Autophagy in Chronic Kidney Diseases


Abstract
:1. Introduction
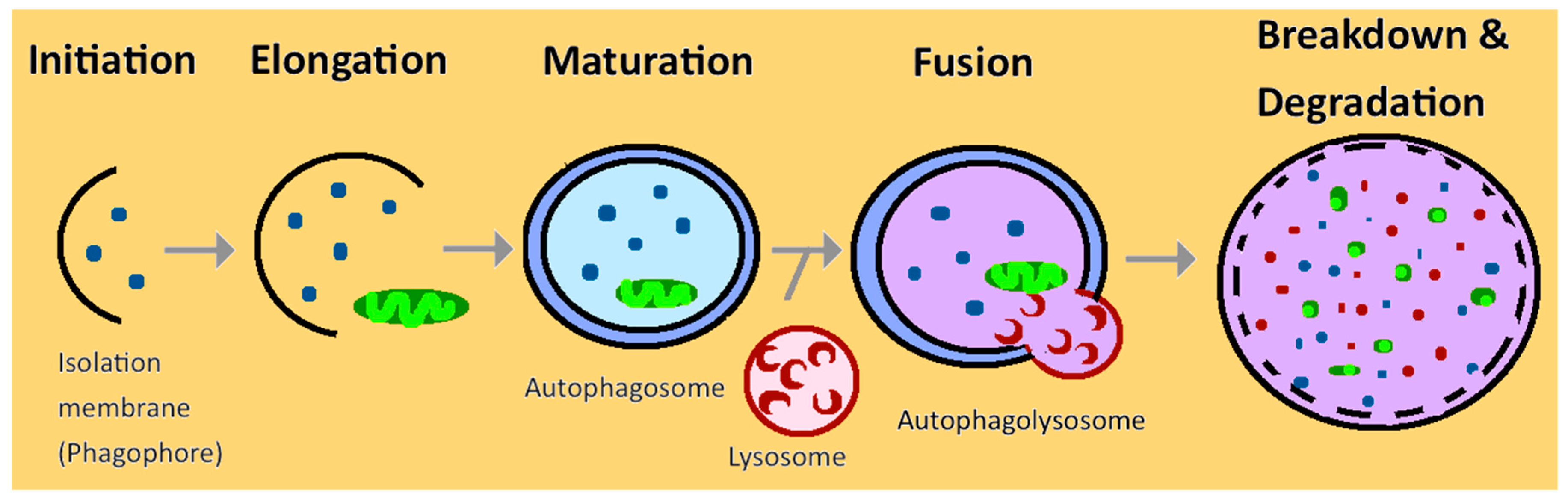
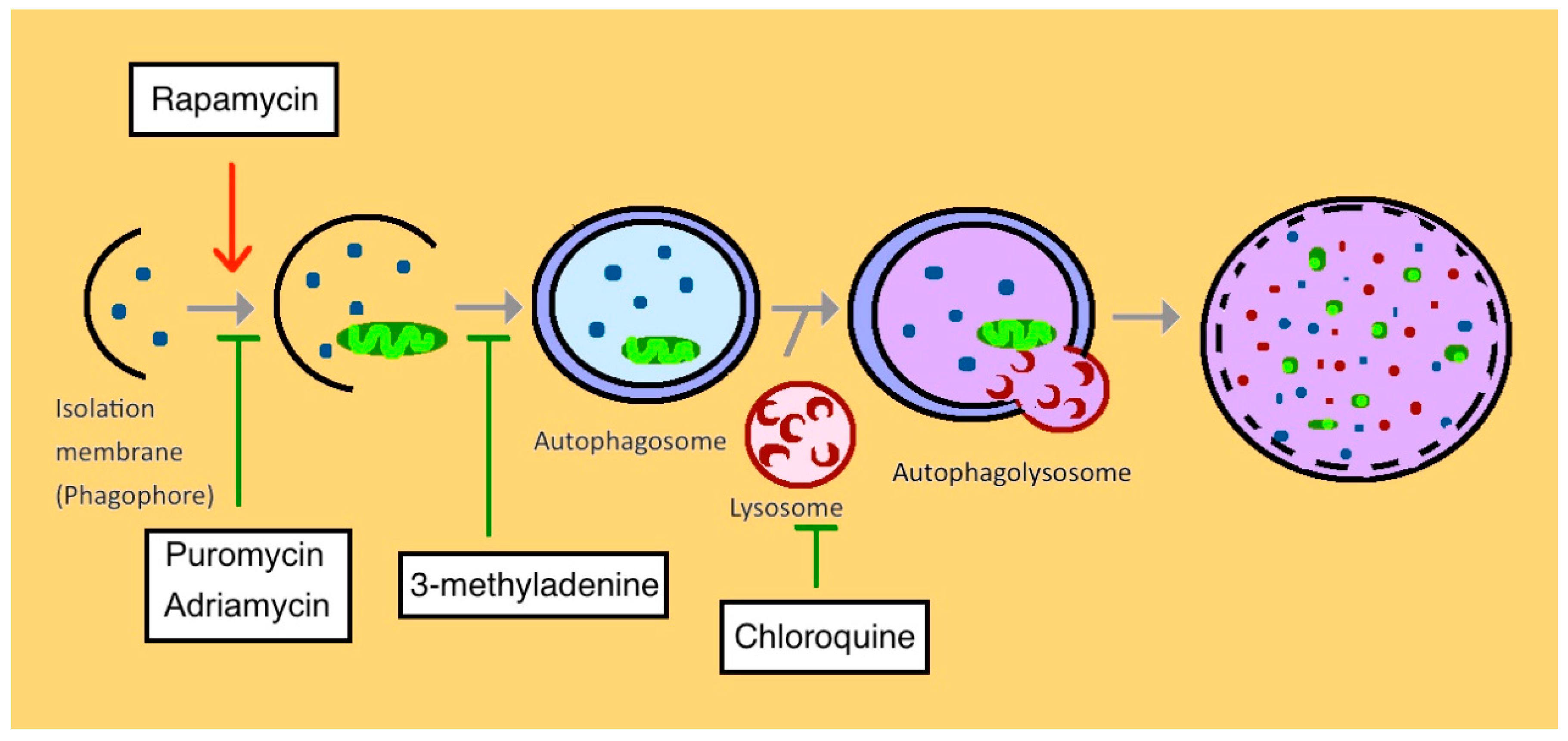
2. Autophagy in Normal Kidney
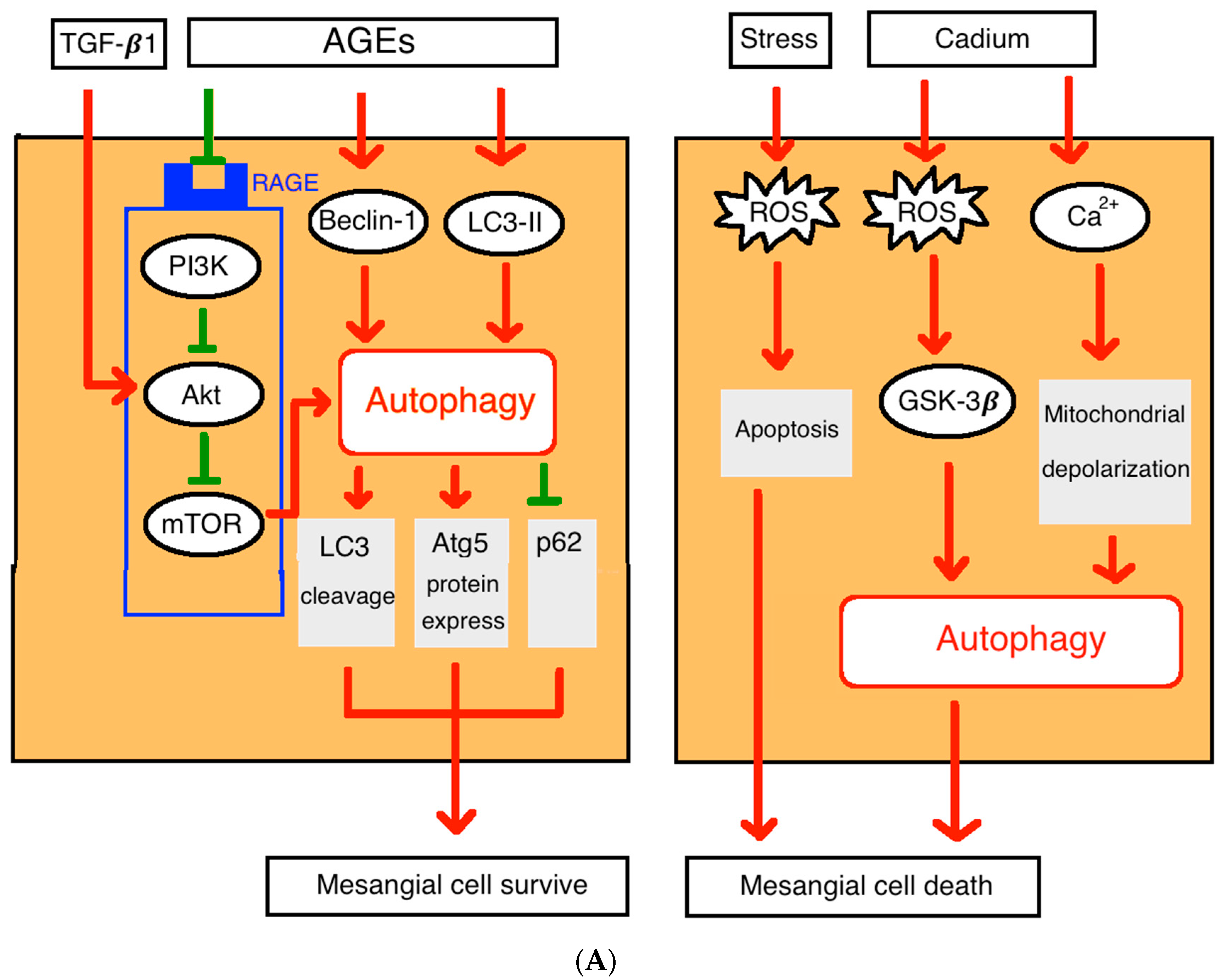
2.1. Autophagy in Glomerular Mesangial Cells
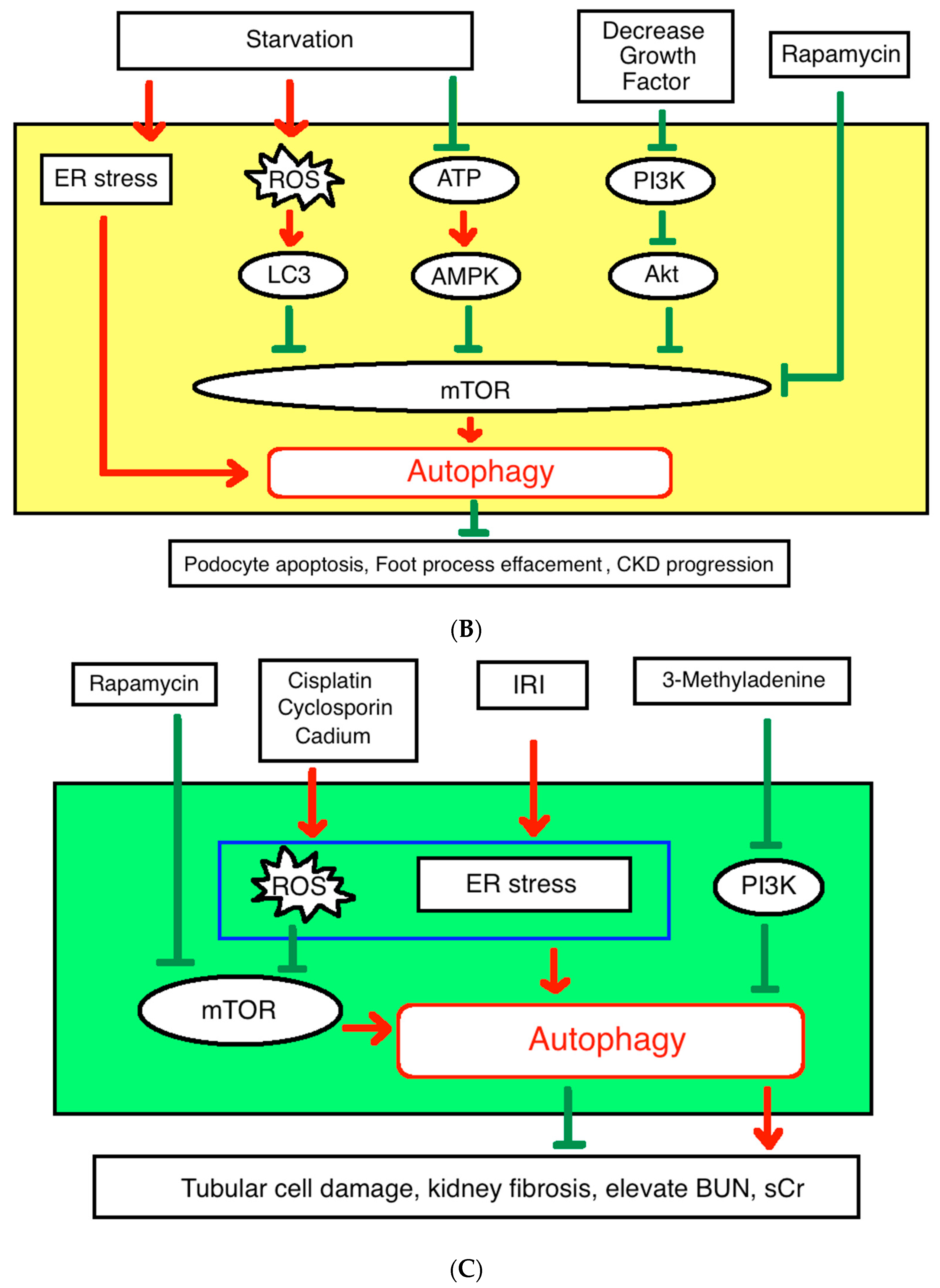
2.2. Autophagy in Podocytes
2.3. Autophagy in Renal Tubular Cells
3. Autophagy in Acute Kidney Injury
3.1. The Kidney Ischemia-Reperfusion Injury and Autophagy
3.2. Autophagy Protects the Renal Cells from Acute Injury
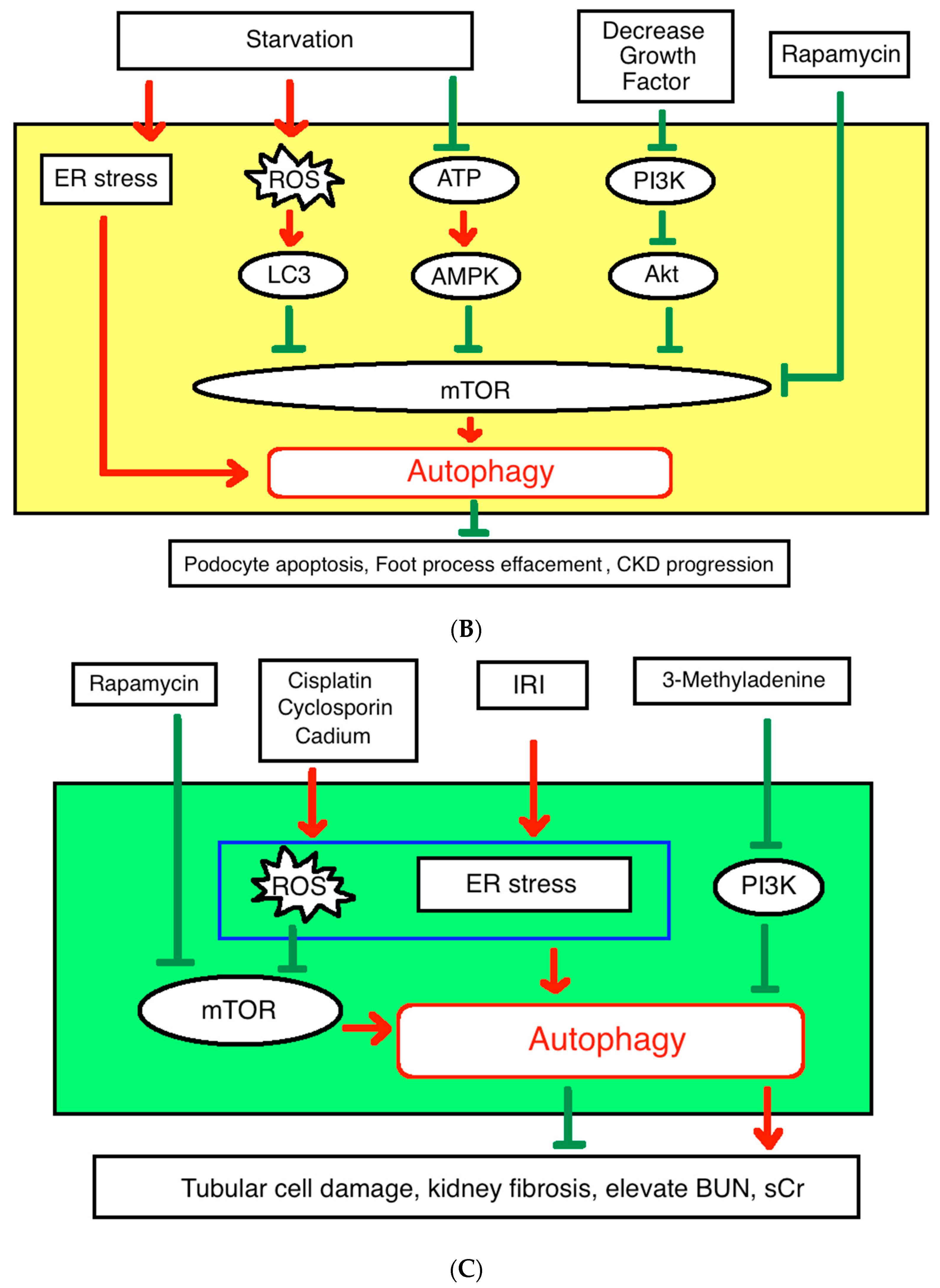
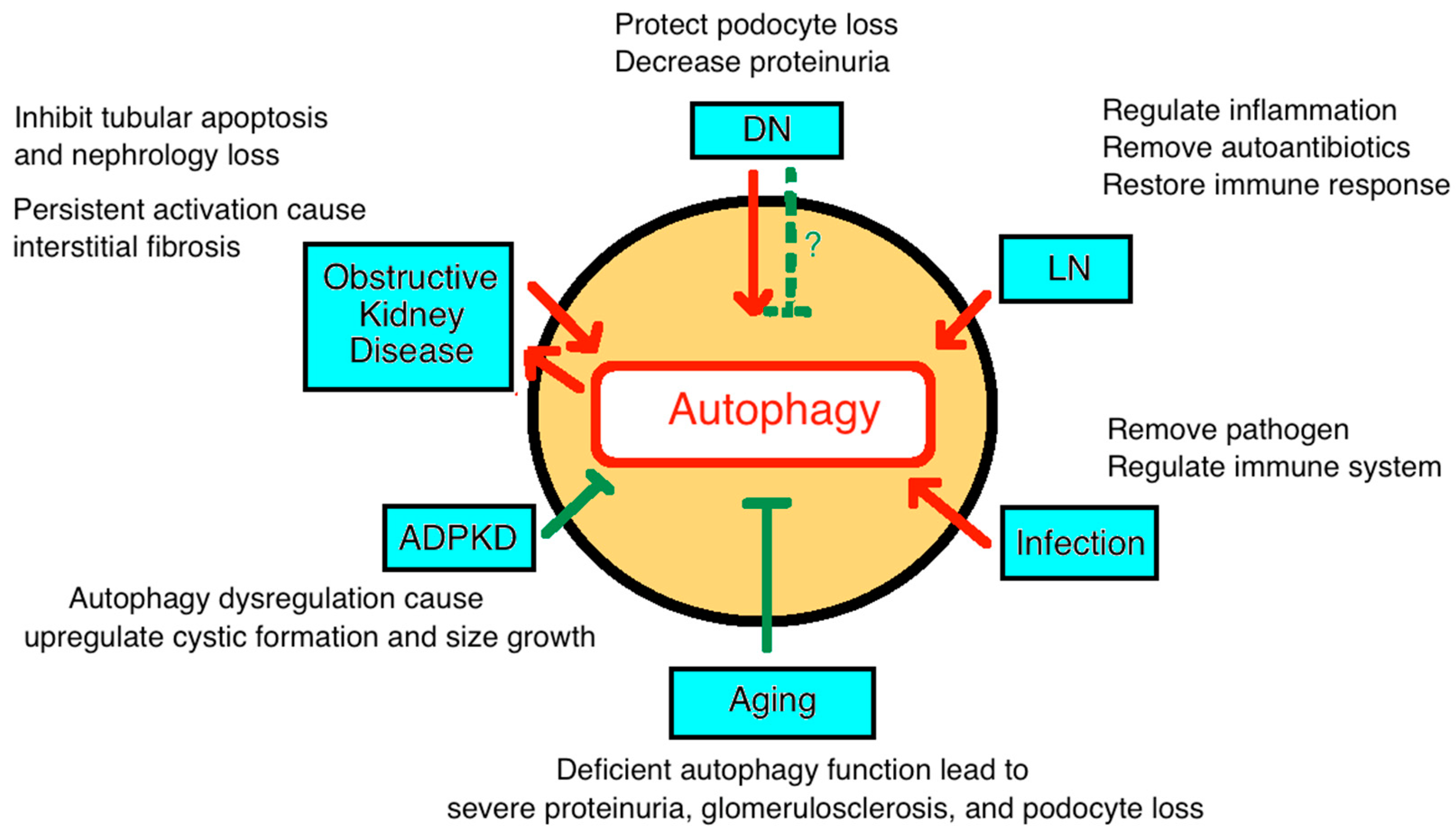
4. Autophagy in Chronic Kidney Disease
4.1. Pathophysiology
4.2. Our Previous Research
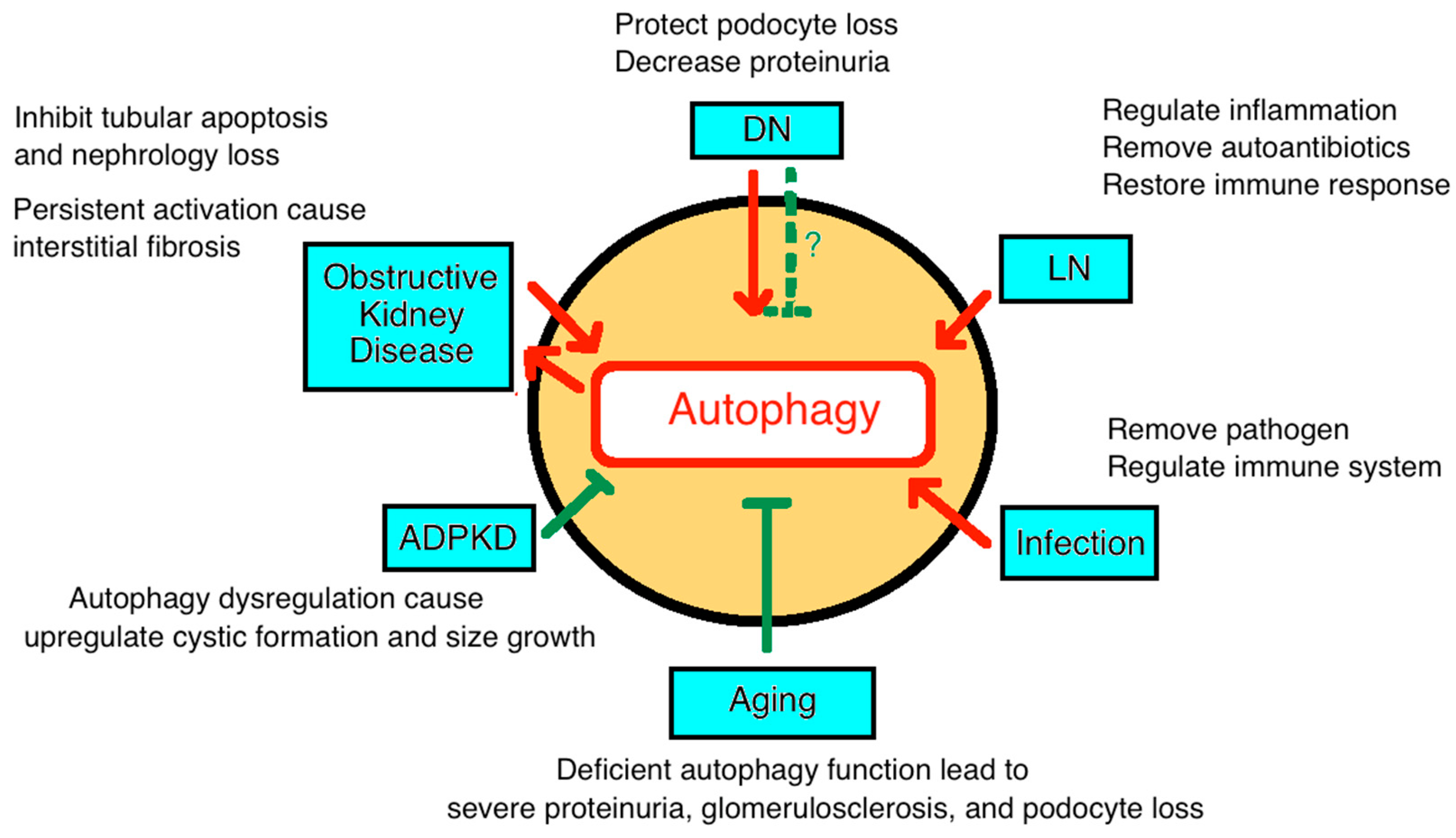
4.3. Diabetic Nephropathy
4.4. Autoimmune Kidney Disease
4.5. Infection
4.6. Renal Tubulointerstitial Diseases and Ureter Obstruction
4.7. Toxic Effects of Drugs
4.8. Cystic Disease (Polycystic Kidney Disease)
4.9. Autophagy in Aging-Associated CKD
5. Autophagy in Dialysis and Renal Transplantation
6. Therapeutic Consideration and Conclusion
6.1. Future Therapeutic Considerations
6.2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Rechter, S.; Decuypere, J.P.; Ivanova, E.; van den Heuvel, L.P.; De Smedt, H.; Levtchenko, E.; Mekahli, D. Autophagy in renal diseases. Pediatr. Nephrol. 2016, 31, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, M.E. Autophagy in kidney health and disease. Antioxid. Redox Signal. 2014, 20, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell. Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-deficient Mutants and Conditions for its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Qian, Y.; Shen, Y.; Zhang, L.; Du, Y.; Dai, H.; Qian, J.; Yan, Y. Autophagy protects renal tubular cells against ischemia/reperfusion injury in a time-dependent manner. Cell. Physiol. Biochem. 2015, 36, 285–298. [Google Scholar] [CrossRef]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: An overview. Am. J. Physiol. Cell Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Tharaux, P.L.; Huber, T.B. Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int. 2016, 90, 950–964. [Google Scholar] [CrossRef]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury and repair. Nephron Clin. Pract. 2014, 127, 56–60. [Google Scholar] [CrossRef]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angoorani, P.; Ejtahed, H.S.; Mirmiran, P.; Mirzaei, S.; Azizi, F. Dietary consumption of advanced glycation end products and risk of metabolic syndrome. Int. J. Food Sci. Nutr. 2016, 67, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Wang, C.C.; Lu, T.F.; Huang, K.H.; Sheu, M.L.; Liu, S.H.; Hung, K.Y. Involvement of Endoplasmic Reticulum Stress, Autophagy, and Apoptosis in Advanced Glycation End Products-Induced Glomerular Mesangial Cell Injury. Sci. Rep. 2016, 6, 34167. [Google Scholar] [CrossRef]
- Hou, X.; Hu, Z.; Xu, H.; Xu, J.; Zhang, S.; Zhong, Y.; He, X.; Wang, N. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc. Diabetol. 2014, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the Kidney: Implications for Ischemia-Reperfusion Injury and Therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef]
- Kreidberg, J.A. Podocyte Differentiation and Glomerulogenesis. J. Am. Soc. Nephrol. 2013, 14, 806–814. [Google Scholar] [CrossRef]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Yanagihara, T.; Ghazizadeh, M.; Ishizaki, M.; Adachi, A.; Sasaki, Y.; Igarashi, T.; Fukunaga, Y. Correlation of Autophagy Type in Podocytes with Histopathological Diagnosis of IgA Nephropathy. Pathobiology 2009, 76, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.; Rodrigues, C.E.; Gomes, S.A.; Noronha, I.L. Acute Kidney Injury as a Condition of Renal Senescence. Cell Transplant. 2018, 27, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Cina, D.P.; Onay, T.; Paltoo, A.; Li, C.; Maezawa, Y.; De Arteaga, J.; Jurisicova, A.; Quaggin, S.E. Inhibition of MTOR disrupts autophagic flux in podocytes. J. Am. Soc. Nephrol. 2012, 23, 412–420. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Pontrelli, P.; Gigante, M.; Montemurno, E.; Loverre, A.; Rossini, M.; Schena, FP.; Grandaliano, G.; Gesualdo, L. Sirolimus and proteinuria in renal transplant patients: Evidence for a dose-dependent effect on slit diaphragm-associated proteins. Transplantation 2011, 91, 997–1004. [Google Scholar] [CrossRef]
- Lin, F. Autophagy in renal tubular injury and repair. Acta Physiol. 2017, 220, 229–237. [Google Scholar] [CrossRef]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, Z.V.; Hill, J.A.; Lin, F. New Autophagy Reporter Mice Reveal Dynamics of Proximal Tubular Autophagy. J. Am. Soc. Nephrol. 2013, 25, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Martin, J.L.; Gruszczyk, A.V.; Beach, T.E.; Murphy, M.P.; Saeb-Parsy, K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr. Nephrol. 2018, 1–8. [Google Scholar] [CrossRef]
- Melk, A.; Baisantry, A.; Schmitt, R. The yin and yang of autophagy in acute kidney injury. Autophagy 2016, 12, 596–597. [Google Scholar] [CrossRef]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Liu, K.; Luo, J.; Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 2010, 176, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takahashi, A.; Takabatake, Y.; Namba, T.; Yamamoto, T.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; Niimura, F.; Matsusaka, T.; et al. Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy 2013, 9, 1876–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coresh, J.; Byrd-Holt, D.; Astor, B.C.; Briggs, J.P.; Eggers, P.W.; Lacher, D.A.; Hostetter, T.H. Chronic kidney disease awareness, prevalence, and trends among U.S. adults, 1999 to 2000. J. Am. Soc. Nephrol. 2005, 16, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Song, Y.; Tao, Q.; Yu, L.; Li, L.; Bai, T.; Song, X.; Hu, H.; Li, Y.; Tan, X. Activation of autophagy contributes to the renoprotective effect of postconditioning on acute kidney injury and renal fibrosis. Biochem. Biophys. Res. Commun. 2018, 504, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Leventhal, J.S.; Wyatt, C.M.; Ross, M.J. Recycling to discover something new: The role of autophagy in kidney disease. Kidney Int. 2017, 91, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Jasiek, M.; Henique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Chen, W.T.; Hung, K.C.; Wen, M.S.; Hsu, P.Y.; Chen, T.H.; Wang, H.D.; Fang, J.T.; Shie, S.S.; Wang, C.Y. Impaired leukocytes autophagy in chronic kidney disease patients. Cardiorenal Med. 2013, 3, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Blume, G.G.; Mcleod, C.J.; Barnes, M.E.; Seward, J.B.; Pellikka, P.A.; Bastiansen, P.M.; Tsang, T.S. Left atrial function: Physiology, assessment, and clinical implications. Eur. J. Echocardiogr. 2011, 12, 421–430. [Google Scholar] [CrossRef]
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef]
- Liu, N.; Xu, L.; Shi, Y.; Zhuang, S. Podocyte Autophagy: A Potential Therapeutic Target to Prevent the Progression of Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 3560238. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhu, J.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. High glucose induces autophagy in podocytes. Exp. Cell. Res. 2013, 319, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda-Yamahara, M.; Kume, S.; Tagawa, A.; Maegawa, H.; Uzu, T. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015, 11, 2385–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-Induced Reactive Oxygen Species Cause Apoptosis of Podocytes and Podocyte Depletion at the Onset of Diabetic Nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Xin, W.; Li, Z.; Xu, Y.; Yu, Y.; Zhou, Q.; Chen, L.; Wan, Q. Autophagy protects human podocytes from high glucose-induced injury by preventing insulin resistance. Metabolism 2016, 65, 1307–1315. [Google Scholar] [CrossRef]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Kitada, M.; Takeda, A.; Nagai, T.; Ito, H.; Kanasaki, K.; Koya, D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: A model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 908185. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Regulating Autophagy as a Therapeutic Target for Diabetic Nephropathy. Curr. Diabetes Rep. 2017, 17, 53. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef]
- Martinez, J.; Cunha, L.D.; Park, S.; Yang, M.; Lu, Q.; Orchard, R.; Li, Q.Z.; Yan, M.; Janke, L.; Guy, C.; et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016, 533, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.J.; Zhang, H. Autophagy in immunity: Implications in etiology of autoimmune/autoinflammatory diseases. Autophagy 2012, 8, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Autophagy: A new concept in autoimmunity regulation and a novel therapeutic option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Maroz, N.; Segal, M.S. Lupus Nephritis and End-stage Kidney Disease. Am. J. Med. Sci. 2013, 346, 319–323. [Google Scholar] [CrossRef]
- Kimura, T.; Isaka, Y.; Yoshimori, T. Autophagy and kidney inflammation. Autophagy 2017, 13, 997–1003. [Google Scholar] [CrossRef]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef]
- Leventhal, J.S.; Ross, M.J. LAPping up dead cells to prevent lupus nephritis: A novel role for noncanonical autophagy in autoimmunity. Kidney Int. 2016, 90, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Ye, M.; Zhao, L.; Zou, W.; Shen, W.; Zhang, H.; Gong, J.; He, Q. The novel involvement of podocyte autophagic activity in the pathogenesis of lupus nephritis. Histol. Histopathol. 2018, 33, 803–814. [Google Scholar]
- Wang, L.; Law, H.K. The Role of Autophagy in Lupus Nephritis. Int. J. Mol. Sci. 2015, 16, 25154–25167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.; Zhang, H.; Wu, T.T.; Yao, Y.M. Autophagy: A Potential Therapeutic Target for Reversing Sepsis-Induced Immunosuppression. Front. Immunol. 2017, 8, 1832. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.W.; Tsai, K.L.; Wang, L.F.; Chen, Y.H.; Chiang, P.C.; Chuang, S.M.; Hsu, C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 2012, 37, 289–296. [Google Scholar] [CrossRef]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef]
- Ding, Y.; Kim, S.L.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy Regulates TGF- Expression and Suppresses Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Zhao, F.; Tang, C.Y.; Luo, M.; Yang, S.K.; Cheng, W.; Li, X.W.; Duan, S.B. Mitophagy Plays a Protective Role in Iodinated Contrast-Induced Acute Renal Tubular Epithelial Cells Injury. Cell. Physiol. Biochem. 2018, 46, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef]
- Inoue, K.; Kuwana, H.; Shimamura, Y.; Ogata, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T.; Morita, T.; Sasaki, S.; et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin. Exp. Nephrol. 2010, 14, 112–122. [Google Scholar] [CrossRef]
- Ning, Y.; Shi, Y.; Chen, J.; Song, N.; Cai, J.; Fang, Y.; Yu, X.; Ji, J.; Ding, X. Necrostatin-1 Attenuates Cisplatin-Induced Nephrotoxicity Through Suppression of Apoptosis and Oxidative Stress and Retains Klotho Expression. Front. Pharmacol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKalpha-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, L.; Liu, Y.; Livingston, MJ.; Chen, JK.; Nahman NS, Jr.; Liu, F.; Dong, Z. Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am. J. Physiol. Renal Physiol. 2017, 313, F74–F84. [Google Scholar] [CrossRef]
- Asanuma, K.; Tanida, I.; Shirato, I.; Ueno, T.; Takahara, H.; Nishitani, T.; Kominami, E.; Tomino, Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003, 17, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. The cytoprotective role of autophagy in puromycin aminonucleoside treated human podocytes. Biochem. Biophys. Res. Commun. 2014, 443, 628–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.; Fan, Y.; Wu, J.; Shi, S.; Chen, Z.; Zhong, Y.; Zhang, C.; Zen, K.; Liu, Z. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J. Pathol. 2014, 234, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Peintner, L.; Borner, C. Role of apoptosis in the development of autosomal dominant polycystic kidney disease (ADPKD). Cell Tissue Res. 2017, 369, 27–39. [Google Scholar] [CrossRef]
- Ravichandran, K.; Edelstein, C.L. Polycystic kidney disease: A case of suppressed autophagy? Semin. Nephrol. 2014, 34, 27–33. [Google Scholar] [CrossRef]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell. Biol. 2009, 29, 2359–2371. [Google Scholar] [CrossRef]
- Chang, M.Y.; Ong, A.C.M. Targeting new cellular disease pathways in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2017, 33, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Cebotaru, V.; Cebotaru, L.; Kim, H.; Chiaravalli, M.; Boletta, A.; Qian, F.; Guggino, W.B. Polycystin-1 Negatively Regulates Polycystin-2 Expression via the Aggresome/Autophagosome Pathway. J. Biol. Chem. 2014, 289, 6404–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Nyengaard, J.R.; Bendtsen, T.F. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat. Rec. 1992, 232, 194–201. [Google Scholar] [CrossRef]
- Wang, X.; Bonventre, J.V.; Parrish, A.R. The aging kidney: Increased susceptibility to nephrotoxicity. Int. J. Mol. Sci. 2014, 15, 15358–15376. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, L.; Minutolo, R.; Chiodini, P.; Borrelli, S.; Zoccali, C.; Postorino, M.; Iodice, C.; Nappi, F.; Fuiano, G.; Gallo, C.; et al. The effect of increasing age on the prognosis of non-dialysis patients with chronic kidney disease receiving stable nephrology care. Kidney Int. 2012, 82, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.; Okada, K.; Yanai, M.; Takahashi, S. Aging and chronic kidney disease. Kidney Blood Press Res. 2013, 38, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A cellular regulator interface in health and disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Silachev, D.N.; Andrianova, N.V.; Pevzner, I.B.; Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Zorov, D.B. Aged kidney: Can we protect it? Autophagy, mitochondria and mechanisms of ischemic preconditioning. Cell Cycle 2018, 17, 1291–1309. [Google Scholar] [CrossRef]
- Andrianova, N.V.; Jankauskas, S.S.; Zorova, L.D.; Pevzner, I.B.; Popkov, V.A.; Silachev, D.N.; Plotnikov, E.Y.; Zorov, D.B. Mechanisms of Age-Dependent Loss of Dietary Restriction Protective Effects in Acute Kidney Injury. Cells 2018, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xing, C.; Zhang, L.; Mao, H.; Chen, X.; Liang, M.; Wang, F.; Ren, H.; Cui, H.; Jiang, A.; et al. Autophagy promotes fibrosis and apoptosis in the peritoneum during long-term peritoneal dialysis. J. Cell. Mol. Med. 2018, 22, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Lin, C.Y.; Su, S.H.; Chuang, C.T.; Chang, Y.L.; Lee, T.Y.; Lee, S.C.; Chang, C.J. Activation of salt-inducible kinase 2 promotes the viability of peritoneal mesothelial cells exposed to stress of peritoneal dialysis. Cell Death Dis. 2016, 7, 2290–2298. [Google Scholar] [CrossRef]
- Bizargity, P.; Schroppel, B. Autophagy: Basic principles and relevance to transplant immunity. Am. J. Transplant. 2014, 14, 1731–1739. [Google Scholar] [CrossRef]
- Nakagawa, S.; Nishihara, K.; Inui, K.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol 2012, 696, 143–154. [Google Scholar] [CrossRef] [Green Version]
- da Silva, J.B.; de Melo Lima, M.H.; Secoli, S.R. Influence of cyclosporine on the occurrence of nephrotoxicity after allogeneic hematopoietic stem cell transplantation: A systematic review. Revista Brasileira de Hematologia e Hemoterapia 2014, 36, 363–368. [Google Scholar] [CrossRef]
- Lim, S.W.; Hyoung, B.J.; Piao, S.G.; Doh, K.C.; Chung, B.H.; Yang, C.W. Chronic cyclosporine nephropathy is characterized by excessive autophagosome formation and decreased autophagic clearance. Transplantation 2012, 94, 218–225. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, X.; Nepovimova, E.; Wang, Y.; Yang, H.; Kuca, K. Mechanism of cyclosporine A nephrotoxicity: Oxidative stress, autophagy, and signalings. Food Chem. Toxicol. 2018, 118, 889–907. [Google Scholar] [CrossRef]
- Schroppel, B.; Legendre, C. Delayed kidney graft function: From mechanism to translation. Kidney Int. 2014, 86, 251–258. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampieri, F.; Afrin, S.; Forbes-Hernandez, T.Y.; Gasparrini, M.; Cianciosi, D.; Reboredo-Rodriguez, P.; Varela-Lopez, A.; Quiles, J.L.; Battino, M. Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 577–634. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, S.; Panigrahi, T.; Shetty, R.; Subramani, M.; Ghosh, A.; Jeyabalan, N. Chloroquine Protects Human Corneal Epithelial Cells from Desiccation Stress Induced Inflammation without Altering the Autophagy Flux. Biomed. Res. Int. 2018, 2018, 7627329. [Google Scholar] [CrossRef]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Ramirez-Tortosa, M.D.C.; Gonzalez-Alonso, A.; Alfonsi, S.; García-Marín, R.; de Miguel, M.; Battino, M. Autophagy in periodontitis patients and gingival fibroblasts: Unraveling the link between chronic diseases and inflammation. BMC Med. 2012, 10, 122. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CKD Categories | Reference | |
|---|---|---|
| Diabetes Nephropathy (DN) | Increase | |
| [47] | |
| [52,53] | |
| [55] | |
| [55] | |
| Decrease | ||
| [56] | |
| [57] | |
| [59,60] | |
| [54,55,61] | |
| Lupus nephritis (LN) | Increase | |
| [64,67] | |
| [68,69] | |
| [70] | |
| [64] | |
| Infection | Increase | |
| [63,72] | |
| [41] | |
| [73] | |
| [66] | |
| Tubulointerstitial injury | Protect | |
| [74] | |
| [75] | |
| [13] | |
| Damage | ||
| [75] | |
| Toxic/Drugs | Iodinated contrast | |
| [77] | |
| Cisplatin | ||
| [78] | |
| [79,81] | |
| [78,79,82] | |
| [83] | |
| ||
| [84,85] | |
| Autosomal dominant polycystic kidney disease (ADPKD) | Dysregulation | |
| [89,90,91] | |
| [1,94] | |
| Aging-Associated CKD | Deficiency | |
| [22] | |
| [99] | |
| [41] | |
| [101,102] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-A.; Wu, V.C.-C.; Wang, C.-Y. Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61. https://doi.org/10.3390/cells8010061
Lin T-A, Wu VC-C, Wang C-Y. Autophagy in Chronic Kidney Diseases. Cells. 2019; 8(1):61. https://doi.org/10.3390/cells8010061
Chicago/Turabian StyleLin, Tien-An, Victor Chien-Chia Wu, and Chao-Yung Wang. 2019. "Autophagy in Chronic Kidney Diseases" Cells 8, no. 1: 61. https://doi.org/10.3390/cells8010061
APA StyleLin, T. -A., Wu, V. C. -C., & Wang, C. -Y. (2019). Autophagy in Chronic Kidney Diseases. Cells, 8(1), 61. https://doi.org/10.3390/cells8010061