Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Rat Primary Microglial Cell Isolation
2.3. Antiretroviral Treatment
2.4. Western Blotting
2.5. Immunocytochemistry
2.6. Quantification of MAP1LC3B and LAMP2 Puncta
2.7. Real-Time qPCR
2.8. CTSD Activity Determination
2.9. Lysosomal Membrane Permeability Assay
2.10. Lysosomal pH Measurement
2.11. MAP1LC3B Turnover and SQSTM1 Degradation Assays
2.12. Plasmids Transfection
2.13. Statistical Analysis
3. Results
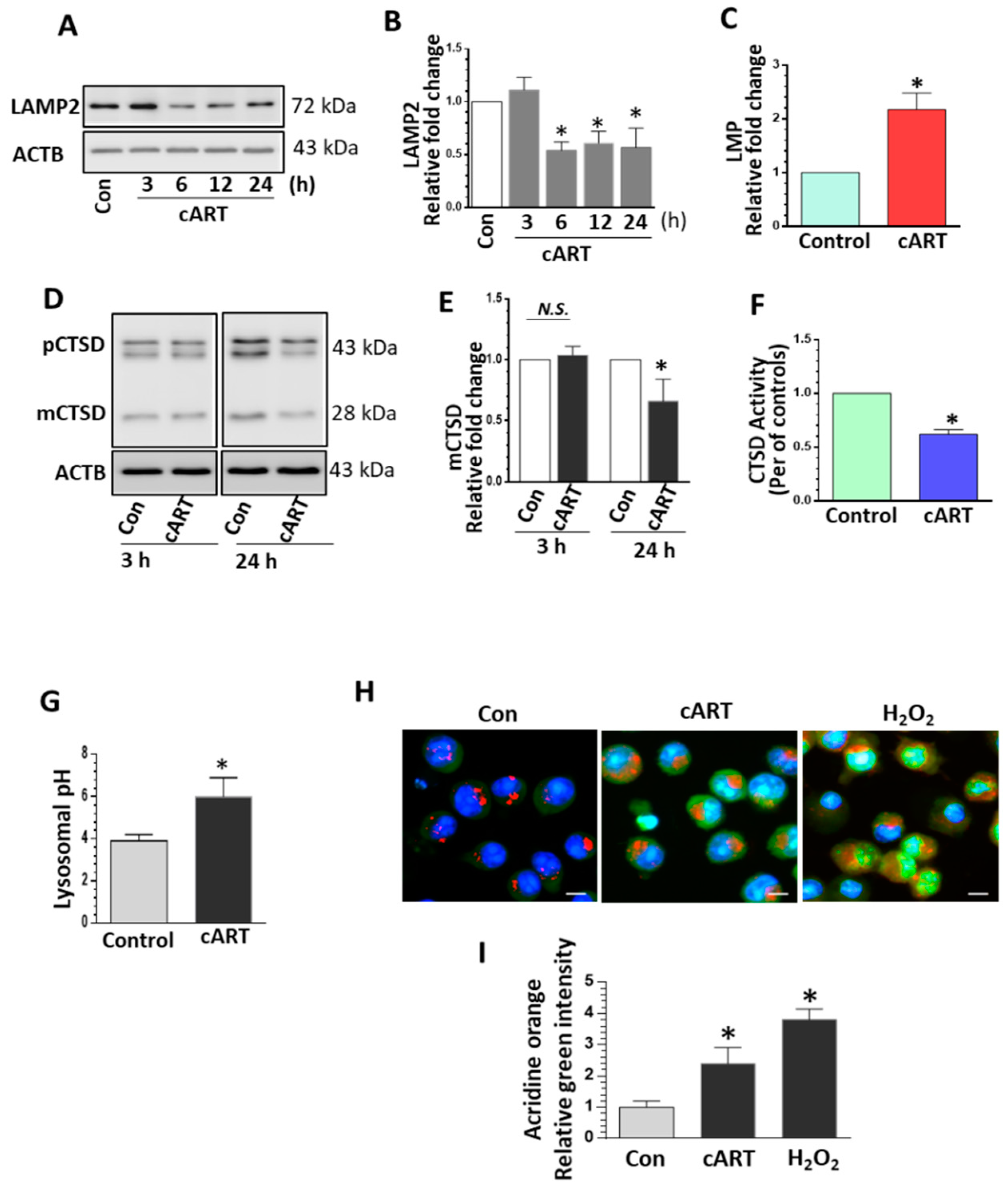
3.1. cART-Mediated Impairment of Lysosomal Function in rPMs
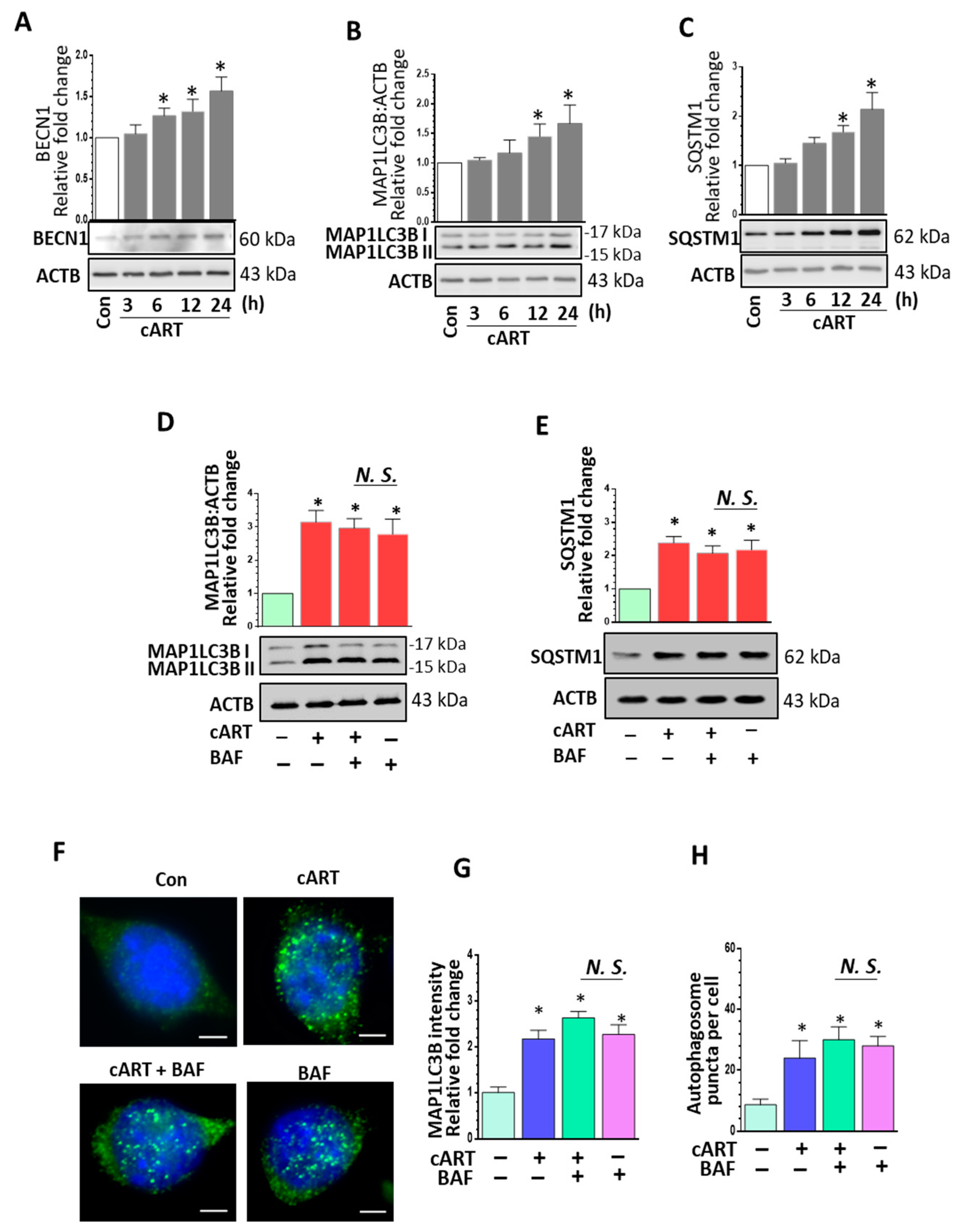
3.2. cART-Mediated Dysregulated Autophagy in rPMs
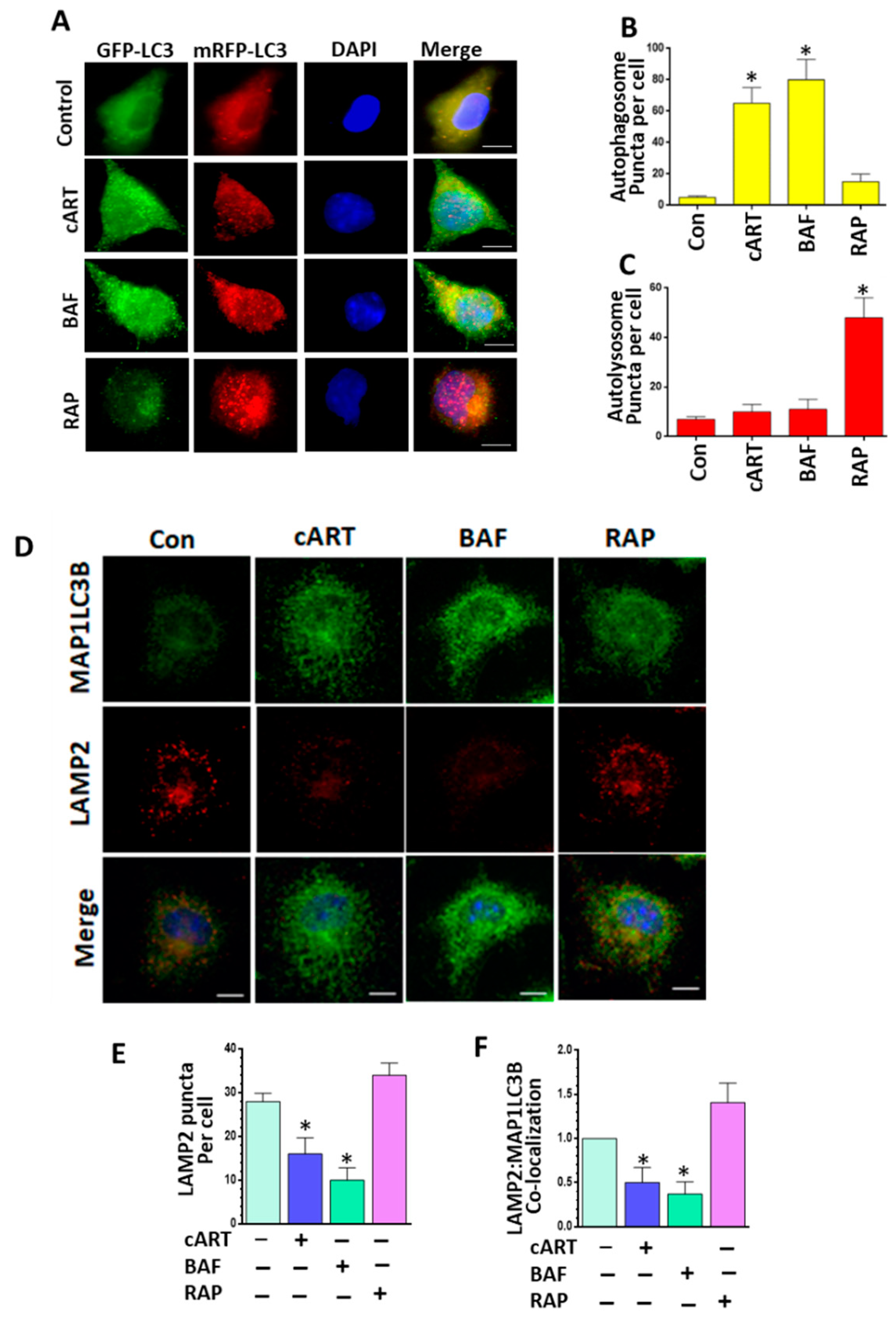
3.3. cART-Mediated Impairment of Autophagosome–Lysosome Fusion
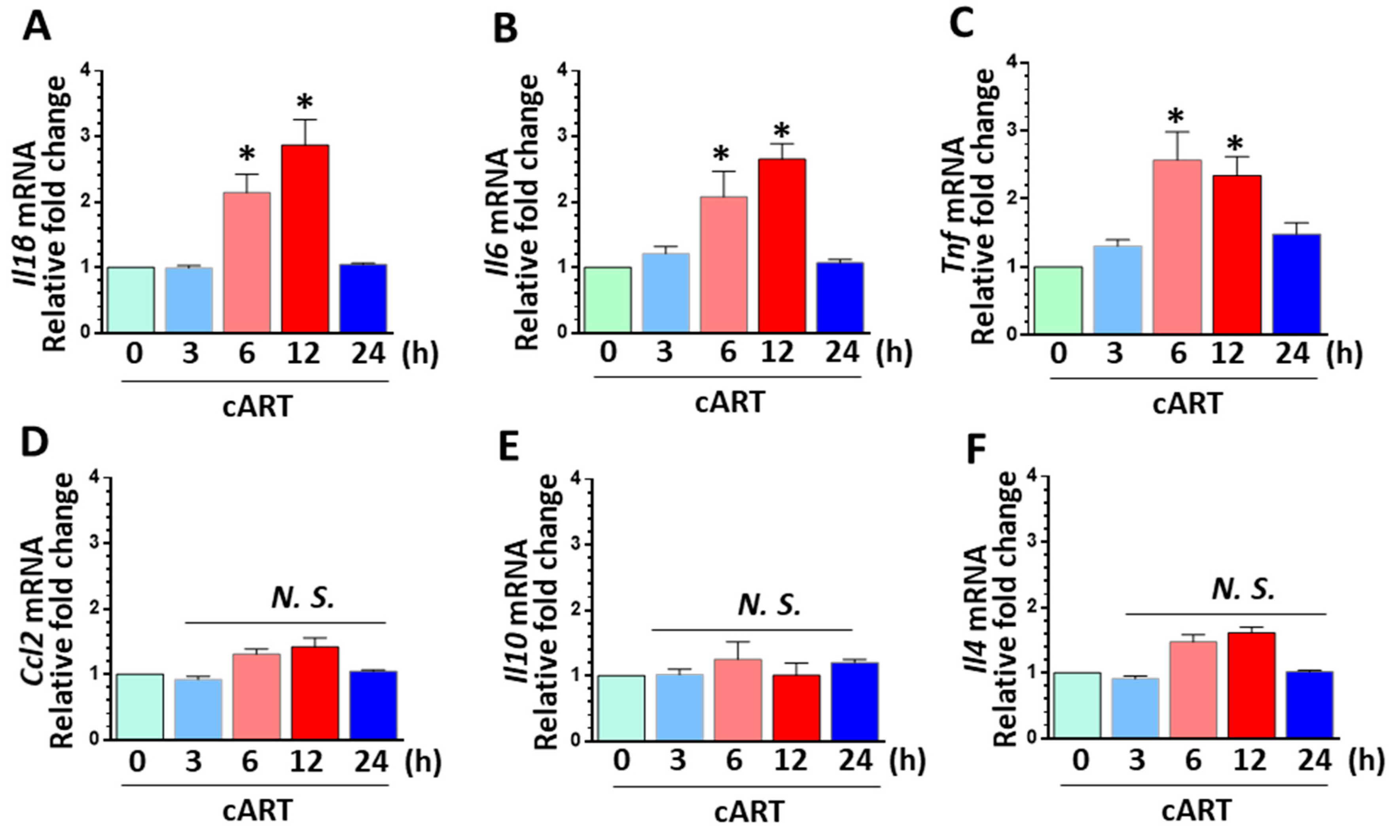
3.4. cART-Mediated Activation of rPMs
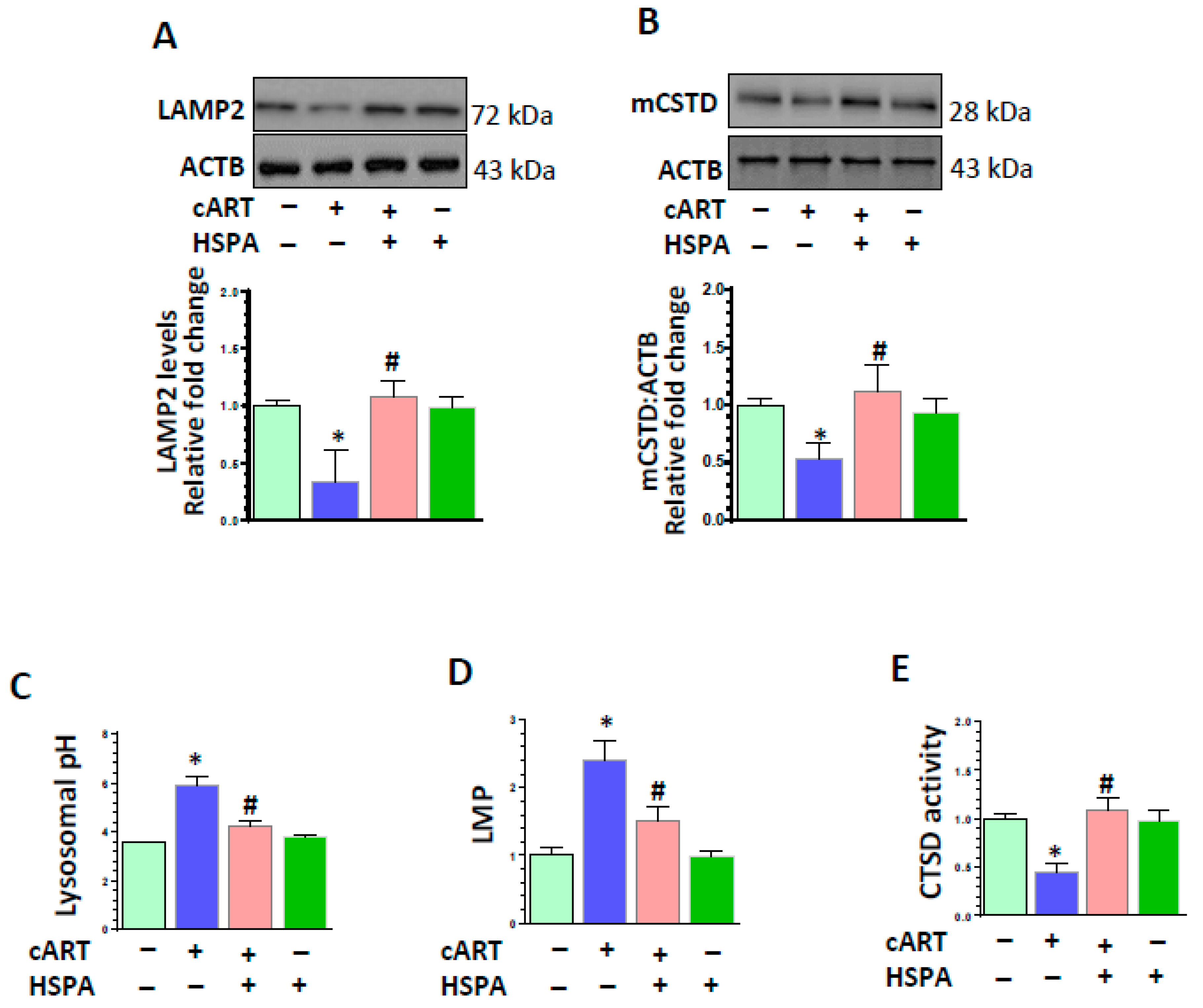
3.5. HSPA Overexpression Abrogated cART-Mediated Impairment of Lysosomal Function
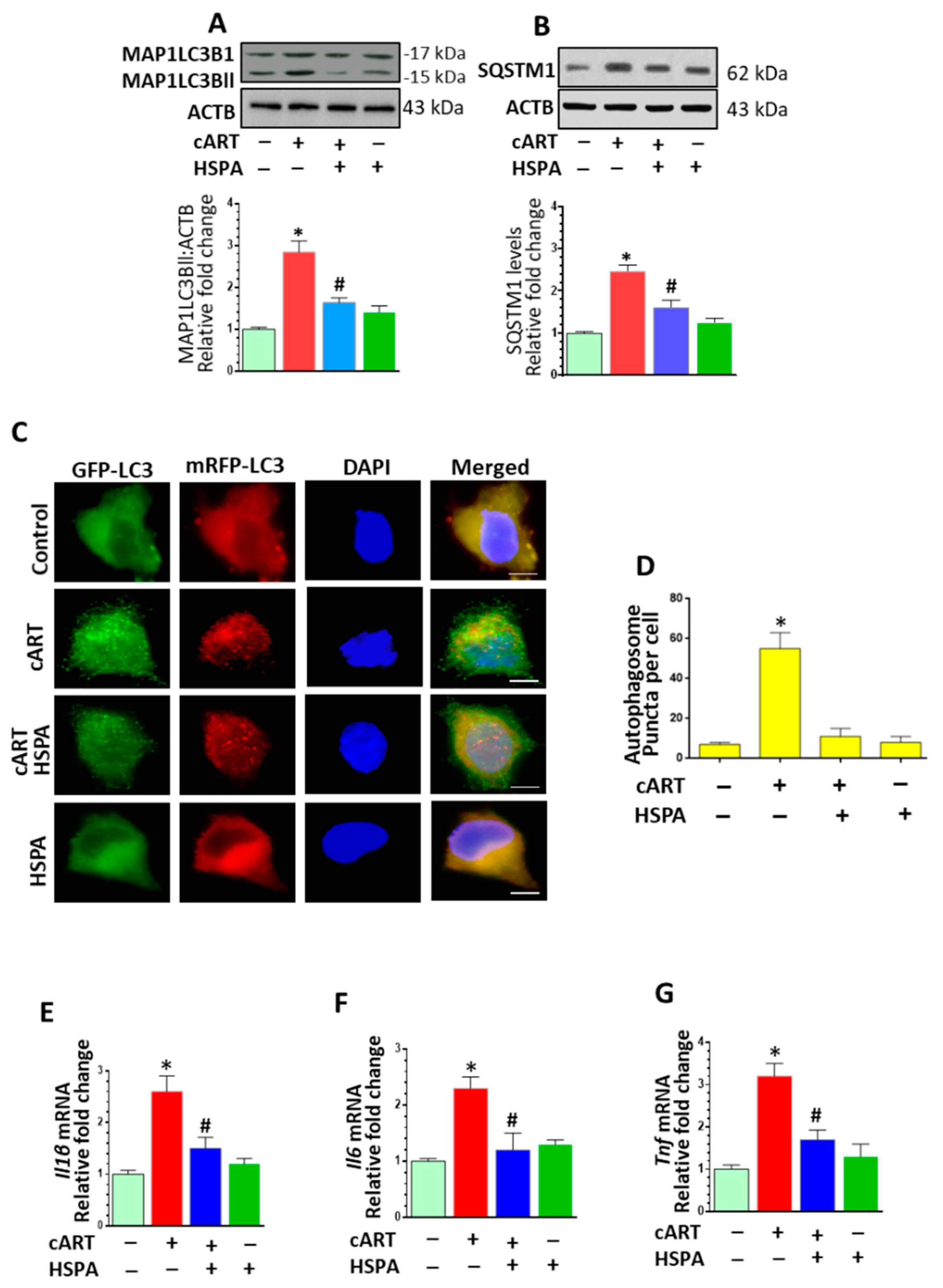
3.6. HSPA Overexpression Abrogated cART-Mediated Autophagy Dysregulation and Microglial Activation
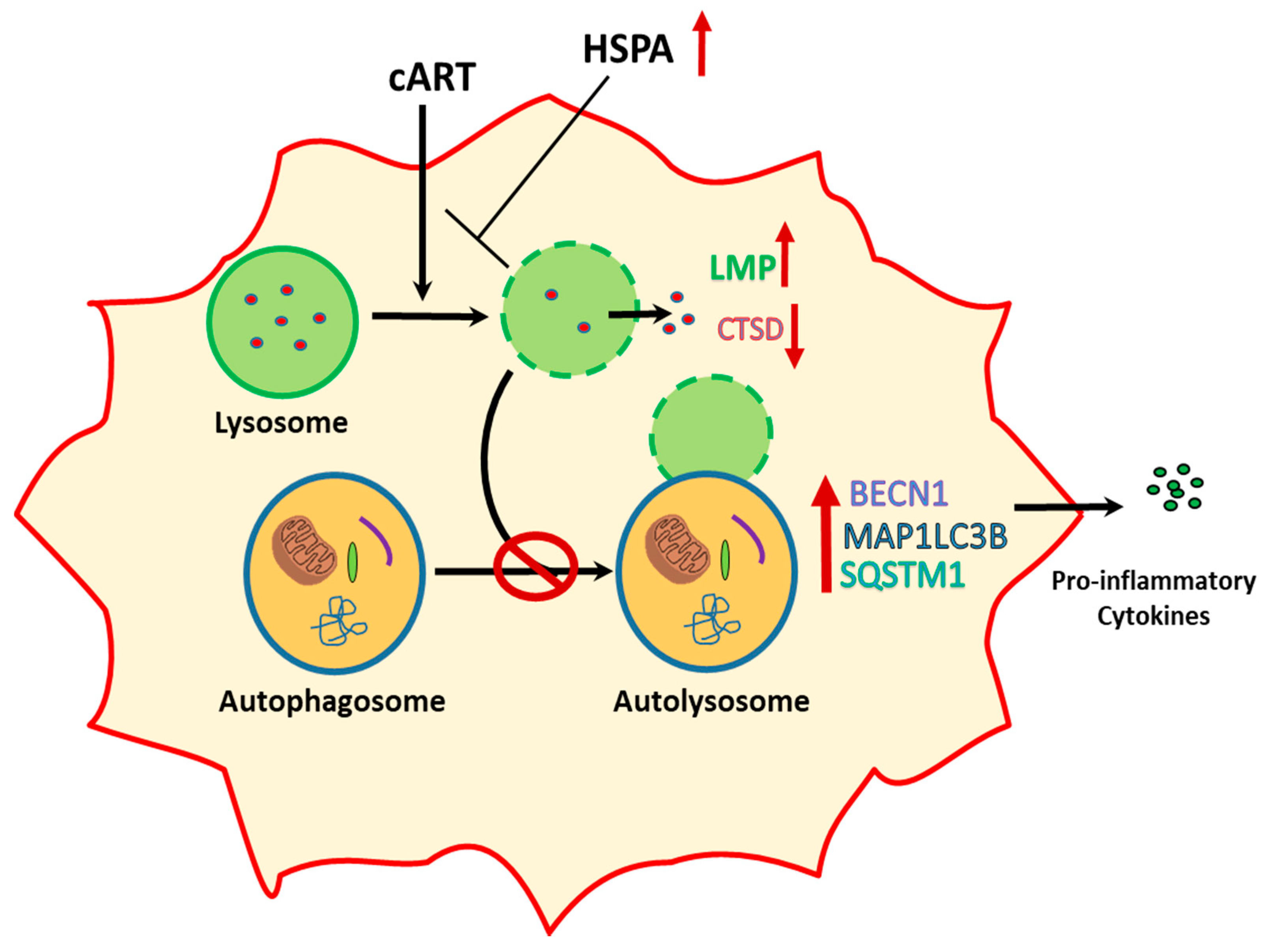
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTB | actin, beta |
| BAF | bafilomycin A1 |
| BECN1 | beclin 1, autophagy related |
| cART | combined antiretroviral therapy |
| CCL2 | C-C motif chemokine ligand 2 |
| CNS | central nervous system |
| CTSD | cathepsin D |
| CTSB | cathepsin B |
| DAPI | 4,6-Diamidino-2-phenylindole |
| DMEM | Dulbecco modified eagle medium |
| DTG | dolutegravir |
| FBS | fetal bovine serum |
| FTC | emtricitabine |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HAND | HIV-1-associated neurocognitive disorders |
| HIV-1 Tat | human immunodeficiency virus-1 transactivator of transcription |
| IL1B | interleukin 1, beta |
| IL6 | interleukin 6 |
| LAMP2 | lysosome associated membrane protein 2 |
| LMP | lysosomal membrane permeabilization |
| M6PR | mannose-6-phosphate receptor |
| MAP1LC3B | microtubule-associated protein 1 light chain 3 beta |
| NRTIs | nucleoside reverse transcriptase inhibitors |
| PBS | phosphate buffered saline |
| RAP | rapamycin |
| rPMs | rat primary microglial cells |
| SQSTM1 | sequestosome 1 |
| TFEB | transcription factor EB |
| TDF | tenofovir disoproxil fumarate |
| TNF | tumor necrosis factor |
References
- Saloner, R.; Cysique, L.A. HIV-Associated Neurocognitive Disorders: A Global Perspective. J. Int. Neuropsychol. Soc. 2017, 23, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Farhadian, S.; Patel, P.; Spudich, S. Neurological Complications of HIV Infection. Curr. Infect. Dis. Rep. 2017, 19, 50. [Google Scholar] [CrossRef]
- Ru, W.; Tang, S.J. HIV-associated synaptic degeneration. Mol. Brain 2017, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Taddei, T.H.; Lo Re, V., 3rd; Justice, A.C. HIV, Aging and Viral Coinfections: Taking the Long View. Curr. HIV/AIDS Rep. 2016, 13, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, D. Demographics of HIV and aging. Curr. Opin. HIV AIDS 2014, 9, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Gougeon, M.L. Alarmins and central nervous system inflammation in HIV-associated neurological disorders. J. Intern. Med. 2017, 281, 433–447. [Google Scholar] [CrossRef]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Cardona, A.E. The myeloid cells of the central nervous system parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Wooten, A.K.; Jackson, J.W.; Maggirwar, S.B.; Kiebala, M. Investigating the role of ankyrin-rich membrane spanning protein in human immunodeficiency virus type-1 Tat-induced microglia activation. J. Neurovirol. 2015, 21, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.J.; Singh, H.D.; Carey, A.N.; McLaughlin, J.P. Exposure to HIV-1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav. Brain Res. 2015, 291, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Guo, M.L.; Hu, G.; Callen, S.; Buch, S. Epigenetic Promoter DNA Methylation of miR-124 Promotes HIV-1 Tat-Mediated Microglial Activation via MECP2-STAT3 Axis. J. Neurosci. 2018, 38, 5367–5383. [Google Scholar] [CrossRef]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef] [Green Version]
- Lisi, L.; Tramutola, A.; Navarra, P.; Dello Russo, C. Antiretroviral agents increase NO production in gp120/IFNgamma-stimulated cultures of rat microglia via an arginase-dependent mechanism. J. Neuroimmunol. 2014, 266, 24–32. [Google Scholar] [CrossRef]
- Lisi, L.; Laudati, E.; Miscioscia, T.F.; Dello Russo, C.; Topai, A.; Navarra, P. Antiretrovirals inhibit arginase in human microglia. J. Neurochem. 2016, 136, 363–372. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G. Autophagosome maturation and lysosomal fusion. Essays Biochem. 2013, 55, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Thompson, C.B. Autophagy: Basic principles and relevance to disease. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.E.; Kielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015, 9, 417. [Google Scholar] [CrossRef] [Green Version]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services Panel On Antiretroviral Guidelines for Adults and Adolescents. Recommendation on Integrase Inhibitor Use in Antiretroviral Treatment-Naïve HIV-Infected Individuals. 30 October 2013. Available online: https://aidsinfo.nih.gov/news/1392/recommendation-on-integrase-inhibitor-use-in-antiretroviral-treatment-naive-hiv-infected-individuals-from-the-hhs-panel-on-antiretroviral-guidelines-for-adults-and-adolescents (accessed on 15 February 2019).
- Antiretroviral Therapy for HIV Infection: Overview, FDA-Approved Antivirals and Regimens, Medscape 2018. Available online: https://emedicine.medscape.com/article/1533218-overview#showall. (accessed on 15 May 2019).
- Saag, M.S.; Benson, C.A.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Mugavero, M.J.; Sax, P.E.; Smith, D.M.; Thompson, M.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel. JAMA 2018, 320, 379–396. [Google Scholar] [CrossRef]
- Baldin, G.; Ciccullo, A.; Capetti, A.; Rusconi, S.; Sterrantino, G.; Cossu, M.V.; Giacomelli, A.; Lagi, F.; Latini, A.; Bagella, P. Efficacy and safety of switching to dolutegravir plus emtricitabine/tenofovir disoproxil fumarate (TDF) or elvitegravir/cobicistat/emtricitabine/TDF in virologically suppressed HIV-infected patients in clinical practice: Results from a multicentre, observational study. HIV Med. 2019, 20, 164–168. [Google Scholar] [CrossRef]
- Stellbrink, H.J.; Arribas, J.R.; Stephens, J.L.; Albrecht, H.; Sax, P.E.; Maggiolo, F.; Creticos, C.; Martorell, C.T.; Wei, X.; Acosta, R.; et al. Co-formulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide for initial treatment of HIV-1 infection: Week 96 results from a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet HIV 2019, 6, e364–e372. [Google Scholar] [CrossRef] [PubMed]
- Cahn, P.; Madero, J.S.; Arribas, J.R.; Antinori, A.; Ortiz, R.; Clarke, A.E.; Hung, C.C.; Rockstroh, J.K.; Girard, P.M.; Sievers, J.; et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): Week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet 2019, 393, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Guo, M.L.; Buch, S. Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy 2016, 12, 1310–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.C.; Bhasin, S.; Wu, X.; Lee, J.G.; Maffi, S.; Nichols, C.J.; Lee, K.J.; Taylor, J.P.; Greene, L.E.; Eisenberg, E. Hsp70 dynamics in vivo: Effect of heat shock and protein aggregation. J. Cell Sci. 2004, 117, 4991–5000. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, A.; Di Perri, G.; Bonora, S. Pharmacokinetics and pharmacodynamics of antiretrovirals in the central nervous system. Clin. Pharmacokinet. 2014, 53, 891–906. [Google Scholar] [CrossRef]
- Patel, S.H.; Ismaiel, O.A.; Mylott, W.R., Jr.; Yuan, M.; McClay, J.L.; Paris, J.J.; Hauser, K.F.; McRae, M. Cell-type specific differences in antiretroviral penetration and the effects of HIV-1 Tat and morphine among primary human brain endothelial cells, astrocytes, pericytes, and microglia. Neurosci. Lett. 2019, 712, 134475. [Google Scholar] [CrossRef]
- Cattaneo, D.; Minisci, D.; Cozzi, V.; Riva, A.; Meraviglia, P.; Clementi, E.; Galli, M.; Gervasoni, C. Dolutegravir plasma concentrations according to companion antiretroviral drug: Unwanted drug interaction or desirable boosting effect? Antivir. Ther. 2017, 22, 353–356. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Ishidoh, K.; Kominami, E. Processing and activation of lysosomal proteinases. Biol. Chem. 2002, 383, 1827–1831. [Google Scholar] [CrossRef]
- Erickson, A.H. Biosynthesis of lysosomal endopeptidases. J. Cell. Biochem. 1989, 40, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Canuel, M.; Korkidakis, A.; Konnyu, K.; Morales, C.R. Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun. 2008, 373, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef]
- Bollmann, F.M. Telomerase inhibition may contribute to accelerated mitochondrial aging induced by anti-retroviral HIV treatment. Med. Hypotheses 2013, 81, 285–287. [Google Scholar] [CrossRef]
- Brinkman, K.; Smeitink, J.A.; Romijn, J.A.; Reiss, P. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 1999, 354, 1112–1115. [Google Scholar] [CrossRef]
- Manda, K.R.; Banerjee, A.; Banks, W.A.; Ercal, N. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood-brain barrier endothelial cells. Free Radic. Biol. Med. 2011, 50, 801–810. [Google Scholar] [CrossRef]
- Tavazzi, E.; Morrison, D.; Sullivan, P.; Morgello, S.; Fischer, T. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr. HIV Res. 2014, 12, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.M.; Wang, F.; Qi, D.; Liu, W.W.; Gu, C.; Mao, C.J.; Yang, Y.P.; Zhao, Z.; Hu, L.F.; Liu, C.F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and Non-Canonical Autophagy in HIV-1 Replication Cycle. Viruses 2017, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, E.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS ONE 2008, 3, e2906. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Oh, J.E.; Yi, J.K.; Kim, R.H.; Shin, K.H.; Mitsuyasu, R.; Park, N.H.; Kang, M.K. Efavirenz induces autophagy and aberrant differentiation in normal human keratinocytes. Int. J. Mol. Med. 2013, 31, 1305–1312. [Google Scholar] [CrossRef]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Gortat, A.; Blas-Garcia, A.; Esplugues, J.V. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology 2011, 54, 1009–1019. [Google Scholar] [CrossRef]
- Wallace, Z.R.; Sanderson, S.; Simon, A.K.; Dorrell, L. Exposure to zidovudine adversely affects mitochondrial turnover in primary T cells. Antivir. Res. 2016, 133, 178–182. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Pinti, M.; Nasi, M.; Riccio, M.; Carnevale, G.; Cavallini, G.M.; Sala de Oyanguren, F.J.; O’Connor, J.E.; Mussini, C.; et al. The protease inhibitor atazanavir triggers autophagy and mitophagy in human preadipocytes. AIDS 2012, 26, 2017–2026. [Google Scholar] [CrossRef]
- McLean, K.; VanDeVen, N.A.; Sorenson, D.R.; Daudi, S.; Liu, J.R. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol. Oncol. 2009, 112, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, X.; Haughey, N.J.; Geiger, J.D. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 2012, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Cesen, M.H.; Turk, B. The Use of Lysosomotropic Dyes to Exclude Lysosomal Membrane Permeabilization. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic (Cph. Den.) 2018, 19, 918–931. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.-L.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168. https://doi.org/10.3390/cells8101168
Tripathi A, Thangaraj A, Chivero ET, Periyasamy P, Callen S, Burkovetskaya ME, Guo M-L, Buch S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells. 2019; 8(10):1168. https://doi.org/10.3390/cells8101168
Chicago/Turabian StyleTripathi, Ashutosh, Annadurai Thangaraj, Ernest T. Chivero, Palsamy Periyasamy, Shannon Callen, Maria E. Burkovetskaya, Ming-Lei Guo, and Shilpa Buch. 2019. "Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction" Cells 8, no. 10: 1168. https://doi.org/10.3390/cells8101168