
Taking a Bad Turn: Compromised DNA Damage Response in Leukemia


Abstract
:1. Introduction
2. DNA Double-Strand Break Repair
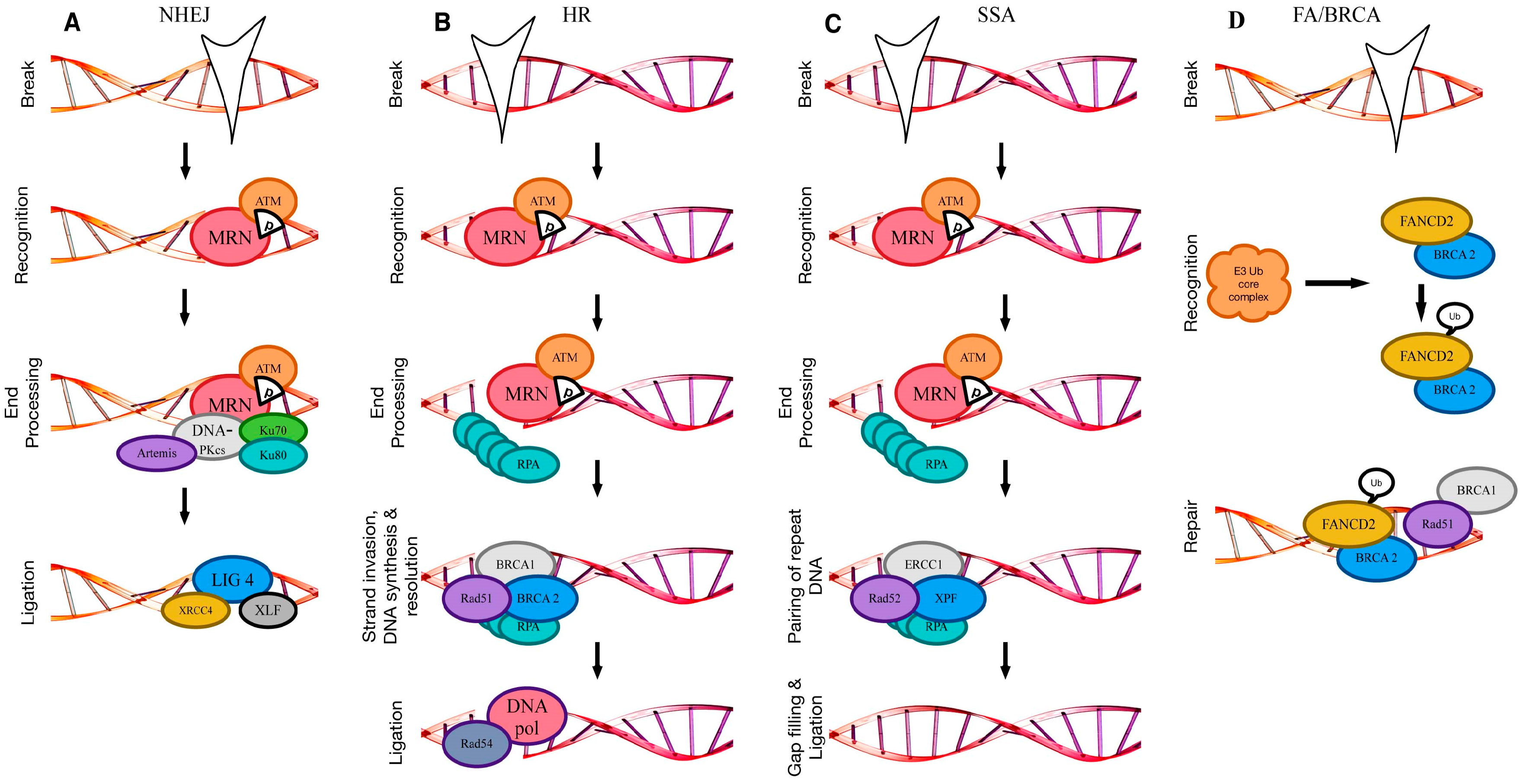
2.1. Non-Homologous End Joining
2.2. Homologous Recombination
2.3. Single-Strand Annealing
2.4. Alternative Non-Homologous End Joining/Microhomology-Mediated End Joining (Alt-NHEJ/MMEJ)
2.5. Crosslink Repair (Fanconi Anaemia/BRCA Pathway)
3. Compromised Double Strand Break Repair in Leukemia
3.1. Altered Non-Homologous End Joining
3.2. Altered Alternative Non-Homologous End Joining
3.3. Altered Homologous Recombination
3.4. Altered Single-Strand Annealing
3.5. Alterations in Other Repair Pathways
3.6. Indirect Effects on DDR
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [Google Scholar] [CrossRef] [PubMed]
- O′Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Soutoglou, E. Nuclear compartmentalization of DNA repair. Curr. Opin. Genet. Dev. 2016, 37, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Tauchi, H.; Nakamura, A.; Kondo, N.; Sakamoto, S.; Endo, S.; Smeets, D.; Solder, B.; Belohradsky, B.H.; Der Kaloustian, V.M.; et al. Positional cloning of the gene for nijmegen breakage syndrome. Nat. Genet. 1998, 19, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to pi-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Kastan, M.B. DNA damage responses: Mechanisms and roles in human disease: 2007 G.H.A. Clowes memorial award lecture. Mol. Cancer Res. 2008, 6, 517–524. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.T.; So, C.W. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Villarreal, D.; Shim, E.Y.; Lee, S.E. Risky business: Microhomology-mediated end joining. Mutat. Res. 2016, 788, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. Rpa antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. Ctip-brca1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome maintenance in response to DNA damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Schuermann, D.; Weber, A.R.; Schar, P. Active DNA demethylation by DNA repair: Facts and uncertainties. DNA Repair 2016, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Di Micco, R.; d′Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The mrn complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Dickey, J.S.; Redon, C.E.; Nakamura, A.J.; Baird, B.J.; Sedelnikova, O.A.; Bonner, W.M. H2ax: Functional roles and potential applications. Chromosoma 2009, 118, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone h2ax phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53bp1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Ishino, T.; Ishii, Y.; Takeda, S.; Koyama, H. DNA ligase iv-deficient cells are more resistant to ionizing radiation in the absence of ku70: Implications for DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2001, 98, 12109–12113. [Google Scholar] [CrossRef] [PubMed]
- Mari, P.O.; Florea, B.I.; Persengiev, S.P.; Verkaik, N.S.; Bruggenwirth, H.T.; Modesti, M.; Giglia-Mari, G.; Bezstarosti, K.; Demmers, J.A.; Luider, T.M.; et al. Dynamic assembly of end-joining complexes requires interaction between ku70/80 and xrcc4. Proc. Natl. Acad. Sci. USA 2006, 103, 18597–18602. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Jackson, S.P. The DNA-dependent protein kinase. Genes Dev. 1999, 13, 916–934. [Google Scholar] [CrossRef] [PubMed]
- Niewolik, D.; Pannicke, U.; Lu, H.; Ma, Y.; Wang, L.C.; Kulesza, P.; Zandi, E.; Lieber, M.R.; Schwarz, K. DNA-pkcs dependence of artemis endonucleolytic activity, differences between hairpins and 5′ or 3′ overhangs. J. Biol. Chem. 2006, 281, 33900–33909. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Morotomi-Yano, K.; Wang, S.Y.; Uematsu, N.; Lee, K.J.; Asaithamby, A.; Weterings, E.; Chen, D.J. Ku recruits xlf to DNA double-strand breaks. EMBO Rep. 2008, 9, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.A.; Rothstein, R. At loose ends: Resecting a double-strand break. Cell 2009, 137, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The cdk regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [PubMed]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require cdk1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors rad50, mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. DNA end resection: Many nucleases make light work. DNA Repair 2009, 8, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Chi, P.; Sehorn, M.G.; Etchin, J.; Krejci, L.; Sung, P. Recombination mediator and rad51 targeting activities of a human brca2 polypeptide. J. Biol. Chem. 2006, 281, 11649–11657. [Google Scholar] [CrossRef] [PubMed]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.D. Rad54, a swi2/snf2-like recombinational repair protein, disassembles rad51:Dsdna filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Hiramoto, T.; Nakanishi, T.; Sumiyoshi, T.; Fukuda, T.; Matsuura, S.; Tauchi, H.; Komatsu, K.; Shibasaki, Y.; Inui, H.; Watatani, M.; et al. Mutations of a novel human rad54 homologue, rad54b, in primary cancer. Oncogene 1999, 18, 3422–3426. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Ormonde, P.A.; Pero, R.; Chen, Y.; Lian, L.; Salada, G.; Berry, S.; Lawrence, Q.; Dayananth, P.; Ha, P.; et al. Characterization of a carboxy-terminal brca1 interacting protein. Oncogene 1998, 17, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.R.; Roskelley, C.D. Regulation of brca1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. 2003, 5, 45–52. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in brca1 and brca2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene brca2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Hannay, J.A.; Liu, J.; Zhu, Q.S.; Bolshakov, S.V.; Li, L.; Pisters, P.W.; Lazar, A.J.; Yu, D.; Pollock, R.E.; Lev, D. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: A role for p53/activator protein 2 transcriptional regulation. Mol. Cancer Ther. 2007, 6, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Schoenmakers, E.F.; Huysmans, C.; Van de Ven, W.J. Allelic knockout of novel splice variants of human recombination repair gene rad51b in t(12;14) uterine leiomyomas. Cancer Res. 1999, 59, 19–23. [Google Scholar] [PubMed]
- Mohaghegh, P.; Hickson, I.D. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum. Mol. Genet. 2001, 10, 741–746. [Google Scholar] [CrossRef] [PubMed]
- German, J.; Bloom, D.; Passarge, E. Bloom’s syndrome. V. Surveillance for cancer in affected families. Clin. Genet. 1977, 12, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human ctip promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human ctip mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Role of rad52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.; Grimme, J.M.; Spies, M.; Ha, T. Human rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 20274–20279. [Google Scholar] [CrossRef] [PubMed]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the xpf/ercc1 endonuclease and hrad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini-Denchi, E.; Sfeir, A. Stop pulling my strings-what telomeres taught us about the DNA damage response. Nat. Rev. Mol. Cell Biol. 2016, 17, 364–378. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. Mmej repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair 2014, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D′Andrea, A.D. The fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D′Andrea, A.D. The fanconi anemia/brca pathway: New faces in the crowd. Genes Dev. 2005, 19, 2925–2940. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, A.; Grompe, M. Repair kinetics of genomic interstrand DNA cross-links: Evidence for DNA double-strand break-dependent activation of the fanconi anemia/brca pathway. Mol. Cell Biol. 2004, 24, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of fancd2 and nbs1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the fanconi anemia proteins and brca1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Taniguchi, T.; Garcia-Higuera, I.; Andreassen, P.R.; Gregory, R.C.; Grompe, M.; D’Andrea, A.D. S-phase-specific interaction of the fanconi anemia protein, fancd2, with brca1 and rad51. Blood 2002, 100, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.; Drapkin, R.; Zhang, F.; Lin, Y.; Han, J.; Pamidi, S.; Livingston, D.M. The brca1-associated protein bach1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Johnson, M.R.; Rabe, K.; Boardman, L.; McWilliams, R.; de Andrade, M.; Petersen, G. Germ line fanconi anemia complementation group c mutations and pancreatic cancer. Cancer Res. 2005, 65, 383–386. [Google Scholar] [PubMed]
- van der Heijden, M.S.; Brody, J.R.; Gallmeier, E.; Cunningham, S.C.; Dezentje, D.A.; Shen, D.; Hruban, R.H.; Kern, S.E. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am. J. Pathol. 2004, 165, 651–657. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the world health organization (who) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- El Rouby, S.; Thomas, A.; Costin, D.; Rosenberg, C.R.; Potmesil, M.; Silber, R.; Newcomb, E.W. P53 gene mutation in b-cell chronic lymphocytic leukemia is associated with drug resistance and is independent of mdr1/mdr3 gene expression. Blood 1993, 82, 3452–3459. [Google Scholar] [PubMed]
- Blaise, R.; Alapetite, C.; Masdehors, P.; Merle-Beral, H.; Roulin, C.; Delic, J.; Sabatier, L. High levels of chromosome aberrations correlate with impaired in vitro radiation-induced apoptosis and DNA repair in human b-chronic lymphocytic leukaemia cells. Int. J. Radiat. Biol. 2002, 78, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar] [PubMed]
- Deriano, L.; Guipaud, O.; Merle-Beral, H.; Binet, J.L.; Ricoul, M.; Potocki-Veronese, G.; Favaudon, V.; Maciorowski, Z.; Muller, C.; Salles, B.; et al. Human chronic lymphocytic leukemia b cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood 2005, 105, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Christodoulopoulos, G.; Salles, B.; Panasci, L. DNA-dependent protein kinase activity correlates with clinical and in vitro sensitivity of chronic lymphocytic leukemia lymphocytes to nitrogen mustards. Blood 1998, 92, 2213–2219. [Google Scholar] [PubMed]
- Bouley, J.; Saad, L.; Grall, R.; Schellenbauer, A.; Biard, D.; Paget, V.; Morel-Altmeyer, S.; Guipaud, O.; Chambon, C.; Salles, B.; et al. A new phosphorylated form of ku70 identified in resistant leukemic cells confers fast but unfaithful DNA repair in cancer cell lines. Oncotarget 2015, 6, 27980–28000. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. Bcr-abl down-regulates the DNA repair protein DNA-pkcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of wrn and DNA ligase iiialpha in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Tobin, L.A.; Robert, C.; Rapoport, A.P.; Gojo, I.; Baer, M.R.; Tomkinson, A.E.; Rassool, F.V. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene 2013, 32, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. Bcr/abl oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Johnston, C.; Reeves, W.H.; Carter, T.; Wyche, J.H.; Hendrickson, E.A. Characterization of a ku86 variant protein that results in altered DNA binding and diminished DNA-dependent protein kinase activity. J. Biol. Chem. 1996, 271, 14098–14104. [Google Scholar] [PubMed]
- Paillard, S.; Strauss, F. Site-specific proteolytic cleavage of ku protein bound to DNA. Proteins 1993, 15, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Critchlow, S.E.; Teo, S.H.; Doherty, A.J.; Priestley, A.; Broughton, B.; Kysela, B.; Beamish, H.; Plowman, N.; Arlett, C.F.; et al. Identification of a defect in DNA ligase iv in a radiosensitive leukaemia patient. Curr. Biol. 1999, 9, 699–702. [Google Scholar] [CrossRef]
- Cheng, W.H.; von Kobbe, C.; Opresko, P.L.; Fields, K.M.; Ren, J.; Kufe, D.; Bohr, V.A. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol. Cell Biol. 2003, 23, 6385–6395. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing flt3/itd mutations exhibit elevated repair errors generated through alternative nhej pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hefferin, M.L.; Chen, L.; Shim, E.Y.; Tseng, H.M.; Kwon, Y.; Sung, P.; Lee, S.E.; Tomkinson, A.E. Role of dnl4-lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat. Struct. Mol. Biol. 2007, 14, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Pixley, F.J. Csf-1 signaling in macrophages: Pleiotrophy through phosphotyrosine-based signaling pathways. Crit. Rev. Clin. Lab. Sci. 2012, 49, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of flt3 (flt3/itd) induces increased ros production, DNA damage, and misrepair: Implications for poor prognosis in aml. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Brown, P.; Smith, B.D.; Stine, A.; Pham, R.; Stone, R.; Deangelo, D.; Galinsky, I.; Giles, F.; Estey, E.; et al. Plasma inhibitory activity (pia): A pharmacodynamic assay reveals insights into the basis for cytotoxic response to flt3 inhibitors. Blood 2006, 108, 3477–3483. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Levis, M.; Beran, M.; Giles, F.; Kantarjian, H.; Berg, K.; Murphy, K.M.; Dauses, T.; Allebach, J.; Small, D. Single-agent cep-701, a novel flt3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 2004, 103, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; Chen, Y.B. The role of flt3 inhibitors in the treatment of flt3-mutated acute myeloid leukemia. Eur. J. Haematol. 2017, 98, 330–336. [Google Scholar] [CrossRef] [PubMed]
- .Dimitrov, L.; Hong, C.S.; Yang, C.; Zhuang, Z.; Heiss, J.D. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int. J. Med. Sci. 2015, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein sir2 is an nad-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [PubMed]
- Lin, Y.H.; Yuan, J.; Pei, H.; Liu, T.; Ann, D.K.; Lou, Z. Kap1 deacetylation by sirt1 promotes non-homologous end-joining repair. PLoS ONE 2015, 10, e0123935. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. Sirt1 promotes DNA repair activity and deacetylation of ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, H.; Yang, M.; Ye, S.; Li, L.; Zhang, H.; Hu, J.; Wang, X.; Xu, J.; Linag, A. Sirt1 inhibition impairs non-homologous end joining DNA damage repair by increasing ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 2015, 7, 13538–13550. [Google Scholar]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. Brca1, parp1 and gammah2ax in acute myeloid leukemia: Role as biomarkers of response to the parp inhibitor olaparib. Biochim. Biophys. Acta 2015, 1852, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Standal, R.; Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997, 325 Pt 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(adp-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by parp inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Sitwala, K.V.; Dandekar, M.N.; Hess, J.L. Hox proteins and leukemia. Int. J. Clin. Exp. Pathol. 2008, 1, 461–474. [Google Scholar] [PubMed]
- Wang, Z.; Iwasaki, M.; Ficara, F.; Lin, C.; Matheny, C.; Wong, S.H.; Smith, K.S.; Cleary, M.L. Gsk-3 promotes conditional association of creb and its coactivators with meis1 to facilitate hox-mediated transcription and oncogenesis. Cancer Cell 2010, 17, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small gtp-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [PubMed]
- Scholl, C.; Frohling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic kras dependency and stk33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Hahnel, P.S.; Enders, B.; Sasca, D.; Roos, W.P.; Kaina, B.; Bullinger, L.; Theobald, M.; Kindler, T. Targeting components of the alternative nhej pathway sensitizes kras mutant leukemic cells to chemotherapy. Blood 2014, 123, 2355–2366. [Google Scholar] [CrossRef] [PubMed]
- Christodoulopoulos, G.; Malapetsa, A.; Schipper, H.; Golub, E.; Radding, C.; Panasci, L.C. Chlorambucil induction of hsrad51 in b-cell chronic lymphocytic leukemia. Clin. Cancer Res. 1999, 5, 2178–2184. [Google Scholar] [PubMed]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. Bcr/abl regulates mammalian reca homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Huang, Y.; Nakada, S.; Ishiko, T.; Utsugisawa, T.; Datta, R.; Kharbanda, S.; Yoshida, K.; Talanian, R.V.; Weichselbaum, R.; Kufe, D.; et al. Role for caspase-mediated cleavage of rad51 in induction of apoptosis by DNA damage. Mol. Cell Biol. 1999, 19, 2986–2997. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Stark, J.M.; Ommundsen, M.; Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004, 23, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene brca1 is required for subnuclear assembly of rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Seedhouse, C.; Faulkner, R.; Ashraf, N.; Das-Gupta, E.; Russell, N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin. Cancer Res. 2004, 10, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Scardocci, A.; Guidi, F.; D’Alo, F.; Gumiero, D.; Fabiani, E.; Diruscio, A.; Martini, M.; Larocca, L.M.; Zollino, M.; Hohaus, S.; et al. Reduced brca1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br. J. Cancer 2006, 95, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Jarrousse, S.; Buet, D.; Dugray, A.; Bonnet, M.L.; Vozenin-Brotons, M.C.; Guilhot, F.; Turhan, A.G.; Feunteun, J.; Bourhis, J. Down-regulation of brca1 in bcr-abl-expressing hematopoietic cells. Blood 2003, 101, 4583–4588. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, M.A.; De Jesus Pizarro, R.E.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2014, 28, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.M.T.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2009, 68, 6884–6888. [Google Scholar] [CrossRef] [PubMed]
- Mattarucchi, E.; Guerini, V.; Rambaldi, A.; Campiotti, L.; Venco, A.; Pasquali, F.; Lo Curto, F.; Porta, G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosomes Canc. 2008, 47, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi’s anemia and malignancies. Am. J. Hematol. 1996, 53, 99–110. [Google Scholar] [CrossRef]
- Tischkowitz, M.D.; Morgan, N.V.; Grimwade, D.; Eddy, C.; Ball, S.; Vorechovsky, I.; Langabeer, S.; Stoger, R.; Hodgson, S.V.; Mathew, C.G. Deletion and reduced expression of the fanconi anemia fanca gene in sporadic acute myeloid leukemia. Leukemia 2004, 18, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Condie, A.; Powles, R.L.; Hudson, C.D.; Shepherd, V.; Bevan, S.; Yuille, M.R.; Houlston, R.S. Analysis of the fanconi anaemia complementation group a gene in acute myeloid leukaemia. Leukemia 2002, 1849–1853. [Google Scholar]
- Rischewski, J.R.; Clausen, H.; Leber, V.; Niemeyer, C.; Ritter, J.; Schindler, D.; Schneppenheim, R. A heterozygous frameshift mutation in the fanconi anemia c gene in familial t-all and secondary malignancy. Klin. Padiatr. 2000, 212, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Iqbal, J.; Lemonnier, F.; Kucuk, C.; de Leval, L.; Jais, J.P.; Parrens, M.; Martin, A.; Xerri, L.; Brousset, P.; et al. Idh2 mutations are frequent in angioimmunoblastic t-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Lemonnier, F.; Mak, T.W. Roles of idh1/2 and tet2 mutations in myeloid disorders. Int. J. Hematol. 2016, 103, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. Tet1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite d-2-hydroxyglutarate inhibits alkbh DNA repair enzymes and sensitizes idh mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic idh1 and idh2 mutations result in a hypermethylation phenotype, disrupt tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, L.Y.; Chen, X.H.; et al. Fda approval: Belinostat for the treatment of patients with relapsed or refractory peripheral t-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Lonial, S.; Hungria, V.; Moreau, P.; Einsele, H.; Lee, J.H.; Yoon, S.; Corradini, P.; Jedrzejczak, W.W.; Tan, D.C.; et al. Panorama1: A randomized, double-blind, placebo controlled phase iii study of panobinostat in combination with bortezomib and dexamethasone in patients with relapsed multiple myeloma. J. Clin. Oncol. 2011, 29, TPS227. [Google Scholar] [CrossRef]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. An integrated database of genes responsive to the myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Preudhomme, C.; Lai, J.L.; Quiquandon, I.; Jonveaux, P.; Vanrumbeke, M.; Sartiaux, C.; Morel, P.; Loucheux-Lefebvre, M.H.; Bauters, F.; et al. Mutations of the p53 gene in b-cell chronic lymphocytic leukemia: A report on 39 cases with cytogenetic analysis. Leukemia 1992, 6, 246–250. [Google Scholar] [PubMed]
- Cheng, J.; Haas, M. Frequent mutations in the p53 tumor suppressor gene in human leukemia t-cell lines. Mol. Cell Biol. 1990, 10, 5502–5509. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, J.; Andersen, M.K.; Andersen, M.T.; Christiansen, D.H. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008, 22, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Austen, B.; Skowronska, A.; Baker, C.; Powell, J.E.; Gardiner, A.; Oscier, D.; Majid, A.; Dyer, M.; Siebert, R.; Taylor, A.M.; et al. Mutation status of the residual atm allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J. Clin. Oncol. 2007, 25, 5448–5457. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Repair Pathway | Affected Component | Leukemia Subtype |
|---|---|---|
| Non-homologous end joining | DNA-PK, Ku70/80 | B-CLL, CML, AML, CLL, PML |
| DNA ligase IV, Artemis | CML | |
| Mre11A | t-AML | |
| SIRT1 | CML, AML | |
| Homologous recombination | BRCA1/2 | CML, AML |
| Rad51 | B-CLL, CML, de novo and t-AML | |
| Mre11A | t-AML | |
| Alternative non-homologous end joining | K-RAS | T-ALL, AML |
| Fanconi anemia | FANCA | AML |
| FANCC | T-ALL | |
| Base excision repair | PARP1/2 | CML, AML |
| DNA ligase III | CML | |
| Non-specific | IDH1/2 | CMML, AML |
| ATM | CLL | |
| MYC | t-AML | |
| TP53 | B-CLL, ALL, t-AML |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilles, N.; Fahrenkrog, B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells 2017, 6, 11. https://doi.org/10.3390/cells6020011
Nilles N, Fahrenkrog B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells. 2017; 6(2):11. https://doi.org/10.3390/cells6020011
Chicago/Turabian StyleNilles, Nadine, and Birthe Fahrenkrog. 2017. "Taking a Bad Turn: Compromised DNA Damage Response in Leukemia" Cells 6, no. 2: 11. https://doi.org/10.3390/cells6020011
APA StyleNilles, N., & Fahrenkrog, B. (2017). Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells, 6(2), 11. https://doi.org/10.3390/cells6020011