Coagulation, Microenvironment and Liver Fibrosis


Abstract
:1. Introduction
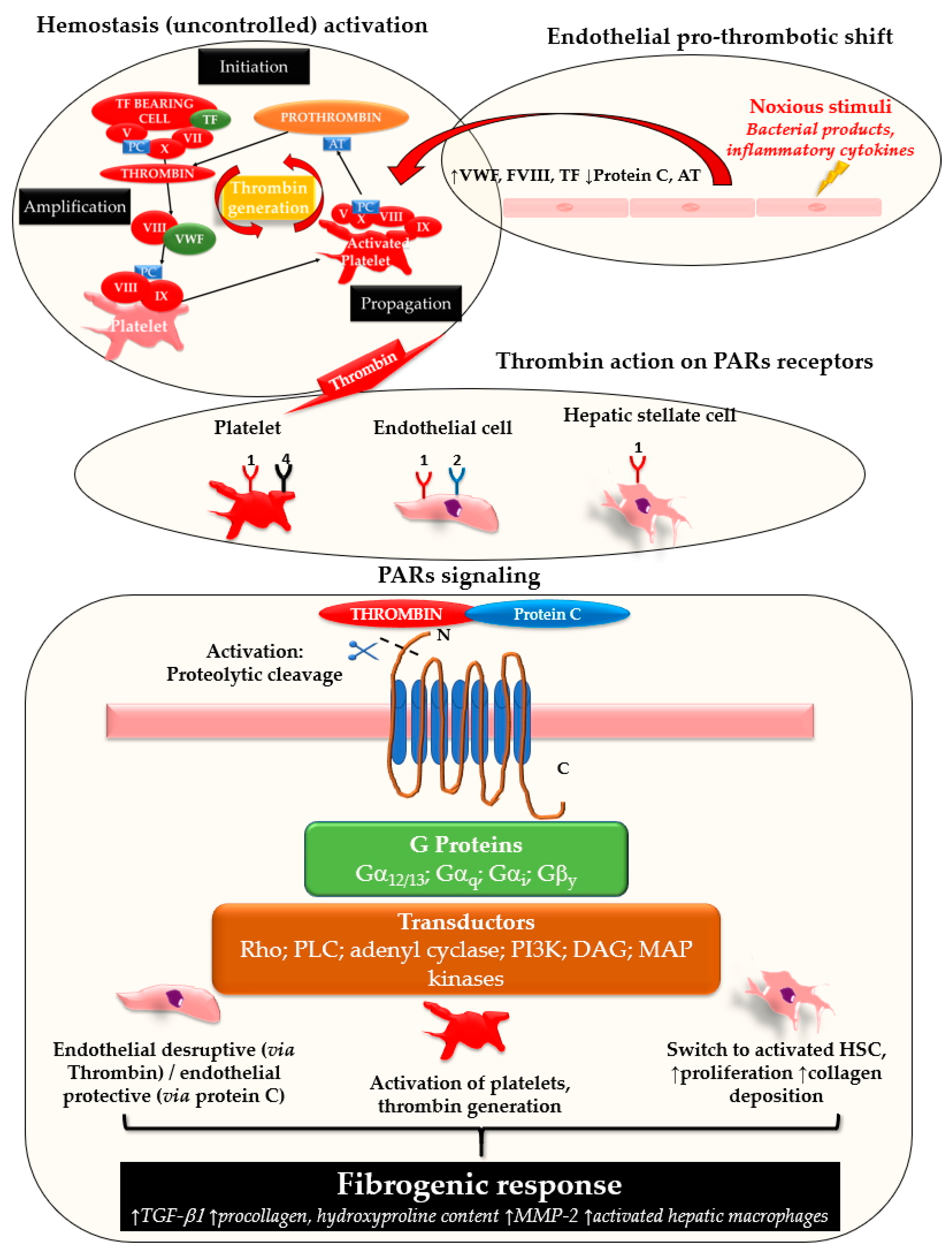
2. Coagulation in Fibrosis and Disease Progression
2.1. Hepatic Stellate Cells, Endothelium and Fibrosis: Role of PARs
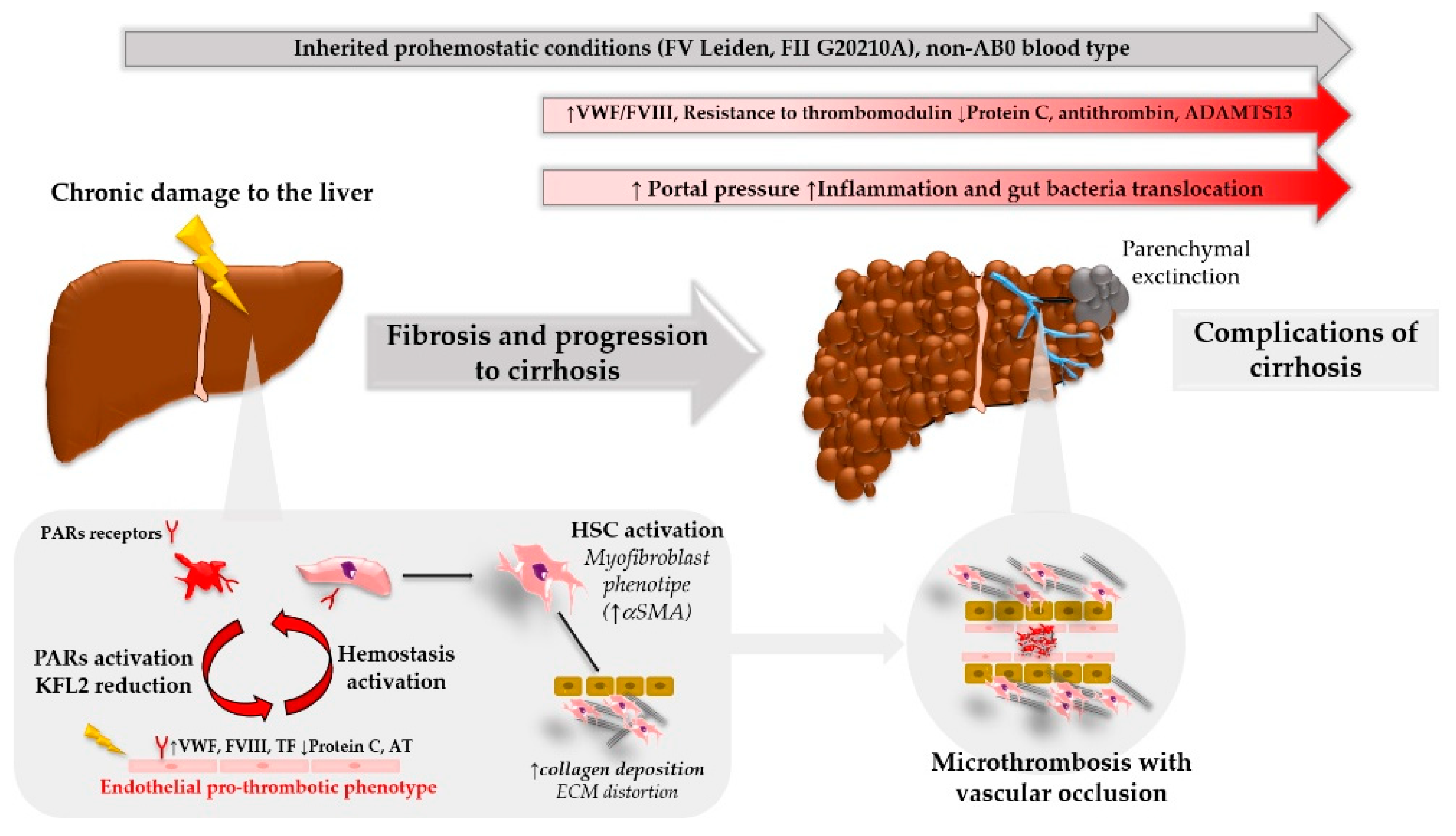
2.2. Parenchymal Extinction: From Clot Generation to Liver Damage
2.3. Procoagulant Imbalance and Disease Progression: Clinical Observations
2.3.1. Common Inherited Pro-Hemostatic Genotype and Risk of Fibrosis Development
2.3.2. Hemostatic Balance in Advanced Liver Disease
3. Anticoagulation as Anti-Fibrotic Strategy
3.1. Heparin
3.2. Oral Anticoagulants: From Vitamin K Antagonists to Direct Oral Anticoagulants (DOACs)
4. Future Directions: Hemostasis as Immune Response
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoffman, M.; Monroe, D.M. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [PubMed]
- Weksler, B.B.; Ley, C.W.; Jaffe, E.A. Stimulation of Endothelial Cell Prostacyclin Production by Thrombin, Trypsin, and the Ionophore A 23187. J. Clin. Investig. 1978, 62, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, S.M.; Zimmerman, G.A.; McIntyre, T.M. Human endothelial cells in culture produce platelet-activating factor (1-alkyl-2-acetyl-sn-glycero-3-phosphocholine) when stimulated with thrombin. Proc. Natl. Acad. Sci. USA 1984, 81, 3534–3538. [Google Scholar] [CrossRef] [PubMed]
- Sugama, Y.; Tiruppathi, C.; offakidevi, K.; Andersen, T.T.; Fenton, J.W.; Malik, A.B. Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: A mechanism for stabilizing neutrophil adhesion. J. Cell Biol. 1992, 119, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Laurens, N.; Koolwijk, P.; de Maat, M.P.M. Fibrin structure and wound healing. J. Thromb. Haemost. 2006, 4, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.-K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Martorell, L.; Martínez-González, J.; Rodríguez, C.; Gentile, M.; Calvayrac, O.; Badimon, L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb. Haemost. 2008, 99, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Hamilton, J.R.; McKemy, D.D.; Camerer, E.; Zheng, Y.-W.; Cheng, A.; Griffin, C.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood 2003, 102, 3224–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, R.; Noorbakhsh, F.; Defea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-S.; Kim, Y.; Park, M.-K.; Rezaie, A.R. Concentration dependent dual effect of thrombin in endothelial cells via PAR-1 and PI3 kinase. J. Cell. Physiol. 2009, 219, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Wiisanen, M.E.; Moliterno, D.J. Platelet protease-activated receptor antagonism in cardiovascular medicine. Coron. Artery Dis. 2012, 23, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Isermann, B. Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors. J. Thromb. Haemost. 2017, 15, 1273–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: How Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J. Hepatol. 2013, 58, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2014. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A.; Balasubramanian, V.; García-Cardeña, G.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 2005, 96, e48–e57. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Tatsumi, K.; Matsumoto, M.; Fujimoto, M.; Matsuyama, T.; Ishikawa, M.; Iwamoto, T.-A.; Mori, T.; Wanaka, A.; Fukui, H.; et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood 2005, 106, 922–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, M.; Fujimura, Y.; Matsumoto, M.; Ishizashi, H.; Kato, S.; Matsuyama, T.; Isonishi, A.; Ishikawa, M.; Yagita, M.; Morioka, C.; et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb. Haemost. 2008, 99, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Scully, M.; Singer, M. The role of ADAMTS-13 in the coagulopathy of sepsis. J. Thromb. Haemost. 2018. [Google Scholar] [CrossRef] [PubMed]
- García-Pagán, J.-C.; Gracia-Sancho, J.; Bosch, J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J. Hepatol. 2012, 57, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Dabbagh, K.; McAnulty, R.J.; Gray, A.J.; Blanc-Brude, O.P.; Laurent, G.J. Thrombin stimulates fibroblast procollagen production via proteolytic activation of protease-activated receptor 1. Biochem. J. 1998, 333, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaça, M.D.A.; Zhou, X.; Benyon, R.C. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J. Hepatol. 2002, 36, 362–369. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Severino, B.; Fiorentina, R.; Baldoni, M.; Caliendo, G.; Santagada, V.; Morelli, A.; Cirino, G. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology 2004, 39, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Dhar, A.; Thursz, M.R. The role of hypercoagulability in liver fibrogenesis. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Duplantier, J.G.; Dubuisson, L.; Senant, N.; Freyburger, G.; Laurendeau, I.; Herbert, J.-M.; Desmoulière, A.; Rosenbaum, J. A role for thrombin in liver fibrosis. Gut 2004, 53, 1682–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahara, T.; Furui, K.; Funaki, J.; Nakayama, Y.; Itoh, H.; Miyabayashi, C.; Sato, H.; Seiki, M.; Ooshima, A.; Watanabe, A. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995, 21, 787–795. [Google Scholar] [PubMed]
- Knight, V.; Lourensz, D.; Tchongue, J.; Correia, J.; Tipping, P.; Sievert, W. Cytoplasmic domain of tissue factor promotes liver fibrosis in mice. World J. Gastroenterol. 2017, 23, 5692–5699. [Google Scholar] [CrossRef] [PubMed]
- Knight, V.; Tchongue, J.; Lourensz, D.; Tipping, P.; Sievert, W. Protease-activated receptor 2 promotes experimental liver fibrosis in mice and activates human hepatic stellate cells. Hepatology 2012, 55, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Rullier, A.; Gillibert-Duplantier, J.; Costet, P.; Cubel, G.; Haurie, V.; Petibois, C.; Taras, D.; Dugot-Senant, N.; Deleris, G.; Bioulac-Sage, P.; et al. Protease-activated receptor 1 knockout reduces experimentally induced liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G226–G235. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.P.; Weinreb, P.H.; Violette, S.M.; Luyendyk, J.P. The Coagulation System Contributes to αVβ6 Integrin Expression and Liver Fibrosis Induced by Cholestasis. Am. J. Pathol. 2010, 177, 2837–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, R.; Fader, K.A.; Kopec, A.K.; Harkema, J.R.; Zacharewski, T.R.; Luyendyk, J.P. From the Cover: Coagulation-Driven Hepatic Fibrosis Requires Protease Activated Receptor-1 (PAR-1) in a Mouse Model of TCDD-Elicited Steatohepatitis. Toxicol. Sci. 2016, 154, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.; Knapp, S.; Anstee, Q.; Worku, M.; Tommasi, A.; Zucoloto, S.; Goldin, R.; Thursz, M. Effect of a thrombin receptor (protease-activated receptor 1, PAR-1) gene polymorphism in chronic hepatitis C liver fibrosis. J. Gastroenterol. Hepatol. 2008, 23, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Role of Tissue Factor in Hemostasis, Thrombosis, and Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautou, P.-E.; Tatsumi, K.; Antoniak, S.; Owens, A.P.; Sparkenbaugh, E.; Holle, L.A.; Wolberg, A.S.; Kopec, A.K.; Pawlinski, R.; Luyendyk, J.P.; et al. Hepatocyte Tissue Factor Contributes to the Hypercoagulable State in a Mouse Model of Chronic Liver Injury. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gillibert-Duplantier, J.; Neaud, V.; Blanc, J.-F.; Bioulac-Sage, P.; Rosenbaum, J. Thrombin inhibits migration of human hepatic myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G128–G136. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and portal vein thrombosis in cirrhosis: Possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995, 21, 1238–1247. [Google Scholar] [PubMed]
- Hou, P.C.; Mcfadzean, A.J. Thrombosis and Intimal Thickening in the Portal System in Cirrhosis of the Liver. J. Pathol. Bacteriol. 1965, 89, 473–480. [Google Scholar] [PubMed]
- Wanless, I.R.; Liu, J.J.; Butany, J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis). Hepatology 1995, 21, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Simonetto, D.A.; Yang, H.; Yin, M.; de Assuncao, T.M.; Kwon, J.H.; Hilscher, M.; Pan, S.; Yang, L.; Bi, Y.; Beyder, A.; et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology 2015, 61, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ridder, G.G.; Lundblad, R.L.; Pizzo, S.V. Actions of thrombin in the interstitium. J. Thromb. Haemost. 2016, 14, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.-H.; Han, J.H.; Yeom, Y.I.; Kim, D.-S. Thrombin-dependent MMP-2 activity is regulated by heparan sulfate. J. Biol. Chem. 2010, 285, 41270–41279. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Gaxiola, M.; Arreola, J.L.; Ramírez, R.; Jara, P.; D’Armiento, J.; Richards, T.; Selman, M.; Pardo, A. Overexpression of MMP9 in macrophages attenuates pulmonary fibrosis induced by bleomycin. Int. J. Biochem. Cell Biol. 2007, 39, 2324–2338. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Cammà, C.; Marietta, M.; Luongo, M.; Critelli, R.; Colopi, S.; Tata, C.; Zecchini, R.; Gitto, S.; Petta, S.; et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012, 143, 1253–1260.e4. [Google Scholar] [CrossRef] [PubMed]
- La Mura, V.; Braham, S.; Tosetti, G.; Branchi, F.; Bitto, N.; Moia, M.; Fracanzani, A.L.; Colombo, M.; Tripodi, A.; Primignani, M. Harmful and Beneficial Effects of Anticoagulants in Patients With Cirrhosis and Portal Vein Thrombosis. Clin. Gastroenterol. Hepatol. 2018, 16, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.K.; Hankey, G.J.; Quinlan, D.J.; Eikelboom, J.W. Risk of recurrent venous thromboembolism in patients with common thrombophilia: A systematic review. Arch. Intern. Med. 2006, 166, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, A.; Mosena, L.; Prins, M.H.; Prandoni, P. The risk of recurrent venous thromboembolism among heterozygous carriers of factor V Leiden or prothrombin G20210A mutation. A systematic review of prospective studies. Haematologica 2007, 92, 1107–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, J.M. Thrombophilia Testing and Venous Thrombosis. N. Engl. J. Med. 2017, 377, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Binetti, B.M.; Tripodi, A.; Chantarangkul, V.; Semeraro, N. Hyperprothrombinemia associated with prothrombin G20210A mutation inhibits plasma fibrinolysis through a TAFI-mediated mechanism. Blood 2004, 103, 2157–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cott, E.M.; Khor, B.; Zehnder, J.L. Factor V Leiden. Am. J. Hematol. 2016, 91, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.; Goldin, R.; Hellier, S.; Knapp, S.; Frodsham, A.; Hennig, B.; Hill, A.; Apple, R.; Cheng, S.; Thomas, H.; et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut 2003, 52, 1206–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharshak, N.; Halfon, P.; Deutsch, V.; Peretz, H.; Berliner, S.; Fishman, S.; Zelber-Sagi, S.; Rozovski, U.; Leshno, M.; Oren, R. Increased fibrosis progression rates in hepatitis C patients carrying the prothrombin G20210A mutation. World J. Gastroenterol. 2011, 17, 5007–5013. [Google Scholar] [CrossRef] [PubMed]
- Plompen, E.P.C.; Murad, S.D.; Hansen, B.E.; Loth, D.W.; Schouten, J.N.L.; Taimr, P.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H.; Janssen, H.L.A.; et al. Prothrombotic genetic risk factors are associated with an increased risk of liver fibrosis in the general population: The Rotterdam Study. J. Hepatol. 2015, 63, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Bekirov, I.; Mejritsky, Y.; Solomon, L.; Szvalb, S.; Hussein, O. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J. Gastroenterol. 2005, 11, 5834–5839. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Pasta, F.; Pasta, L. Thrombophilic genetic factors PAI-1 4G-4G and MTHFR 677TT as risk factors of alcohol, cryptogenic liver cirrhosis and portal vein thrombosis, in a Caucasian population. Gene 2015, 568, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Poujol-Robert, A.; Rosmorduc, O.; Serfaty, L.; Coulet, F.; Poupon, R.; Robert, A. Genetic and acquired thrombotic factors in chronic hepatitis C. Am. J. Gastroenterol. 2004, 99, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Goulding, C.; O’Brien, C.; Egan, H.; Hegarty, J.E.; McDonald, G.; O’Farrelly, C.; White, B.; Kelleher, D.; Norris, S. The impact of inherited prothrombotic risk factors on individuals chronically infected with hepatitis C virus from a single source. J. Viral Hepat. 2007, 14, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Poujol-Robert, A.; Boëlle, P.-Y.; Wendum, D.; Poupon, R.; Robert, A. Association between ABO blood group and fibrosis severity in chronic hepatitis C infection. Dig. Dis. Sci. 2006, 51, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
- Shavakhi, A.; Hajalikhani, M.; Minakari, M.; Norian, A.; Riahi, R.; Azarnia, M.; Liaghat, L. The association of non-O blood group and severity of liver fibrosis in patients with chronic hepatitis C infection. J. Res. Med. Sci. 2012, 17, 466–469. [Google Scholar] [PubMed]
- Koster, T.; Vandenbroucke, J.P.; Rosendaal, F.R.; Briët, E.; Rosendaal, F.R.; Blann, A.D. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995, 345, 152–155. [Google Scholar] [CrossRef]
- O’Donnell, J.; Laffan, M.A. The relationship between ABO histo-blood group, factor VIII and von Willebrand factor. Transfus. Med. 2001, 11, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Mannucci, P.M. The coagulopathy of chronic liver disease. N. Engl. J. Med. 2011, 365, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ratnoff, O.D.; Patek, A.J. The Natural History of Laennec’s Cirrhosis of the Liver an Analysis of 386 Cases. Medicine 1942, 21, 207–268. [Google Scholar] [CrossRef]
- La Mura, V.; Nicolini, A.; Tosetti, G.; Primignani, M. Cirrhosis and portal hypertension: The importance of risk stratification, the role of hepatic venous pressure gradient measurement. World J. Hepatol. 2015, 7, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Salerno, F.; Chantarangkul, V.; Clerici, M.; Cazzaniga, M.; Primignani, M.; Mannuccio Mannucci, P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005, 41, 553–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, T.; Porte, R.J. Rebalanced hemostasis in patients with liver disease: Evidence and clinical consequences. Blood 2010, 116, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Mannucci, P.M.; Caldwell, S.H. Changing Concepts of Cirrhotic Coagulopathy. Am. J. Gastroenterol. 2017, 112, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Chantarangkul, V.; Dell’Era, A.; Clerici, M.; de Franchis, R.; Colombo, M.; Mannucci, P.M. An imbalance of pro- vs. anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology 2009, 137, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Lemma, L.; Chantarangkul, V.; Mannucci, P.M. Evidence that low protein C contributes to the procoagulant imbalance in cirrhosis. J. Hepatol. 2013, 59, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Tarantino, L.; Minno, G.D.; Paternoster, M.; Graziano, V.; Petitto, M.; Nasto, A.; Minno, M.N.D.D. The risk of venous thromboembolism in patients with cirrhosis. Thromb. Haemost. 2017, 117, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Nonami, T.; Yokoyama, I.; Iwatsuki, S.; Starzl, T.E. The Incidence of Portal Vein Thrombosis at Liver Transplantation. Hepatology 1992, 16, 1195–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsochatzis, E.A.; Senzolo, M.; Germani, G.; Gatt, A.; Burroughs, A.K. Systematic review: Portal vein thrombosis in cirrhosis. Aliment. Pharmacol. Ther. 2010, 31, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Northup, P.G.; McMahon, M.M.; Ruhl, A.P.; Altschuler, S.E.; Volk-Bednarz, A.; Caldwell, S.H.; Berg, C.L. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am. J. Gastroenterol. 2006, 101, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Søgaard, K.K.; Horváth-Puhó, E.; Grønbaek, H.; Jepsen, P.; Vilstrup, H.; Sørensen, H.T. Risk of venous thromboembolism in patients with liver disease: A nationwide population-based case-control study. Am. J. Gastroenterol. 2009, 104, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Nguyen, G.C. Liver cirrhosis is associated with venous thromboembolism among hospitalized patients in a nationwide US study. Clin. Gastroenterol. Hepatol. 2010, 8, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.; Quintarelli, C.; Lattuada, A.; Leo, R.; Alessandroni, M.; Mannucci, P.M.; Violi, F. High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: Relationship to endotoxemia. Hepatology 1996, 23, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albornoz, L.; Alvarez, D.; Otaso, J.C.; Gadano, A.; Salviú, J.; Gerona, S.; Sorroche, P.; Villamil, A.; Mastai, R. Von Willebrand factor could be an index of endothelial dysfunction in patients with cirrhosis: Relationship to degree of liver failure and nitric oxide levels. J. Hepatol. 1999, 30, 451–455. [Google Scholar] [CrossRef]
- La Mura, V.; Reverter, J.C.; Flores-Arroyo, A.; Raffa, S.; Reverter, E.; Seijo, S.; Abraldes, J.G.; Bosch, J.; García-Pagán, J.C. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut 2011, 60, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Ferlitsch, M.; Reiberger, T.; Hoke, M.; Salzl, P.; Schwengerer, B.; Ulbrich, G.; Payer, B.A.; Trauner, M.; Peck-Radosavljevic, M.; Ferlitsch, A. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012, 56, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- La Mura, V.; Tripodi, A.; Tosetti, G.; Cavallaro, F.; Chantarangkul, V.; Colombo, M.; Primignani, M. Resistance to thrombomodulin is associated with de novo portal vein thrombosis and low survival in patients with cirrhosis. Liver Int. 2016, 36, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Kalambokis, G.N.; Oikonomou, A.; Baltayiannis, G.; Christou, L.; Kolaitis, N.I.; Tsianos, E.V. Thrombin generation measured as thrombin-antithrombin complexes predicts clinical outcomes in patients with cirrhosis. Hepatol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kalambokis, G.N.; Oikonomou, A.; Christou, L.; Kolaitis, N.I.; Tsianos, E.V.; Christodoulou, D.; Baltayiannis, G. von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. J. Hepatol. 2016, 65, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Maieron, A.; Salzl, P.; Peck-Radosavljevic, M.; Trauner, M.; Hametner, S.; Schöfl, R.; Ferenci, P.; Ferlitsch, M. Von Willebrand Factor as a new marker for non-invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2014, 39, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Hametner, S.; Ferlitsch, A.; Ferlitsch, M.; Etschmaier, A.; Schöfl, R.; Ziachehabi, A.; Maieron, A. The VITRO Score (Von Willebrand Factor Antigen/Thrombocyte Ratio) as a New Marker for Clinically Significant Portal Hypertension in Comparison to Other Non-Invasive Parameters of Fibrosis Including ELF Test. PLoS ONE 2016, 11, e0149230. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; Peyvandi, F.; Bertelli, C.; Valenti, L.; Fargion, S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Non-Alcoholic Fatty Liver Disease–Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology 2015. [Google Scholar] [CrossRef]
- Potze, W.; Siddiqui, M.S.; Boyett, S.L.; Adelmeijer, J.; Daita, K.; Sanyal, A.J.; Lisman, T. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Fracanzani, A.L.; Chantarangkul, V.; Primignani, M.; Fargion, S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2017, 66, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Potze, W.; Sanyal, A.J.; Lisman, T. Reply to: “Procoagulant imbalance in patients with non-alcoholic fatty liver disease.”. J. Hepatol. 2017, 66, 250–251. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Di Marco, V.; Iavarone, M.; Roffi, L.; Crosignani, A.; Calvaruso, V.; Aghemo, A.; Cabibbo, G.; Viganò, M.; Boccaccio, V.; et al. Survival of patients with HCV cirrhosis and sustained virologic response is similar to the general population. J. Hepatol. 2016, 64, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Pinzani, M. Anti-fibrotic therapy: Lost in translation? J. Hepatol. 2012, 56 (Suppl. 1), S66–S74. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Levine, M.N. Low molecular weight heparin. Blood 1992, 79, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-J.; Yang, Z.-H.; Shi, X.-L.; Liu, D.-L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J. Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- Abe, W.; Ikejima, K.; Lang, T.; Okumura, K.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J. Hepatol. 2007, 46, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; García-Irigoyen, O.; García-Calderó, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2016, 64, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Goldin, R.D.; Wright, M.; Martinelli, A.; Cox, R.; Thursz, M.R. Coagulation status modulates murine hepatic fibrogenesis: Implications for the development of novel therapies. J. Thromb. Haemost. 2008, 6, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Kassel, K.M.; Sullivan, B.P.; Cui, W.; Copple, B.L.; Luyendyk, J.P. Therapeutic administration of the direct thrombin inhibitor argatroban reduces hepatic inflammation in mice with established fatty liver disease. Am. J. Pathol. 2012, 181, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; García-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; García-Pagán, J.C. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.G.; Sadowski, J.A.; Matschiner, J.T. Mechanism of action of warfarin. Warfarin and metabolism of vitamin K1. Biochemistry 1972, 11, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. Calibration of reference thromboplastins and standardisation of the prothrombin time ratio. Thromb. Haemost. 1983, 49, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.; Hirsh, J.; Hylek, E.; Jacobson, A.; Crowther, M.; Palareti, G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008, 133, 160S–198S. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Kurtz, B. Direct oral anticoagulants: Unique properties and practical approaches to management. Heart 2016, 102, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Lucas, E.; Alexander, G.C.; Goldberger, Z.D. National Trends in Ambulatory Oral Anticoagulant Use. Am. J. Med. 2015, 128, 1300–1305.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intagliata, N.M.; Maitland, H.; Caldwell, S.H. Direct Oral Anticoagulants in Cirrhosis. Curr. Treat. Opt. Gastroenterol. 2016, 14, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, N.M.; Henry, Z.H.; Maitland, H.; Shah, N.L.; Argo, C.K.; Northup, P.G.; Caldwell, S.H. Direct Oral Anticoagulants in Cirrhosis Patients Pose Similar Risks of Bleeding When Compared to Traditional Anticoagulation. Dig. Dis. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hum, J.; Shatzel, J.J.; Jou, J.H.; Deloughery, T.G. The efficacy and safety of direct oral anticoagulants vs. traditional anticoagulants in cirrhosis. Eur. J. Haematol. 2017, 98, 393–397. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Trebicka, J.; Klinger, C.; Plessier, A.; Seijo, S.; Terziroli, B.; Magenta, L.; Semela, D.; Buscarini, E.; Langlet, P.; et al. Antithrombotic treatment with direct-acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017, 37, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Antunes, M.M.; David, B.A.; Pereira, R.V.; Teixeira, M.M.; Menezes, G.B. Imaging liver biology in vivo using conventional confocal microscopy. Nat. Protoc. 2015, 10, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Jenne, C.N.; Zhuo, L.; Kimata, K.; Kubes, P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G797–G806. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C. Liver: Neutrophil extracellular traps mediate bacterial liver damage. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 251. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Arthur, J.F.; Gardiner, E.E. Neutrophil extracellular traps (NETs) and the role of platelets in infection. Thromb. Haemost. 2014, 112, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.D.; Puy, C.; Itakura, A.; Chu, T.; Robinson, D.K.; Bylund, A.; Phillips, K.G.; Gardiner, E.E.; McCarty, O.J.T. Colocalization of neutrophils, extracellular DNA and coagulation factors during NETosis: Development and utility of an immunofluorescence-based microscopy platform. J. Immunol. Methods 2016. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C.; Kubes, P. Platelets and infection. Semin. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Tetaz, T.J.; Andrews, R.K.; Berndt, M.C. Binding of the von Willebrand factor A1 domain to histone. Thromb. Res. 1997, 86, 469–477. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Fuchs, T.A.; Jackson, T.O.; Kremer Hovinga, J.A.; Lämmle, B.; Henke, P.K.; Myers, D.D.; Wagner, D.D.; Wakefield, T.W.; Michigan Research Venous Group. Plasma DNA is Elevated in Patients with Deep Vein Thrombosis. J. Vasc. Surg. Venous Lymphat Disord. 2013, 1. [Google Scholar] [CrossRef]
- Liaw, P.C.; Ito, T.; Iba, T.; Thachil, J.; Zeerleder, S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sun, W.; Cui, W.; Li, X.; Yao, J.; Jia, X.; Li, C.; Wu, H.; Hu, Z.; Zou, X. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 14075–14086. [Google Scholar] [PubMed]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade Body exocytosis. J. Thromb. Haemost. 2016. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017. [Google Scholar] [CrossRef] [PubMed]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taurá, P.; Fuster, J.; García-Valdecasas, J.C.; Lacy, A.; Suárez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; Francés, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Abraldes, J.G.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, M.; Moreau, R.; Angeli, P.; Schnabl, B.; Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 2015, 63, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Ferro, D.; Basili, S.; Saliola, M.; Quintarelli, C.; Alessandri, C.; Cordova, C. Association between low-grade disseminated intravascular coagulation and endotoxemia in patients with liver cirrhosis. Gastroenterology 1995, 109, 531–539. [Google Scholar] [CrossRef]
- Ferro, D.; Basili, S.; Lattuada, A.; Mantovani, B.; Bellomo, A.; Mannucci, P.M.; Violi, F. Systemic clotting activation by low-grade endotoxaemia in liver cirrhosis: A potential role for endothelial procoagulant activation. Ital. J. Gastroenterol. Hepatol. 1997, 29, 434–440. [Google Scholar] [PubMed]
- Raparelli, V.; Basili, S.; Carnevale, R.; Napoleone, L.; Del Ben, M.; Nocella, C.; Bartimoccia, S.; Lucidi, C.; Talerico, G.; Riggio, O.; et al. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 2016. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Raparelli, V.; Nocella, C.; Bartimoccia, S.; Novo, M.; Severino, A.; De Falco, E.; Cammisotto, V.; Pasquale, C.; Crescioli, C.; et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis. J. Hepatol. 2017, 67, 950–956. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Experimental Model | Pathway Explored | Methods | Results |
|---|---|---|---|---|
| Chambers 1998 [24] | human fetal lung fibroblasts | PAR-1 | Exposure to incremental dose of thrombin; TRAPs (thrombin receptor-activating peptide) +/− inhibitors (hirudin/Phe-Pro-ArgCH2CL) | Thrombin ↑ αI-procollagen mRNA through PAR-1 activation |
| Gaça 2002 [25] | Cultured stellate HSEC | Thrombin, tryptase/PAR 1–2 | PAR 1/2 mRNA RT-PCR analysis + northern blotting in lysate of HSEC. Use of PD98059 (kinase inhibitor) | ↑ PAR-1/2 while fibroblast transforms in myofibroblast phenotype ↑ HSC proliferation by PARs |
| Fiorucci et al. 2004 [26] | rat HSC cell line; BDL cirrhotic rat | Thrombin-PARs | type I collagen mRNA expression; quantitative morphometric analysis; hepatic and urinary excretion of hydroxyproline | Thrombin triggers HSC activation and collagen deposition via PARs, prevented by PAR1 antagonist |
| J Gillibert Duplantier et al. 2007 [38] | Human hepatic myofibroblasts | PAR-1; COX-2; Akt-1; platelet derived growth factor (PDGF) | Cell migration; RNA isolation and analysis for Prostaglandin E2 receptor; analysis of Akt-1 phosphorylation and PDGF-receptor phosphorylation. | Thrombin inhibits human hepatic myofibroblast migration via PAR-1; Thrombin inhibits PDGF induced migration (inhibition of PI3K) |
| Martinelli 2007 [35] | Patients with HCV (287 european, 90 brazilian) | PAR1 | Cross-sectional study; fibrosis evaluated by liver biopsy; polymorphism of PAR-1 gene analysis (−1426 C/T, IVS-14, −506 I/D | ↑ fibrosis in TT genotype of 1426 C/T polymorphism |
| Rullier 2008 [32] | PAR-1 −/− and +/− mice exposed to CCL4 | PAR1 | Histology; RT-PCR for type I collagen, MMP-2, PDGFβ-r, MP-1, mRNA | ↓ fibrosis and activated fibrogenic cells ↓ type I collagen, MMP-2, PDGFβ-r mRNA ↓ T lymphoctyes infiltration |
| B. P. Sullivan et al. 2010 [33] | Bile duct epithelial cells (BDECs); PAR1−/−, TF +/−, mice with low levels of human TF expression. All mice were fed with BDEC toxicant (ANIT); Human Liver Samples from patients with PBC/PSC | TF, PAR-1, αVβ6 | Real-Time PCR of snap-frozen liver or adherent cells; immunofluorescence on liver frozen sections for αVβ6 | TF and PAR-1 deficiency ↓ Liver Fibrosis/αVβ6 mRNA ↑ TGF-β1 related αVβ6 expression by PAR-1 αVβ6 inhibition ↓ fibrosis ↑ TF and PAR-1 mRNAs in livers from PBC/PSC patients |
| V. Knight et al. 2012 [31] | HSC cells; HSEC cells; (PAR-2 knockout mice; C57BL/6 mice; CCl4 cirrhotic mice | PARs | Hepatic hydroxyproline content in frozen liver tissue; PCR analysis of MMP-2, TIMP-1 and PAR-1/2; identification of α-SMA, F4/80 and CD68; TGF-β1 Production In Vitro; HSC Proliferation in Response to PAR Activation; Hepatic TGF-β1 Content | PAR-2 Deficiency ↓ Fibrosis/ procollagen mRNA/Hydroxyproline Content/ Stellate Cell Activation/ Hepatic TGF-β1 Expression/MMPs/ Activated Hepatic Macrophages; PAR 1/2 ↑ HSC Collagen Production/TGF-β1 |
| R. Nault et al. 2016 [34] | PAR-1 −/− and +/− mice exposed to to TCDD (progression to NASH) | PAR-1; | Identification of Fibrin(ogen) | TCDD Exposure Activates the Coagulation Cascade; ↓ inflammation and collagen deposition in PAR-1 −/− |
| V. Knight et al. 2017 [30] | PAR-1 −/− mice; HSC cells; CCl4 treated mice | TF and PARs | Hepatic fibrosis assessment; Hepatic collagen content; Gene expression of TGF-β1, MMP-2, TIMP 1, PAR1 and 2; expression TGF-β1 | ↓ fibrosis/MMP2/activated macrophages in TF and PAR-1 −/− |
| Reference | Drug | Animal Model | Fibrosis/Cirrhosis Induction | Fibrosis Assesment | Results |
|---|---|---|---|---|---|
| Duplantier 2004 [28] | Wistars rat | Thrombin antagonist SSR182289 | CCL4 (three or seven week exposure) | Histology; immunohistochemistry (IHC) for αSMA collagen type I, MMP-2, TIMP-1, and TIMP-2 mRNAs by RT-PCR | ↓ 30% fibrosis (7 week CCL4 exposure) Early ↓αSMA positive cells/TIMP-1 mRNA |
| Abe 2007 [99] | Dalteparin | Female Wistars Rats | CCL4 | Histology; IHC | ↓ fibrosis, ↑HGF ↓TGF-β1, COL1A1, αSMA ↓ PDGF induced HSC proliferation |
| Anstee 2008 [101] | Warfarin | FV Leiden mutant mice, C57BL/6 control animals anticoagulated mice | CCL4 | Histology; Liver Hidroxiproline content; αSMA mRNA expression | ↑ fibrosis 80% in male FV mutant Warfarin effect: ↓ Hidroxiproline content ↓ fibrosis scores Effect blunted in FV mutant |
| Kassel 2012 [102] | Argatroban (via micro-osmotic pump) | LDLr−/− mice | Western diet | Histology); real time PCR hepatic mRNA expression of αSMA, COL1A1, PDGFβ, TIMP1/2, TGF-β1; IHC (anti CD68, F4/80, αSMA); MCP-1 Elisa | No change in collagen deposition ↓ αSMA, COL1A1, PDGFβ, TIMP1/2 No ↓TGF-β1 ↓inflammation (↓neutrophil/macrophage accumulation) |
| Cerini 2016 [100] | Enoxaparin | Male Wistars Rats | CCL4 (acute vs short vs long term exposure); TAA | Histology; IHC (anti FBN/αSMA/CD68); expression of procollagen I/ αSMA on isolated HSC | ↓25–26% in short and long term CCL4 exposure; ↓ 41% in TAA ↓PP and HVR ↓αSMA, procollagen I in HSC No change on inflammation |
| Vilaseca 2017 [103] | Rivaroxaban | Cirrhotic wistar rats | CCL4; TAA | Histology; TEM analysis; Liver Hidroxiproline content; IHC (anti fibrinogen/αSMA/CD68) and IF (anti FBN, anti VWF); real time PCR hepatic mRNA expression of αSMA, COL1A1, PDGFβ, TIMP1/2, TGF-β1; in vitro thrombin action on HSC | No ↓in CCL4, ↓25% TAA improved sinusoidal architecture ↓Hidroxiproline content/collagen/fibrin deposition ↓PP and HVR ↓HSC activity of profibrotic genes ↓VWF expression in vasculature No direct activity on HSC (in vitro studies) |
| Li 2017 [98] | Aspirin (low/high dose), enoxaparin | Sprague-Dawley rats | TAA | Histology (METAVIR score) | ↓ in all treatment group (> for high dose aspirin) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitto, N.; Liguori, E.; Mura, V.L. Coagulation, Microenvironment and Liver Fibrosis. Cells 2018, 7, 85. https://doi.org/10.3390/cells7080085
Bitto N, Liguori E, Mura VL. Coagulation, Microenvironment and Liver Fibrosis. Cells. 2018; 7(8):85. https://doi.org/10.3390/cells7080085
Chicago/Turabian StyleBitto, Niccolò, Eleonora Liguori, and Vincenzo La Mura. 2018. "Coagulation, Microenvironment and Liver Fibrosis" Cells 7, no. 8: 85. https://doi.org/10.3390/cells7080085
APA StyleBitto, N., Liguori, E., & Mura, V. L. (2018). Coagulation, Microenvironment and Liver Fibrosis. Cells, 7(8), 85. https://doi.org/10.3390/cells7080085