The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins

 ,
,  , and
, and 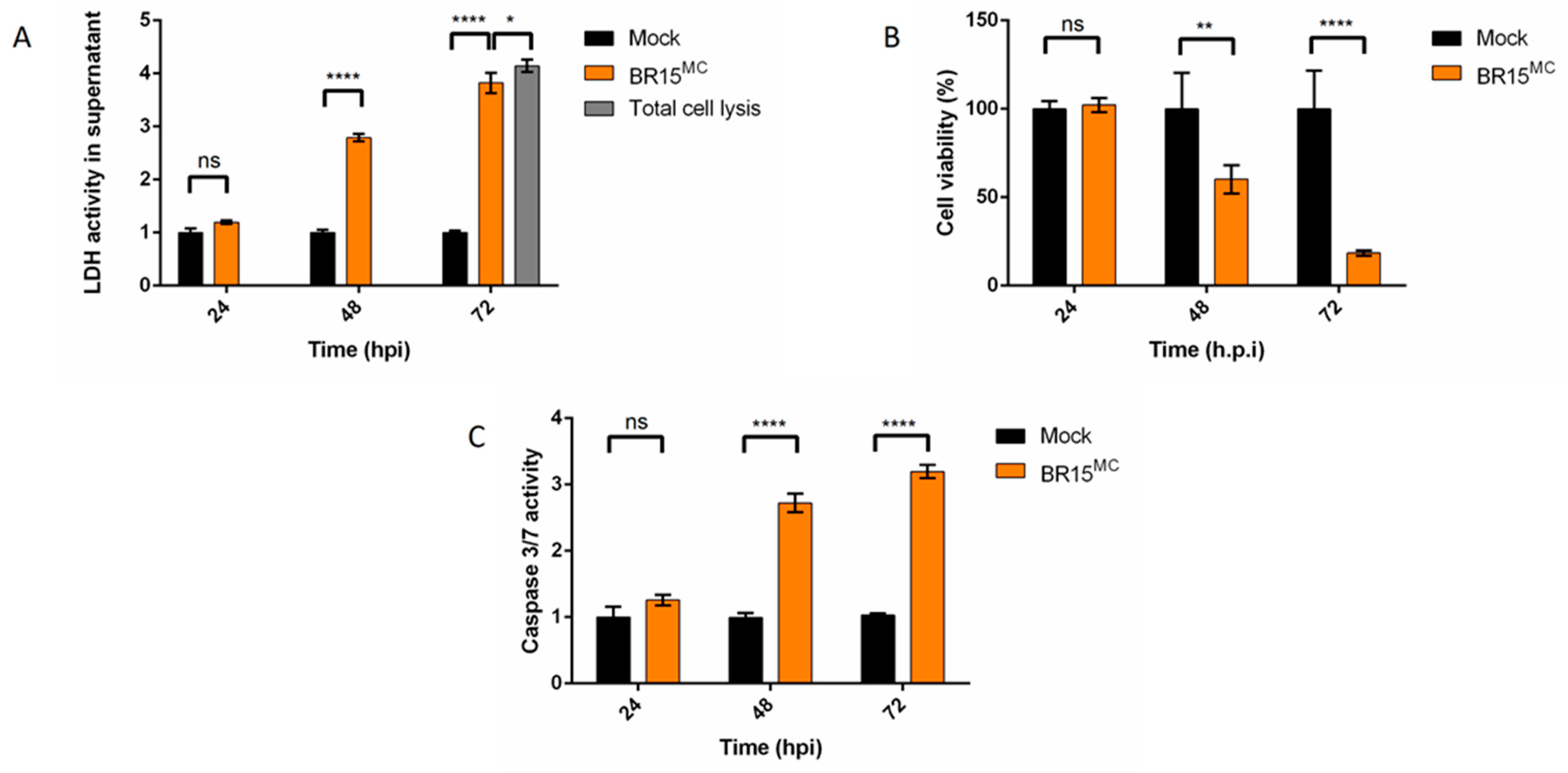{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Cell Lines, Antibodies and Reagents
2.2. Plaque Forming Assay
2.3. Western Blotting (WB)
2.4. Induction of Apoptosis
2.5. Immunofluorescence Assay
2.6. Cytotoxicity Assay
2.7. Cell Viability Assay (MTT)
2.8. Caspase-3/7 Activity
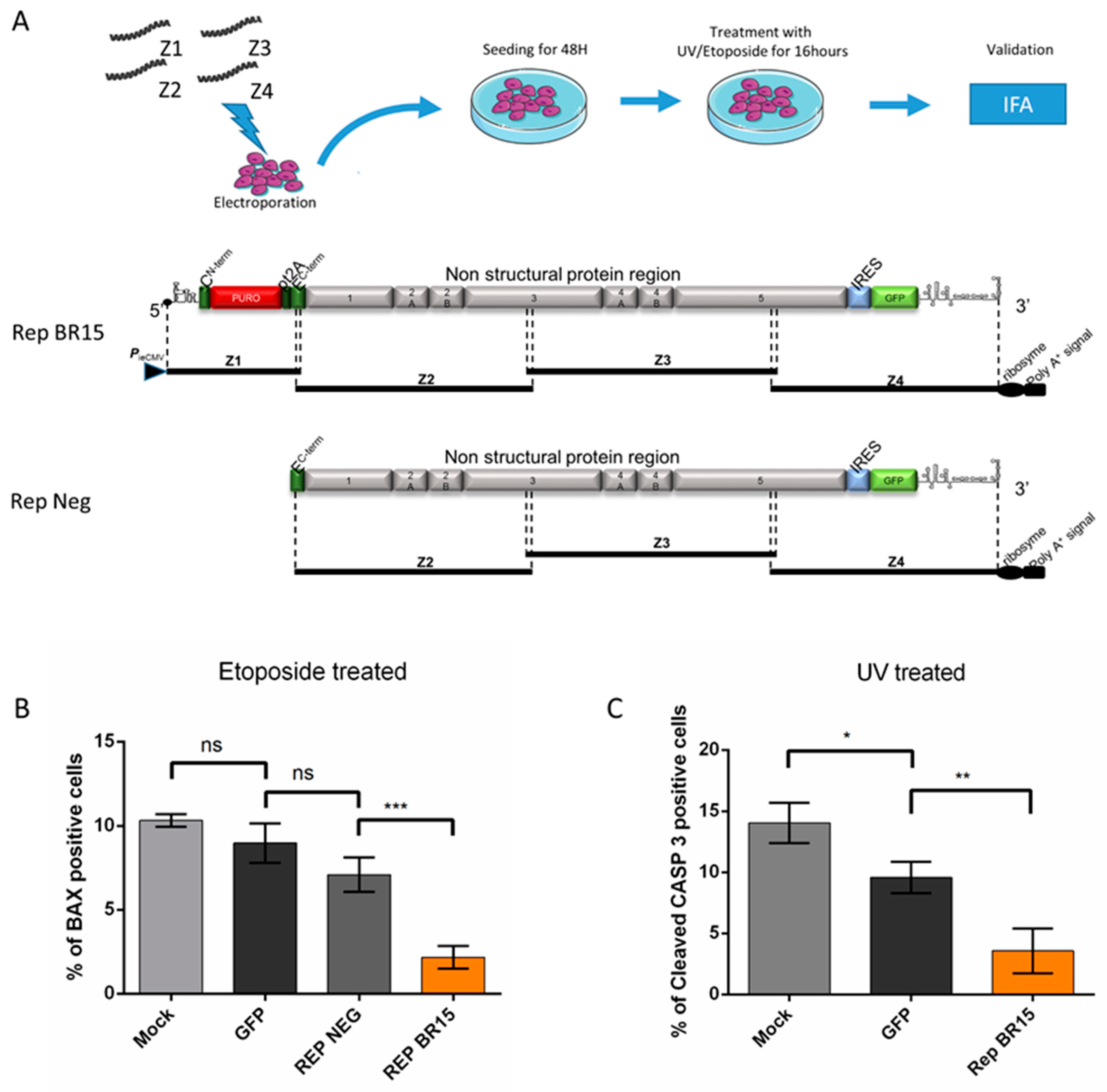
2.9. Generation of ZIKV Replicon by the ISA Method
2.10. Statistical Analysis
3. Results
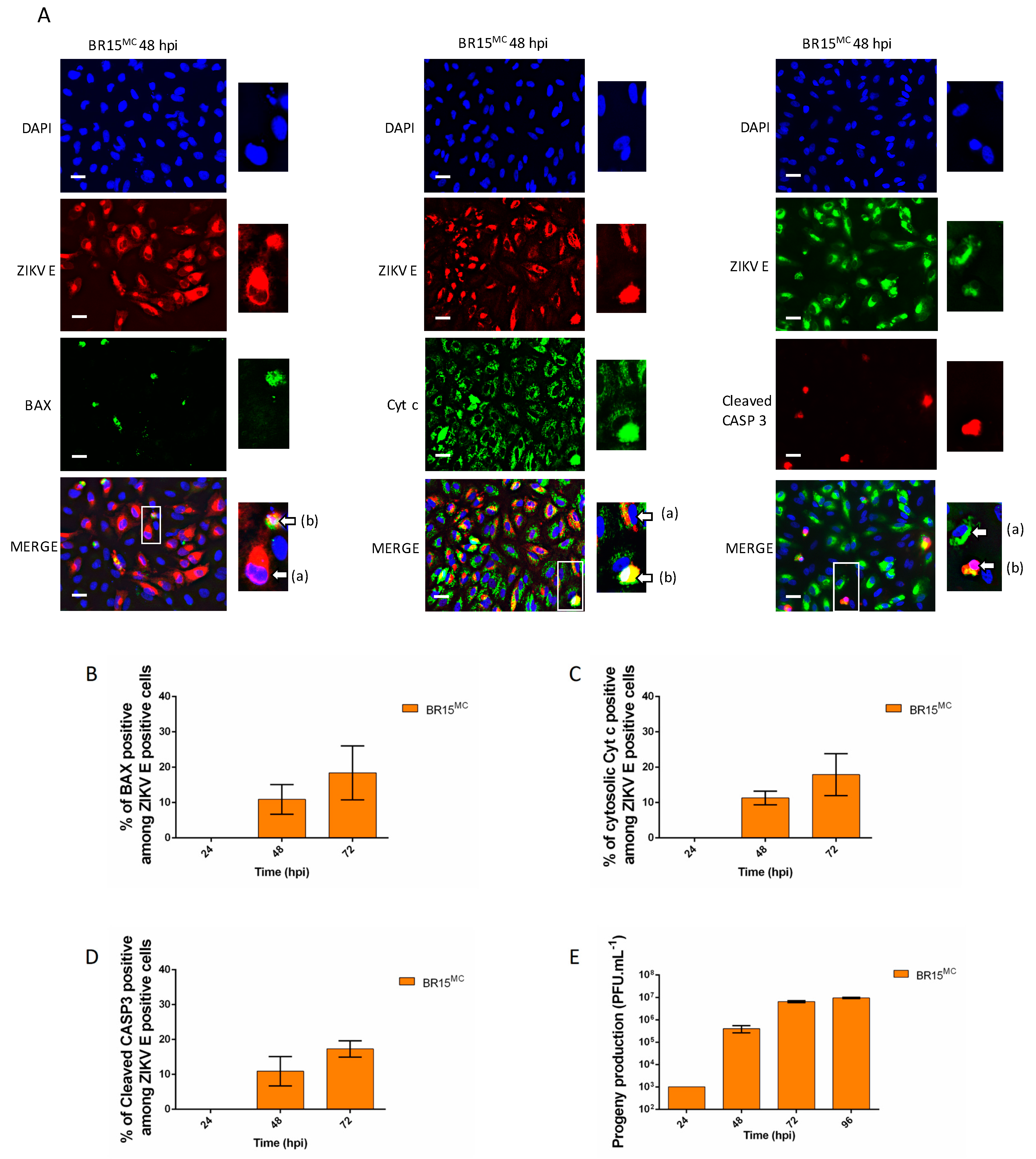
3.1. ZIKV Does Not Trigger Apoptosis Until the Release of Most of its Progeny
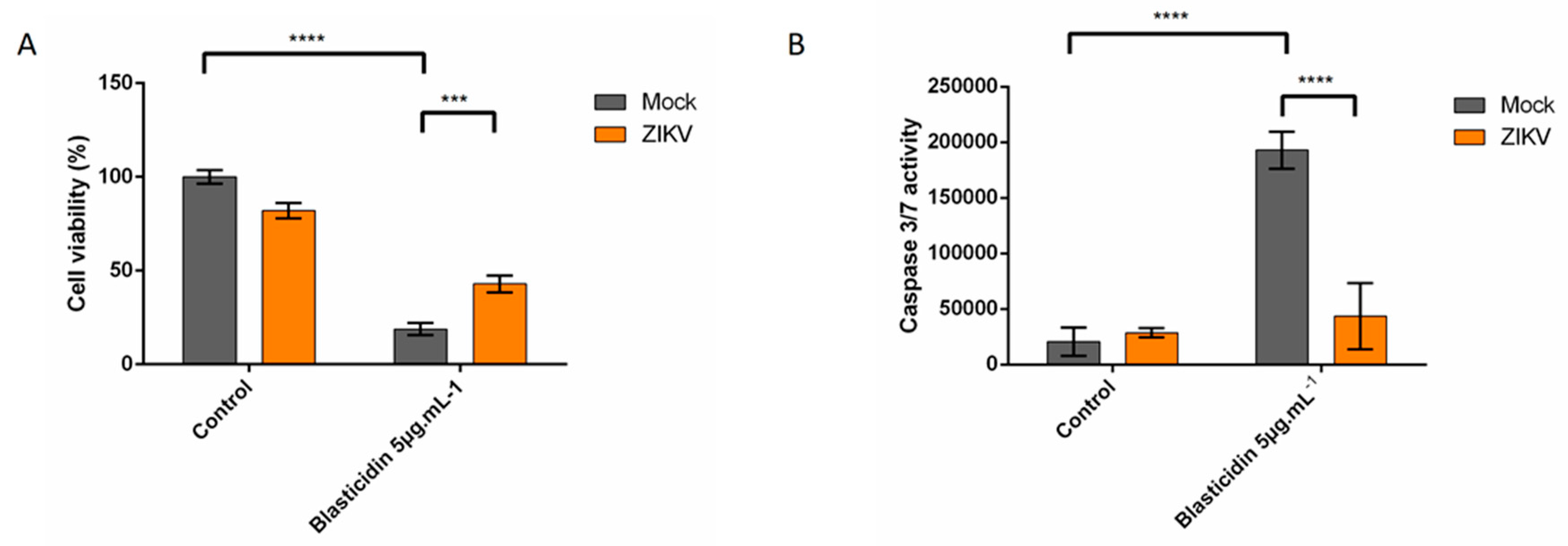
3.2. ZIKV Infected Cells Display Resistance to Apoptosis Inducers
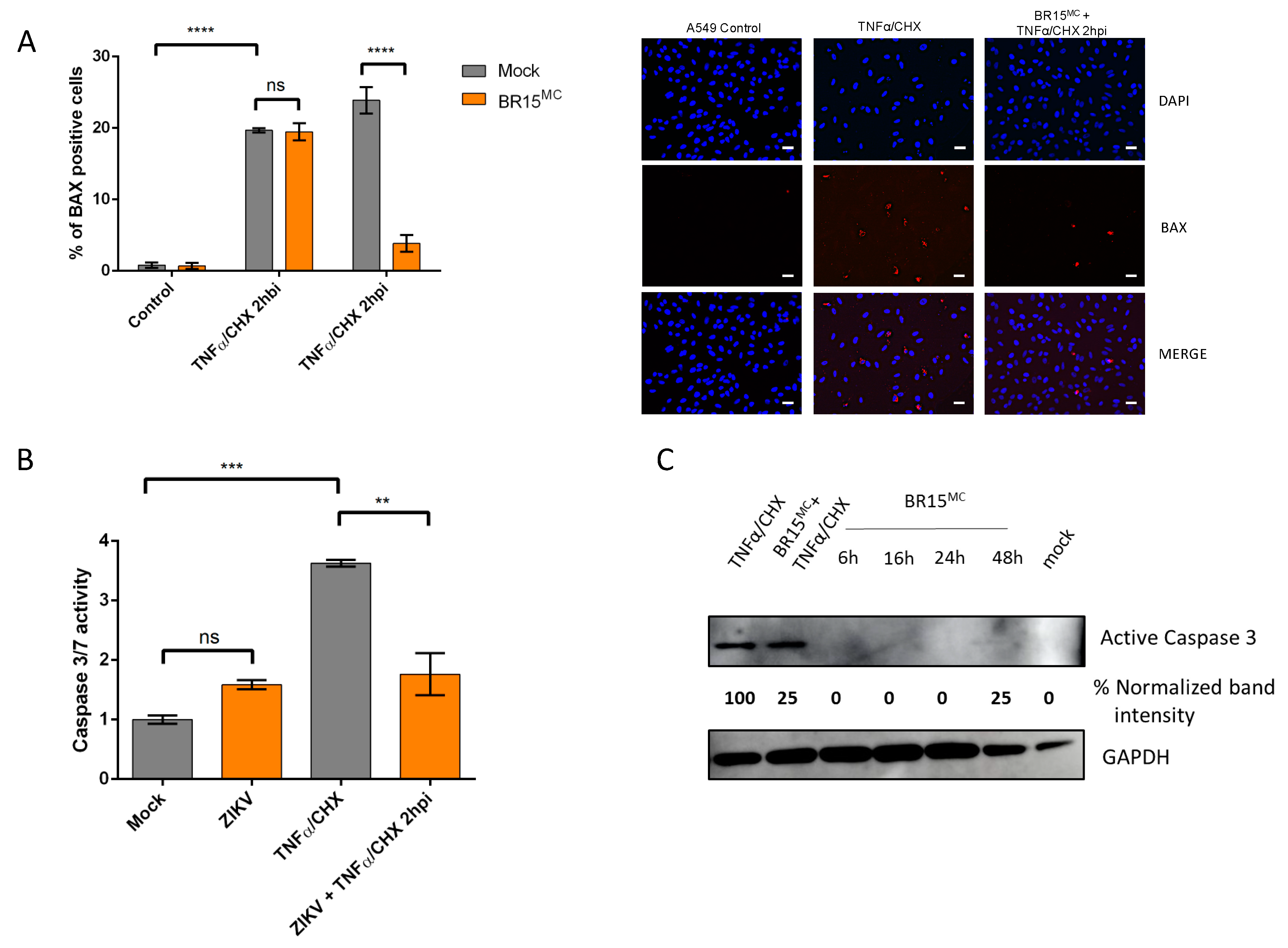
3.2.1. ZIKV Provides Protection Against Death Receptor Mediated Cell Death
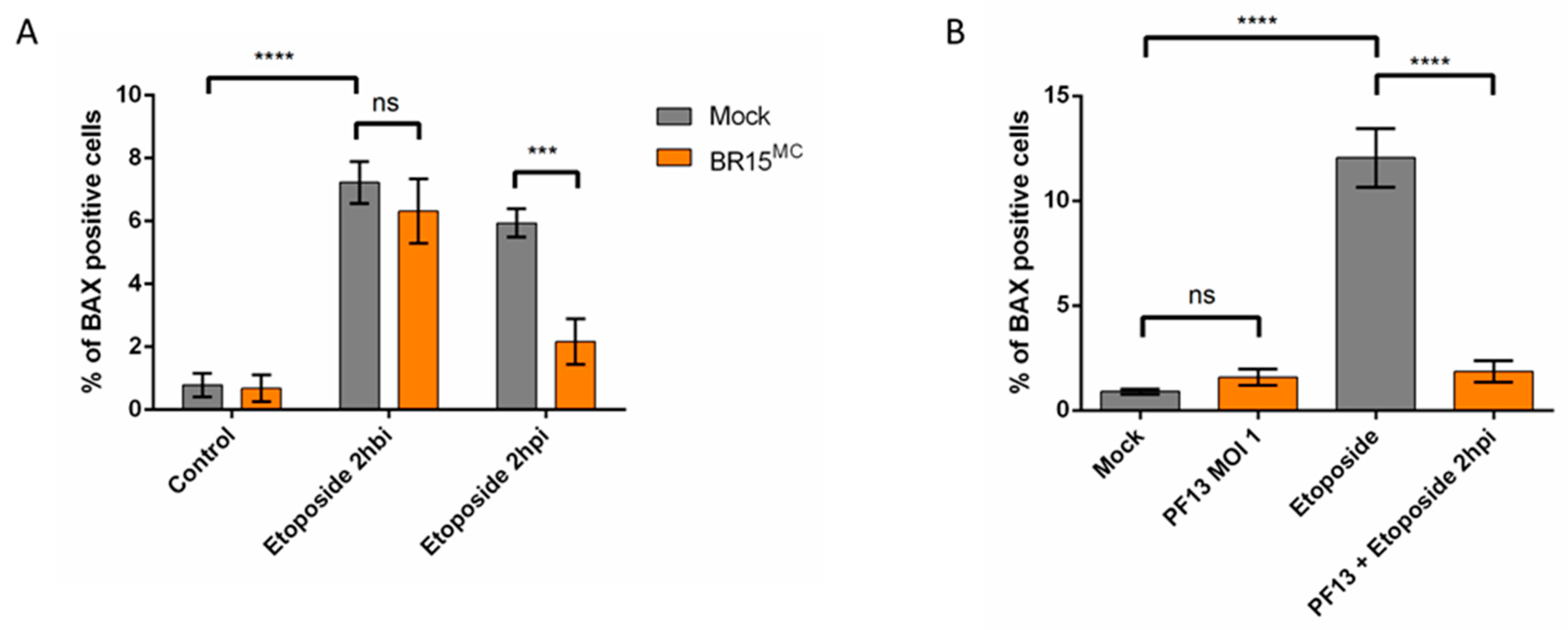
3.2.2. ZIKV Provides Protection Against Intrinsically Induced Cell Death
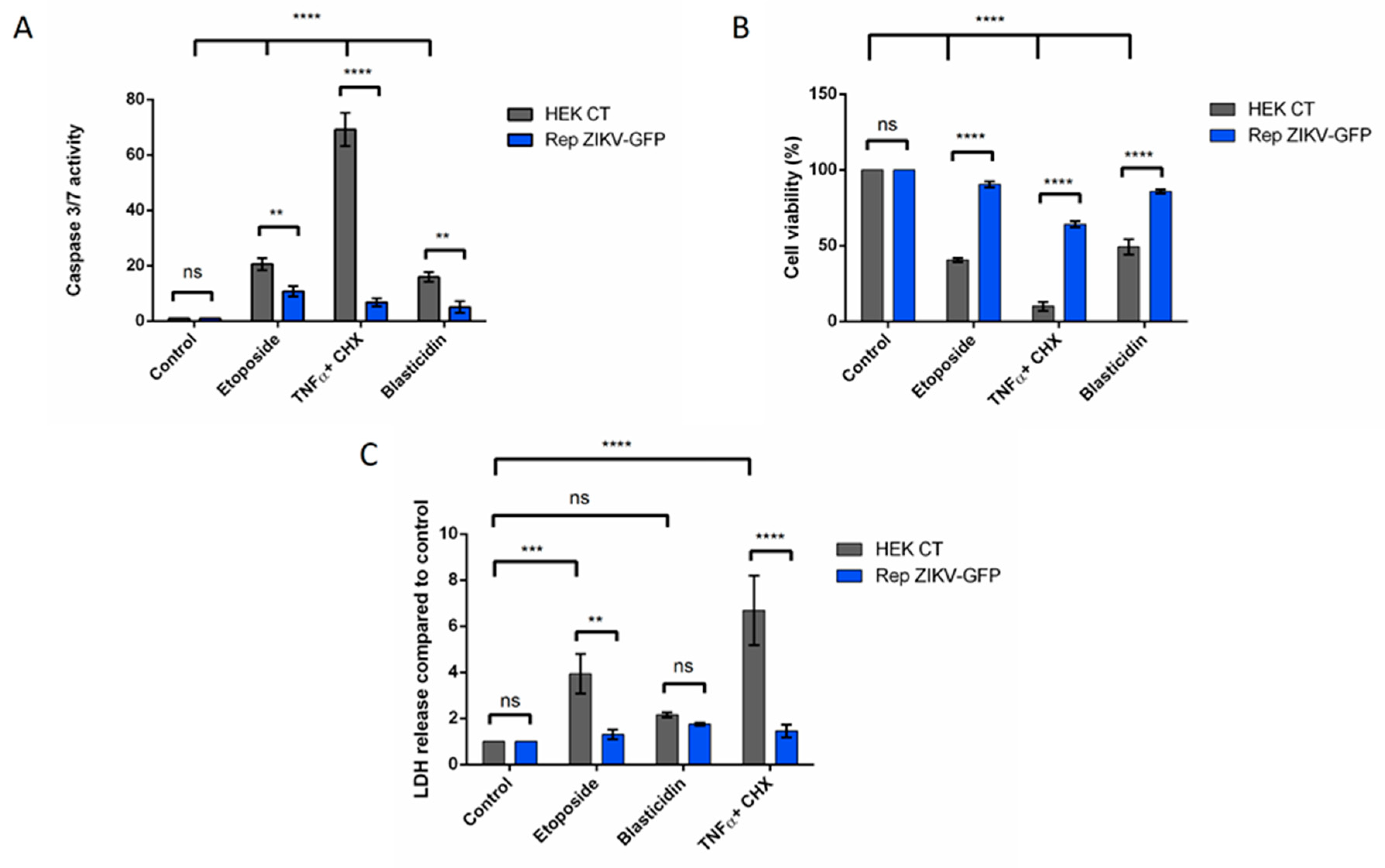
3.3. Apoptosis is Repressed Through ZIKV Replication
3.3.1. Cells Stably Expressing a ZIKV Replicon are Protected from Intrinsically and Extrinsically Mediated Apoptosis
3.3.2. A549 Cells Transiently Expressing a ZIKV Replicon are Protected from Different Intrinsically Induced Apoptosis
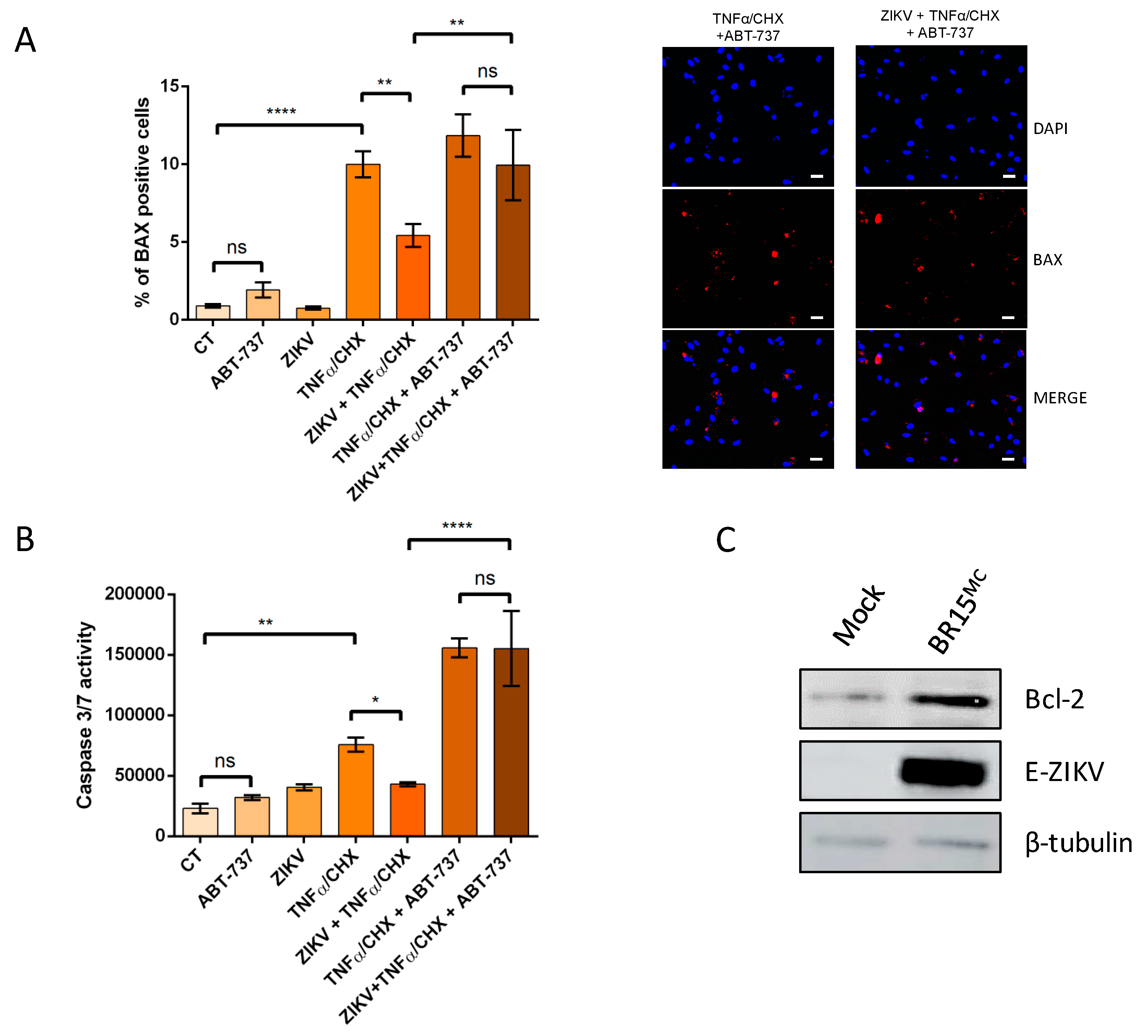
3.4. ZIKV Promotes an Anti-Apoptotic Prevailing Status in Infected Cells Through the Bcl-2 Family Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paixão, E.S.; Barreto, F. History, Epidemiology, and Clinical Manifestations of Zika: A Systematic Review. Am. J. Public Health 2016, 106, 606–612. [Google Scholar]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Lover, A.A. Zika virus and microcephaly. Lancet Infect. Dis. 2016, 16, 1331–1332. [Google Scholar] [CrossRef][Green Version]
- McKenzie, B.A.; Wilson, A.E.; Zohdy, S. Aedes albopictus is a competent vector of Zika virus: A meta-analysis. PLoS ONE 2019. [Google Scholar] [CrossRef]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic Characterization of Zika Virus Strains: Geographic Expansion of the Asian Lineage. PLoS Negl. Trop. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Milano, T.; Alcantara, L.C.J.; Carcangiu, L.; Cella, E.; Lai, A.; Lo Presti, A.; Pascarella, S.; Zehender, G.; Angeletti, S.; et al. Zika Virus spreading in South America: Evolutionary analysis of emerging neutralizing resistant Phe279Ser strains. Asian Pac. J. Trop. Med. 2016, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Roulston, A.; Marcellus, R.C.; Branton, P.E. Viruses and Apoptosis. Annu. Rev. Microbiol. 1999, 53, 577–628. [Google Scholar] [CrossRef] [PubMed]
- Krejbich-Trotot, P.; Denizot, M.; Hoarau, J.-J.; Jaffar-Bandjee, M.-C.; Das, T.; Gasque, P. Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-L.; Stollar, V. Alphaviruses and apoptosis. Int. Rev. Immunol. 2004, 23, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Suzuki, T.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Matsuura, Y. Regulation of Apoptosis during Flavivirus Infection. Viruses 2017. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanakantham, R.; Cheong, Y.K.; Ng, M.-L. West Nile virus capsid protein interaction with importin and HDM2 protein is regulated by protein kinase C-mediated phosphorylation. Microbes Infect. 2010, 12, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, M.D.; Hobman, T.C. The West Nile Virus Capsid Protein Blocks Apoptosis through a Phosphatidylinositol 3-Kinase-Dependent Mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Missé, D.; Gadea, G.; Viranaicken, W.; Desprès, P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-β production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef]
- Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.M.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C.; et al. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018. [Google Scholar] [CrossRef]
- Van der Linden, V.; Pessoa, A.; Dobyns, W.; Barkovich, A.J.; van der Linden Júnior, H.; Filho, E.L.R.; Ribeiro, E.M.; de Carvalho Leal, M.; de Araújo Coimbra, P.P.; de Fátima Viana Vasco Aragão, M.; et al. Description of 13 Infants Born During October 2015-January 2016 With Congenital Zika Virus Infection Without Microcephaly at Birth-Brazil. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1343–1348. [Google Scholar] [CrossRef]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Desprès, P. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef]
- Bos, S.; Viranaicken, W.; Turpin, J.; El-Kalamouni, C.; Roche, M.; Krejbich-Trotot, P.; Desprès, P.; Gadea, G. The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 2018, 516, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Nativel, B.; Marimoutou, M.; Thon-Hon, V.G.; Gunasekaran, M.K.; Andries, J.; Stanislas, G.; Planesse, C.; Da Silva, C.R.; Césari, M.; Iwema, T.; et al. Soluble HMGB1 is a novel adipokine stimulating IL-6 secretion through RAGE receptor in SW872 preadipocyte cell line: Contribution to chronic inflammation in fat tissue. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Viranaicken, W.; Gasmi, L.; Chaumet, A.; Durieux, C.; Georget, V.; Denoulet, P.; Larcher, J.-C. L-Ilf3 and L-NF90 Traffic to the Nucleolus Granular Component: Alternatively-Spliced Exon 3 Encodes a Nucleolar Localization Motif. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Antwerp, D.J.V.; Martin, S.J.; Verma, I.M.; Green, D.R. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998, 8, 107–111. [Google Scholar] [CrossRef]
- El Kalamouni, C.; Frumence, E.; Bos, S.; Turpin, J.; Nativel, B.; Harrabi, W.; Wilkinson, D.A.; Meilhac, O.; Gadea, G.; Desprès, P.; et al. Subversion of the Heme Oxygenase-1 Antiviral Activity by Zika Virus. Viruses 2018, 11. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Souza, B.S.F.; Sampaio, G.L.A.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.R.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.V.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.-F.; Xu, Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ghouzzi, V.E.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-J.; Liao, C.-L.; Lin, Y.-L. Flavivirus Activates Phosphatidylinositol 3-Kinase Signaling To Block Caspase-Dependent Apoptotic Cell Death at the Early Stage of Virus Infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef]
- Liu, J.; Li, Q.; Li, X.; Qiu, Z.; Li, A.; Liang, W.; Chen, H.; Cai, X.; Chen, X.; Duan, X.; et al. Zika Virus Envelope Protein induces G2/M Cell Cycle Arrest and Apoptosis via an Intrinsic Cell Death Signaling Pathway in Neuroendocrine PC12 Cells. Int. J. Biol. Sci. 2018, 14, 1099–1108. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017. [Google Scholar] [CrossRef]
- Barnard, T.R.; Rajah, M.M.; Sagan, S.M. Contemporary Zika Virus Isolates Induce More dsRNA and Produce More Negative-Strand Intermediate in Human Astrocytoma Cells. Viruses 2018. [Google Scholar] [CrossRef]
- Göertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Rooswinkel, R.W.; van de Kooij, B.; de Vries, E.; Paauwe, M.; Braster, R.; Verheij, M.; Borst, J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014, 123, 2806–2815. [Google Scholar] [CrossRef]
- Suzuki, T.; Okamoto, T.; Katoh, H.; Sugiyama, Y.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Fukuhara, T.; Ikawa, M.; et al. Infection with flaviviruses requires BCLXL for cell survival. PLOS Pathog. 2018. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpin, J.; Frumence, E.; Desprès, P.; Viranaicken, W.; Krejbich-Trotot, P. The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins. Cells 2019, 8, 1338. https://doi.org/10.3390/cells8111338
Turpin J, Frumence E, Desprès P, Viranaicken W, Krejbich-Trotot P. The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins. Cells. 2019; 8(11):1338. https://doi.org/10.3390/cells8111338
Chicago/Turabian StyleTurpin, Jonathan, Etienne Frumence, Philippe Desprès, Wildriss Viranaicken, and Pascale Krejbich-Trotot. 2019. "The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins" Cells 8, no. 11: 1338. https://doi.org/10.3390/cells8111338
APA StyleTurpin, J., Frumence, E., Desprès, P., Viranaicken, W., & Krejbich-Trotot, P. (2019). The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins. Cells, 8(11), 1338. https://doi.org/10.3390/cells8111338