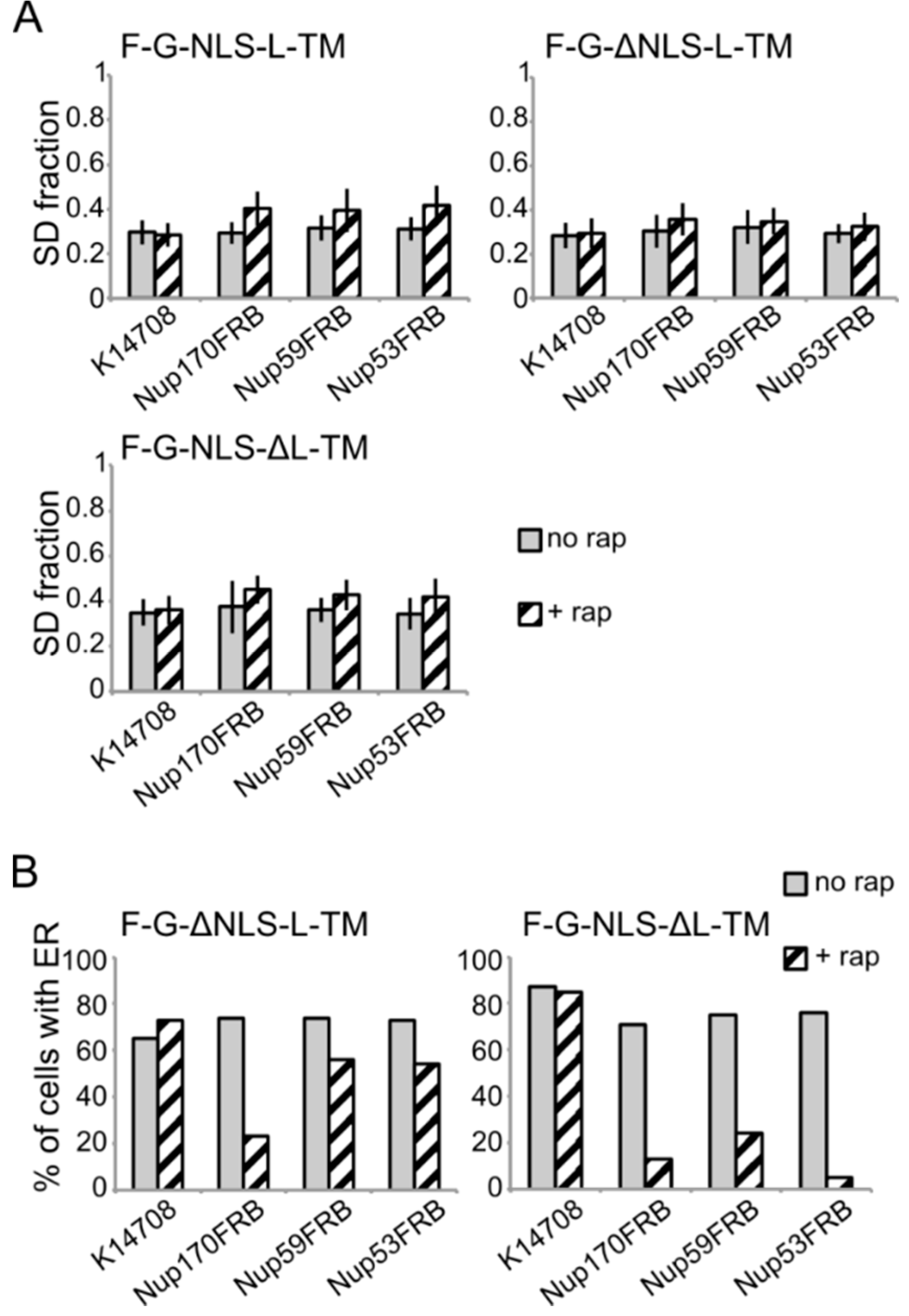
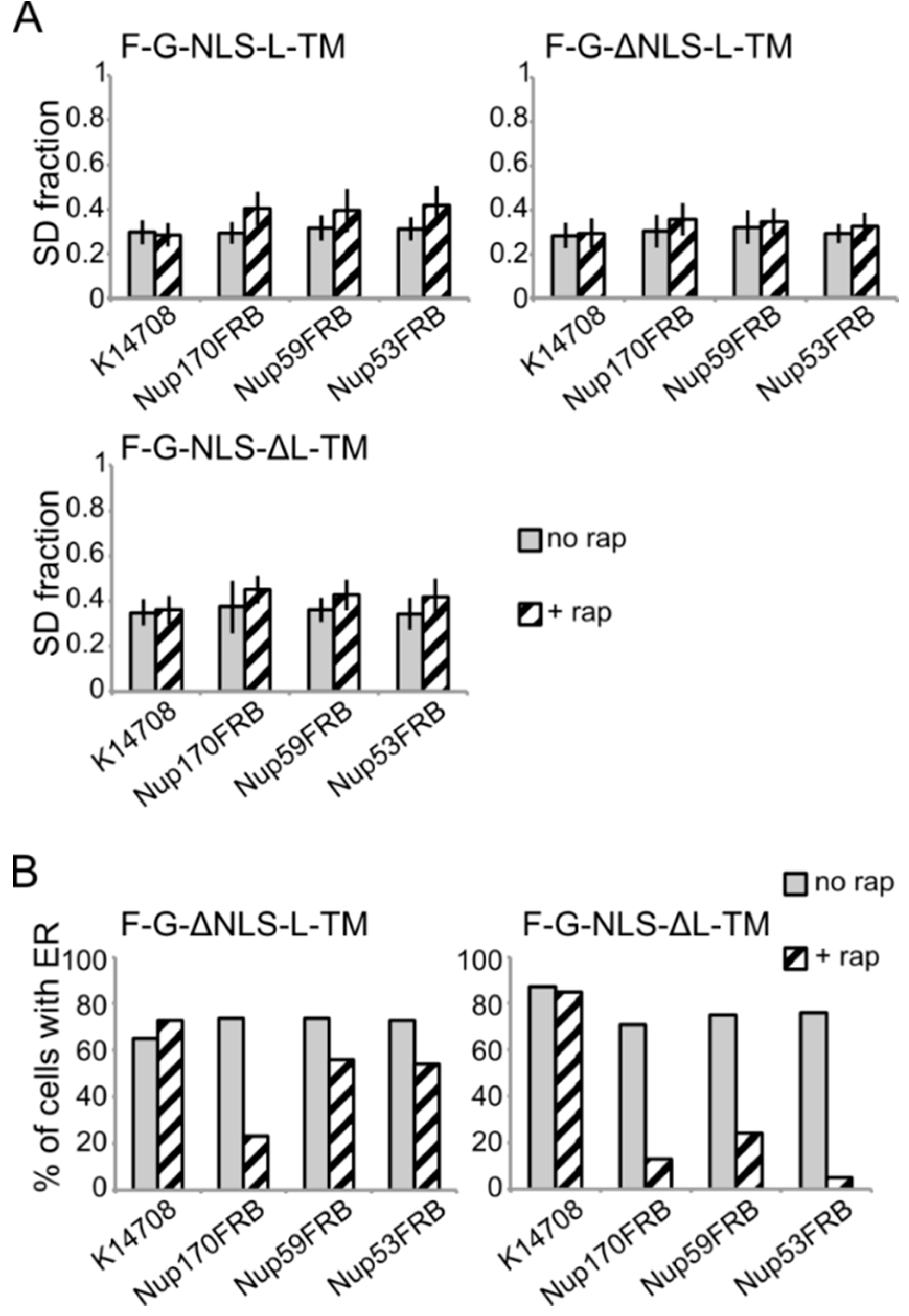
Figure 6.
Analysis of the fluorescence images from
Figure 5. (
A) Comparison of the average SD fraction (as in
Figure 3C) for F-G-NLS-L-TM, F-G-ΔNLS-L-TM, F-G-NLS-ΔL-TM in all the trap strains (Nup53FRB, Nup59FRB, Nup170FRB) and the background strain (K14708). Grey columns, control conditions (no rap); columns with black diagonal stripes, cells incubated with rapamycin (+rap). Number of cells analyzed is between 29 and 55. (
B) Percentage of cells expressing F-G-ΔNLS-L-TM or F-G-NLS-ΔL-TM that display fluorescence in the ER in the trap strains (Nup53FRB, Nup59FRB, Nup170) and the background strain (K14708). The number of cells analyzed is between 31 and 158.
Figure 6.
Analysis of the fluorescence images from
Figure 5. (
A) Comparison of the average SD fraction (as in
Figure 3C) for F-G-NLS-L-TM, F-G-ΔNLS-L-TM, F-G-NLS-ΔL-TM in all the trap strains (Nup53FRB, Nup59FRB, Nup170FRB) and the background strain (K14708). Grey columns, control conditions (no rap); columns with black diagonal stripes, cells incubated with rapamycin (+rap). Number of cells analyzed is between 29 and 55. (
B) Percentage of cells expressing F-G-ΔNLS-L-TM or F-G-NLS-ΔL-TM that display fluorescence in the ER in the trap strains (Nup53FRB, Nup59FRB, Nup170) and the background strain (K14708). The number of cells analyzed is between 31 and 158.
Despite the limitations of the assay, we noted differences in trapping efficiency dependent on the presence of an NLS and dependent on the trap position. The increase in SD fraction seemed smaller in the absence of the NLS (
Figure 6A), possibly reflecting that trapping is less efficient. This NLS-dependent trapping efficiency is more clearly observed when looking at the percentage of cells that show fluorescence at the ER (
Figure 6B). For F-G-NLS-ΔL-TM we see that the percentage of cells that show fluorescence at the ER drops from around 70% to below 20% after trapping at Nup170, Nup53 or Nup59. Trapping of the F-G-ΔNLS-L-TM reporter is less efficient at Nup59 and Nup53 as here still around 55% of the cells show fluorescence at the ER, while this value is around 20% when trapped at Nup170. The difference in trapping efficiency may reflect a different residence time of the reporters in the NPC and suggests increased residence close to Nup53 and Nup59 for reporters with the NLS as compared to those without. While the trap assay is not very suitable for dissecting the native path through the NPC, we can conclude that the extralumenal domains can reach positions in the NPC close to the membrane.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}